

Abstract
Excessive increases in intracellular [Ca2+] in skeletal muscle fibres cause failure of excitation–contraction coupling by disrupting communication between the dihydropyridine receptors in the transverse tubular system and the Ca2+ release channels (RyRs) in the sarcoplasmic reticulum (SR), but the exact mechanism is unknown. Previous work suggested a possible role of Ca2+-dependent proteolysis in this uncoupling process but found no proteolysis of the dihydropyridine receptors, RyRs or triadin. Junctophilin-1 (JP1; ∼90 kDa) stabilizes close apposition of the transverse tubular system and SR membranes in adult skeletal muscle; its C-terminal end is embedded in the SR and its N-terminal associates with the transverse tubular system membrane. Exposure of skeletal muscle homogenates to precisely set [Ca2+] revealed that JP1 undergoes Ca2+-dependent proteolysis over the physiological [Ca2+] range in tandem with autolytic activation of endogenous μ-calpain. Cleavage of JP1 occurs close to the C-terminal, yielding a ∼75 kDa diffusible fragment and a fixed ∼15 kDa fragment. Depolarization-induced force responses in rat skinned fibres were abolished following 1 min exposure to 40 μm Ca2+, with accompanying loss of full-length JP1. Supraphysiological stimulation of rat skeletal muscle in vitro by repeated tetanic stimulation in 30 mm caffeine also produced marked proteolysis of JP1 (and not RyR1). In dystrophic mdx mice, JP1 proteolysis is seen in limb muscles at 4 and not at 10 weeks of age. Junctophilin-2 in cardiac and skeletal muscle also undergoes Ca2+-dependent proteolysis, and junctophilin-2 levels are reduced following cardiac ischaemia–reperfusion. Junctophilin proteolysis may contribute to skeletal muscle weakness and cardiac dysfunction in a range of circumstances.
Key points
If skeletal muscle fibres are subjected to excessive activation, or stretched whilst contracting, they subsequently display long-term reductions in their force response, apparently due in part to structural or molecular changes at the triad junction, where excitation of the surface membrane triggers Ca2+ release from the internal Ca2+ store.
The changes appear to be due to excessive or prolonged increases in intracellular Ca2+ levels, which activate Ca2+-dependent proteases known as calpains, but their target proteins are currently unknown.
This study shows that excessive muscle stimulation, or directly raising intracellular Ca2+ levels, causes calpain activation in tandem with proteolysis of junctophilin, a key protein thought to hold the triad junction together.
Proteolysis of junctophilin is also seen in muscle of mice with muscular dystrophy and in cardiac muscle following ischaemic damage.
Proteolysis of junctophilin may be a major factor causing muscle weakness and cardiac dysfunction in a range of circumstances.
Introduction
Excitation–contraction (EC) coupling in adult skeletal muscle depends on signal transmission across the triad junction, where the transverse tubular (T-) system abuts the terminal cisternae of the sarcoplasmic reticulum (SR). Action potentials activate the dihydropyridine receptors (DHPRs) in the T-system, which by some protein–protein interaction(s) open the Ca2+ release channels/ryanodine receptors (RyRs) in the adjacent SR, leading to a rise in cytoplasmic [Ca2+] and consequent contraction. A range of experiments have shown that excessive or prolonged increases in cytoplasmic [Ca2+] can lead to disruption of coupling between the DHPRs and the RyRs. Elevated intracellular [Ca2+] completely abolishes depolarization-induced and action potential-induced force responses in skinned muscle fibres of the rat and mouse in a concentration- and time-dependent manner, with half-maximal effects seen by applying either comparatively high Ca2+ levels for a short time (e.g. ∼25 μm for 10 s) or lower levels for a longer time (e.g. ∼5 μm for 180 s; Lamb et al. 1995; Verburg et al. 2006). The failure is evidently caused by disrupted coupling between the DHPRs and the RyRs, because the T-system shows no indication of chronic depolarization, and Ca2+ can still be released from the SR by directly activating the RyRs with caffeine or low cytoplasmic [Mg2+]. Furthermore, electron microscopy shows structural alterations in the triad junctions in the Ca2+-treated fibres (Lamb et al. 1995). Coupling can also be disrupted both in intact (Chin & Allen, 1996) and in skinned fibres (Lamb et al. 1995; Verburg et al. 2006) by evoking excessive or prolonged release of SR Ca2+, such as by stimulating fibres simultaneously with caffeine and depolarization. Eccentric (or lengthening) contraction in muscle fibres also produces disrupted coupling between the DHPRs and RyRs (Warren et al. 1993; Balnave & Allen, 1995; Ingalls et al. 1998; Corona et al. 2010) and is associated with increased influx of extracellular Ca2+ and raised resting [Ca2+] (Ingalls et al. 1998; Allen, 2004). Furthermore, eccentric contractions cause Ca2+-dependent disruption of EC coupling more readily in dystrophic mdx mice than in wild-type mice (Yeung et al. 2005) and, interestingly, action potential-induced Ca2+ release in isolated mdx muscle fibres is impaired to some degree even without eccentric stimulation (Woods et al. 2004).
The mechanism underlying the Ca2+-dependent disruption of DHPR–RyR communication is unknown, but a range of evidence suggests possible involvement of the Ca2+-dependent protease, μ-calpain. Firstly, the [Ca2+] range over which the disruption occurs is similar to that for activation of μ-calpain (Murphy et al. 2006a,b). Secondly, low temperature (3°C) and acidic conditions (pH 5.8) greatly slow the rate of coupling disruption (Lamb et al. 1995), similar to their effects on μ-calpain activity (Inomata et al. 1984; Ono et al. 2004), and Sr2+ activates both processes at a similar concentration, which is ∼10- to 20-fold higher than for Ca2+ in both cases (Inomata et al. 1984; Lamb et al. 1995). Thirdly, calpain has been observed to localize near the triadic SR (Gilchrist et al. 1992), and applying exogenous μ-calpain cleaves triad junctions (Kim et al. 1990) and disrupts EC coupling in a Ca2+-dependent manner (Verburg et al. 2009). However, to date no triadic protein has been identified as undergoing Ca2+-dependent proteolysis in tandem with the coupling disruption and, in particular, the DHPR, RyR and triadin were all found to remain intact in fibres where coupling had been fully disrupted (Lamb et al. 1995).
It is currently thought that the junctophilin family of proteins are primarily responsible for mediating the close contact between the SR and the cell surface/T-tubules in both skeletal and cardiac muscle cells (Takeshima et al. 2000; Ito et al. 2001; Komazaki et al. 2002; Hirata et al. 2006). Both the junctophilin-1 (JP1) and junctophilin-2 (JP2) isoforms are localized at the triad junction in skeletal muscle fibres, whereas it is primarily JP2 that is present at diad/triad junctions in cardiac muscle cells (Takeshima et al. 2000; Ito et al. 2001). Knockout of JP2 is embryonically lethal because the SR in the cardiac cells fails to form normal diadic junctions, and the mice experience cardiac arrest (Takeshima et al. 2000). Mice with JP1 knocked out have abnormal skeletal muscle triad formation and die at birth despite the presence of JP2, leading to the proposal that JP2 is sufficient for some degree of coupling between the SR and the cell surface/T-tubule, whereas JP1 is needed for proper formation and maintenance of the tight triad junctions characteristic of mature skeletal muscle (Ito et al. 2001; Komazaki et al. 2002). The structures of JP1 and JP2 are highly conserved across mammalian species, with the proteins observed to run on SDS-PAGE at ∼90 and ∼100 kDa, respectively (Takeshima et al. 2000; Phimister et al. 2007; Golini et al. 2011). In addition to having a structural role at the triad junction in skeletal muscle, JP1 interacts with the RyRs in a conformationally sensitive manner (Phimister et al. 2007), and both JP1 and JP2 interact with the DHPRs (Golini et al. 2011). Both JP1 and JP2 insert into the SR membrane via a transmembrane region in their C-terminal, and their N-terminal ends associate with the surface/T-tubule membrane via so-called ‘MORN’ (membrane occupation and recognition nexus) motifs.
The present study examines whether JP1 and JP2 undergo Ca2+-dependent proteolysis and whether this occurs in conjunction with the disruption of EC coupling in Ca2+-treated skinned fibres and in other intact muscle situations, such as following supraphysiological stimulation of skeletal muscle and ischaemia–reperfusion in heart.
Methods
All animal experiments were carried out in accordance with the Australian National Health and Medical Research Council's ‘Australian code of practice for care and use of animals for scientific purposes’ and with approval of the Animal Ethics Committees of La Trobe University, Victoria University and University of Melbourne. Male Long–Evans hooded rats (12 rats, 5–12 months old) were killed by isoflurane overdose (4% v/v) in a glass chamber and the extensor digitorum longus (EDL) muscle and heart removed. C57BL/10 and mdx (C57BL/10ScSn-Dmdmdx) mice (20 in total; Animal Resource Centre Canning Vale, Western Australia) were killed by cardiac excision while anaesthetized with pentobarbitone sodium (Rhone Merieux, Pinkenba, Queensland, Australia; 40 mg kg−1 i.p.) and the tibialis anterior (TA) and diaphragm muscles removed. For whole-heart preparations, 16 additional adult (16-week-old) Sprague–Dawley rats were anaesthetized with sodium pentobarbitone (60 mg kg−1 i.p.) and injected with sodium heparin (200 IU) via the femoral vein, and the hearts rapidly excised (see below).
Human vastus lateralis biopsy samples from four men aged 18–40 years (two healthy and two diagnosed as having limb girdle muscular dystrophy 2A with dysfunctional calpain-3) were obtained from the Neurogenetic Biospeciman Bank at The Children's Hospital at Westmead (Sydney, NSW, Australia); the results described later were similar for all four cases. All procedures conformed to the Declaration of Helsinki; informed consent was obtained in writing from all subjects, and the Human Research Ethics Committees of the Children's Hospital at Westmead and La Trobe University approved the studies.
Skinned fibre solutions
Chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. Potassium hexamethylene-diamine-tetraacetate (K-HDTA) solution contained (mm): HDTA, 50; EGTA, 0.05; total ATP, 8; creatine phosphate (CrP), 10; Na+, 36; K+, 126; total magnesium, 8.6; total Hepes, 90 (pH 7.1) and 0.1 μm free Ca2+. Skinned fibres were depolarized in Na-HDTA solution (K-HDTA solution with all K+ replaced by Na+). Rigor solution was similar to K-HDTA solution but with 66 mm HDTA, 0.25 mm EGTA, 1.5 mM magnesium and no ATP or CrP, and Ca-rigor solution had 40 μm free Ca2+.
Skinned fibre preparations
Rat EDL muscles were pinned at resting length under paraffin oil at ∼10°C. Individual fibre segments were mechanically skinned and mounted at 120% of resting length on a force transducer (AME801; SensoNor, Horten, Norway), placed in a bath with K-HDTA solution at room temperature (RT), and depolarized and Ca2+ treated as previously (Lamb et al. 1995). Force responses were recorded using Powerlab 4/20 hardware (ADInstruments, Sydney, NSW, Australia).
In vitro stimulation of rat muscles
Extensor digitorum longus muscles were attached to a force transducer in a bath with Krebs–Ringer solution bubbled with 95% O2 and 5% CO2 (pH ∼7.4) at RT, and stimulated by field stimulation (1 ms pulses). Isometric force responses were recorded as for skinned fibres. Supraphysiological stimulation was elicited by adding 30 mm caffeine to the bathing solution and 1 min later applying repeated 50 Hz stimulation (for 0.5 s every 2 s) until tetanic force decreased to <10% of initial (∼3–4 min). Superficial fibres were dissected and Western blotting was performed on groups of three fibre segments. Junctophilin-1 diffusibility was examined, as in the studies of Murphy et al. (2006b, 2011), by skinning fibres and bathing a group of three segments in 10 μl of relaxing solution for 5 min, and analysing the contents of the solution and fibre samples by Western blotting (see Fig. 3E).
Figure 3. Proteolysis of JP1 in intensively stimulated muscles.
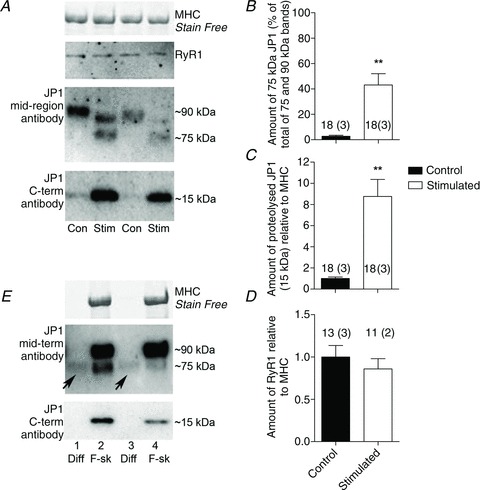
A, Western blot of JP1 in fibres from rat EDL muscles stimulated intensively in vitro in the presence of 30 mm caffeine (Stim) and from non-stimulated contralateral muscles (Con); three fibre segments were pooled in each lane. Proteins were separated on 4–15% Stain Free gels and probed for JP1 using mid-region or C-terminal antibody, and also antibody to RyR1. B, mean (+SEM) amount of 75 kDa JP1 fragment in fibre samples from Stim and Con muscles, expressed as a percentage of the total JP1 detected (i.e. the sum of the 75 and 90 kDa bands). The number of samples is shown with each bar, together with the number of muscles in parentheses (**P < 0.01, Mann–Whitney U test). C, mean (+SEM) amount of 15 kDa JP1 fragment in samples (each value was first normalized to MHC). D, mean (+SEM) amount of RyR1 in samples. E, Western blot of JP1 and the 75 and 15 kDa proteolytic fragments remaining in skinned fibre (F-sk) or diffusing into wash solution (Diff) after bathing skinned fibre segments in wash solution for 5 min (see Methods). Fibres were obtained from stimulated muscle; three skinned segments were used in each of two wash–fibre sets shown. A substantial proportion of the 75 kDa proteolytic fragments diffused into the wash solution (arrows).
Exposure of muscle homogenate to elevated [Ca2+]
Thin (10 μm) cyrosections of human skeletal muscle biopsies (six to eight sections per sample) were prepared in solutions with free Ca2+ strongly buffered to set levels in the range ∼10 nm to 500 μm with 50 mm CaEGTA–EGTA; volume to muscle mass ≥10:1) and kept at RT for 60 min. Pieces of fresh or frozen EDL muscle of rat were homogenized and treated in a similar manner.
Isolated heart preparations
Hearts (n= 8) were retrogradely perfused with oxygenated (95% O2 and 5% CO2) bicarbonate buffer (37°C, pH 7.4) in the non-recirculating Langendorff mode at a constant pressure equivalent to 73 mmHg using an STH pump controller (ADInstruments, Sydney, NSW, Australia). Bicarbonate buffer contained (mm): NaCl, 118.5; KCl, 4.7; KH2PO4, 1.18; NaHCO3, 25.0; MgCl2, 1.2; CaCl2, 1.4; and glucose, 11.1. Left ventricular pressure measurements were performed using a fluid-filled balloon connected to a pressure transducer (MLT844) and recorded on a MacLab data acquisition system (both from ADInstruments, Sydney, NSW, Australia). Following an aerobic stabilization period, hearts were subjected to 20 min global ischaemia (37°C) and 60 min reperfusion, and allowed to beat spontaneously throughout. Control hearts were perfused aerobically throughout. Hearts were frozen in liquid nitrogen at the end of the perfusion protocols and subsequently homogenized (10% w/v) in buffer containing (mm): Tris–HCl, 50.0; EGTA, 5.0; EDTA, 5.0; NaF, 5; Na3VO4, 0.5; and protease inhibitors (Roche, Basel, Switzerland) at 4°C, and added to 2× SDS sample buffer.
Western blotting
Western blotting was performed as previously described (Murphy et al. 2011). Primary antibodies used were as follows: rabbit anti-JP1 (mid-region, 40-5200, 1 in 300 dilution; or C-terminal, 40-5100, 1 in 250 dilution), rabbit anti-JP2 (40-5300, 1 in 250 dilution), mouse anti-μ-calpain antibody (1 in 1000 dilution; all from Invitrogen, Sydney, NSW, Australia), mouse anti-RyR1 (34C, Development Studies Hybridoma Bank, University of Iowa, Iowa City, IA 52242, USA; 1 in 100 dilution) and mouse anti-RyR2 (Badrilla, Leeds, UK; 1 in 300 dilution). Protein bands were visualized using west-femto chemiluminescent substrate (Thermo Fisher Scientific, Scoresby, Vic., Australia, Australia) using a Chemidoc MP (BioRad, Sydney, NSW, Australia) with ImageLab software (BioRad) for collection of images and data analyses. A three- or four-point standard curve was generated on every gel by running a range of homogenate samples encompassing the test sample range (e.g. Fig. 5D) to ensure that any differences in band densities were detectable.
Figure 5. Junctophilin-2 is proteolysed in cardiac muscle with ischaemia–reperfusion.
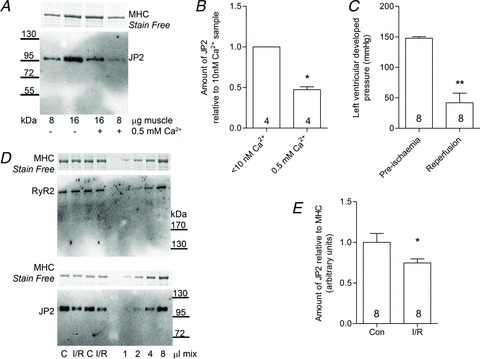
A, Western blot of JP2 in rat cardiac ventricular muscle homogenized in the presence or absence of 0.5 mm free Ca2+ for 60 min at room temperature (4–15% Stain Free gel). B, mean (+SEM) amount of JP2 remaining in ventricular samples following treatments. Data are from four independent gels (*P < 0.01, Student's one-tailed paired t test). C, left ventricular developed pressure in rat hearts before and after ischaemia–reperfusion (I/R; **P < 0.001, Student's one-tailed paired t test). D, Western blots of RyR2 and JP2 in separate portions of the same homogenates of ventricular muscle from control (C) and I/R hearts (4–15 and 10% Stain Free gels). E, mean (+SEM) amount of JP2 in control (Con) and I/R hearts (*P < 0.05, Student's one-tailed unpaired t test).
Results
Ca2+-dependent proteolysis of junctophilins in skeletal muscle
Junctophilin-1 in both human and rat skeletal muscle underwent Ca2+-dependent proteolysis by endogenous proteases when exposed to elevated [Ca2+]. Figure 1A (middle panel) shows a Western blot for JP1 in Ca2+-treated human muscle cryosections. Using an antibody to the mid-region of JP1, skeletal muscle tissue maintained in low-[Ca2+] conditions (e.g. <10 nm Ca2+) for 60 min displayed only a single band running at ∼90 kDa (normal full-length form of JP1), whereas muscle exposed to [Ca2+] higher than ∼0.5 μm showed a progressively greater loss of the 90 kDa band and the appearance of a ∼75 kDa band, which was evidently a proteolytic fragment. Western blotting for μ-calpain in these same preparations showed a matching increase in autolytic activation of μ-calpain over the same [Ca2+] range (Fig. 1A and B). Similar results were seen in four independent human muscle preparations, and also in rat skeletal muscle (not shown). Proteolysis of JP1 could also be detected using a C-terminal antibody, which showed the same Ca2+-dependent loss of the 90 kDa full-length band accompanied by formation of a ∼15 kDa C-terminal fragment (see Supraphysiological stimulation of skeletal muscles also leads to JP1 proteolysis). Junctophilin-2 in skeletal muscle underwent similar Ca2+-dependent proteolysis, with complete loss of full-length JP2 observed following a 60 min exposure to 500 μm Ca2+ at RT in four independent rat muscle preparations (e.g. Fig. 1C), although no proteolytic fragment was detected with the JP2 C-terminal antibody used, possibly because the proteolysis affected the antibody binding site.
Figure 1. Junctophilin-1 (JP1) and junctophilin-2 (JP2) undergo Ca2+-dependent proteolysis by endogenous proteases.
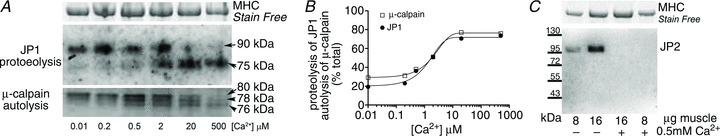
A, Western blot of human skeletal muscle cryosections following exposure to various set levels of [Ca2+] for 60 min at room temperature. Whole muscle was separated on 10% Stain Free gels and probed using a mid-region JP1 antibody, which detects full-length 90 kDa JP1 and proteolysed 75 kDa JP1. Myosin heavy chain (MHC) bands in Stain Free gel were used as a loading control (top panel). A separate gel was run to detect μ-calpain in same homogenate samples (bottom panel). Full-length μ-calpain is detected at 80 kDa, and autolyses to 78 and 76 kDa forms, indicating that it has been activated. B, the percentage proteolysis (JP1) or autolysis (μ-calpain) following exposure to the indicated [Ca2+]; data are mean values from two repeats on separate gels. C, Western blot of JP2 in rat extensor digitorum longus (EDL) muscle homogenates treated without or with 0.5 mm Ca2+ for 60 min at RT, separated on a 4–15% Stain Free gel.
Ca2+-mediated disruption of EC coupling in skinned muscle fibres
As described previously (Lamb et al. 1995; Verburg et al. 2006), depolarization-induced force responses in skinned fibres were completely and irreversibly abolished following a 1 min exposure to a free [Ca2+] of 40 μm or above (e.g. Fig. 2A). Western blotting of such Ca2+-treated fibres (Fig. 2B) revealed significant loss of full-length JP1 in fibre segments treated with 40 μm Ca2+ compared with other fibres treated in a similar manner but exposed to a solution with [Ca2+] kept at <10 nm (Fig. 2B and C). The presence of a 75 kDa proteolytic fragment of JP1 was not detected in the Western blotting of these single-fibre segments, possibly owing to detection limitations in these small samples or to further proteolysis or diffusional loss of the 75 kDa fragment (see The 75 kDa JP1 proteolytic fragment is diffusible). Figure 2D illustrates the possible role of JP1 proteolysis in Ca2+-dependent disruption of EC coupling.
Figure 2. Proteolytic loss of JP1 in skinned fibres after Ca2+ disruption of excitation–contraction (EC) coupling.
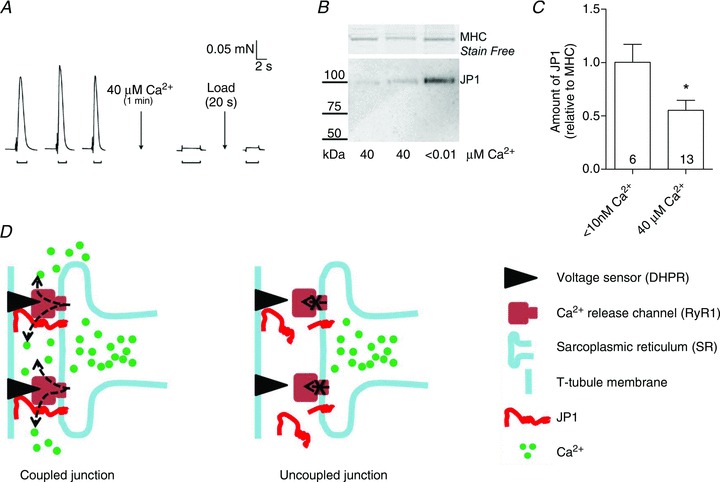
A, depolarization-induced force responses in a skinned EDL fibre from a rat were abolished after 1 min exposure to 40 μm free Ca2+ (applied in rigor conditions; see Methods) and were not restored by additional SR Ca2+ loading. B, Western blot for JP1 in the single-fibre segment shown in A (run in lane 1) and for two other segments treated with 40 μm Ca2+ or in a similar manner with Ca2+ at <10 nm. 10% Stain Free gel, with membrane blotted with mid-region JP1 antibody. C, mean (+SEM) amount of JP1 detected in individual fibre segments exposed to indicated [Ca2+] (the number of fibres is shown inside the bars). The JP1 signal for each fibre was normalized to the MHC band and is expressed relative to the mean value for fibres exposed to 10 nm Ca2+ on the same blot (three on each gel; *P < 0.05, Student's two-tailed unpaired t test). D, diagram depicting a coupled (functional) triad junction in skeletal muscle. After Ca2+ treatment, the junction becomes dysfunctional (uncoupled), possibly owing to cleavage of JP1 and loss of normal communication between the dihydropyridine receptors (DHPRs) and the ryanodine receptors (RyRs).
Supraphysiological stimulation of skeletal muscles also leads to JP1 proteolysis
Figure 3 shows that JP1 proteolysis can also be brought about by raising the intracellular Ca2+ level within intact muscle fibres by stimulation of whole muscles in vitro. Rat EDL muscles were subjected to repeated 50 Hz simulation for ∼4 min in the presence of 30 mm caffeine, in order to elevate the intracellular [Ca2+] substantially for a prolonged period (Chin & Allen, 1996). Western blotting of superficial fibres from these stimulated muscles revealed clear loss of full-length (90 kDa) JP1, accompanied by formation of both the 75 and the 15 kDa proteolytic fragments (Fig. 3A, lower two panels; Western blotting performed with three fibre segments in each lane to improve detection limits). The amount of 75 kDa fragment detected in a given sample, expressed relative to the sum of the 90 and 75 kDa bands, was ∼43% in fibre samples from the stimulated muscles and <5% in fibre samples from the non-stimulated contralateral muscles (Fig. 3B). Western blotting of the lower portion of the same gels with the JP1 C-terminal antibody showed greatly increased amounts of the 15 kDa band in the fibres from the stimulated muscle relative to those from the non-stimulated muscles (Fig. 3A and C), with the amount of the 15 kDa band being highly correlated with the appearance of the 75 kDa band detected with the mid-region antibody in the same fibre samples (r2= 0.70, P < 0.001, n= 17 fibres from three muscles, not shown). In contrast to the very substantial level of JP1 proteolysis, Western blotting of a subset of the same samples showed no evidence of proteolysis of RyR1 (e.g. Fig. 3A; only full-length RyR1 was apparent) nor significant decrease in the amount of RyR1 present in the stimulated samples (Fig. 3D). It is possible, nevertheless, that some proteolysis of RyR1 occurred but that it fell below detection limits.
The 75 kDa JP1 proteolytic fragment is diffusible
In order to investigate whether JP1 and its proteolytic fragments were diffusible or fixed inside muscle fibres, fibre segments from a supraphysiologically stimulated muscle were skinned under paraffin oil and bathed in an intracellular-like solution (‘wash solution’) for 5 min, and then Western blotting was used to examine the presence of JP1 and its fragments in the wash solution and fibre segments (see Methods and Murphy et al. 2006b). Figure 3E displays two examples showing that ∼15 and 40% of the total 75 kDa fragment present had diffused into the wash solution without any detectable washout of either full-length JP1 or the 15 kDa C-terminal fragment. The myosin heavy chain (MHC) bands in the Stain Free gel (top of Fig. 3E) confirm that the fibre segments were fully removed from the wash. These findings are consistent with full-length JP1 being embedded in the SR membrane at its C-terminal end and with proteolysis creating a potentially diffusible 75 kDa N-terminal fragment and a 15 kDa fixed C-terminal fragment (see Fig. 2D).
Proteolysed JP1 and JP2 in limb and diaphragm muscle in mdx mice
In mdx dystrophic mice, limb muscles display an acute period of degeneration and weakness at ∼3 weeks of age but then recover to near normal by ∼12 weeks, whereas the diaphragm muscle shows progressively greater deterioration and weakness throughout adulthood (Dupont-Versteegden & McCarter, 1992). In 28-day-old mdx mice, ∼60% of the JP1 in the TA muscle was in the 75 kDa proteolysed form, whereas in wild-type mice of the same age the percentage was only ∼10% (Fig. 4A and B). There was also marked activation of the μ-calpain in the 28-day-old mdx muscle (not shown). At 70 days, the extent of JP1 proteolysis in the TA muscles of the mdx mice had dropped back to levels close to that in wild-type mice. Likewise, there was a marked loss of full-length JP2 in the TA muscle of mdx mice at 28 days, but it recovered to wild type levels by 70 days of age (Fig. 4C and D). The amounts of both JP1 and JP2 in TA muscles of wild-type mice (expressed relative to MHC) increased roughly twofold between days 28 and 70 (e.g. Fig. 4D), possibly owing to the relative increase in the extensiveness of the T-system in muscle fibres as they become larger with maturity. In contrast to the recovery of JP1 levels in the TA muscle of mdx mice, marked JP1 proteolysis was observed in the diaphragm at 7 months of age, as well as activation of μ-calpain (Fig. 4E).
Figure 4. Proteolysis of JP1 and JP2 in limb mucles of 28-day-old mdx mice.
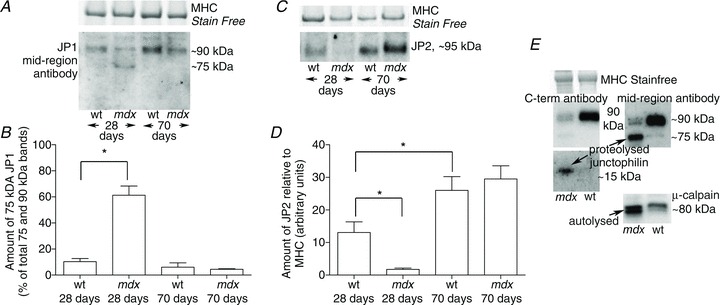
A, Western blot of JP1 in tibialis anterior muscle of mdx and age-matched wild-type (wt) mice at 28 and 70 days of age. B, mean + SEM percentage of JP1 found as the 75 kDa proteolytic fragment (n= 5 mice in each group). C and D, Western blot and mean amounts of JP2 in the same samples. E, Western blot of JP1 and μ-calpain in the diaphragm of an mdx mouse and a wild-type mouse at 7 months. *Significantly different (P < 0.05, Student's two-tailed unpaired t test).
Loss of JP2 in heart muscle following ischaemia–reperfusion
Treatment of rat cardiac ventricular tissue with 0.5 mm Ca2+ for 60 min at RT resulted in ∼50% loss of JP2 (Fig. 5A and B). Interestingly, the loss of JP2 in cardiac muscle was less than that in skeletal tissue run in parallel (e.g. Fig. 1C), perhaps due to differences in the level or location of calpain or its inhibitor, calpastatin, in the two types of tissues. Following ischaemia–reperfusion in rat hearts, left ventricular developed pressure decreased to ∼25% of the initial level (Fig. 5C), and the amount of full-length JP2 found in the hearts was significantly lower (∼25%) than in matched control hearts (Fig. 5D and E). In contrast, there was no detectable proteolytic cleavage or loss of RyR2 in the same samples (e.g. Fig. 5D).
Discussion
This study demonstrates that both JP1 and JP2 in skeletal muscle undergo Ca2+-dependent proteolysis by endogenous proteases when the intracellular [Ca2+] is raised within the physiological range for a sustained period (Fig. 1). This is the first demonstration that any members of the junctophilin family undergo such proteolysis. The proteolysis was probably brought about by the ubiquitous calpains, in particular by μ-calpain, which was found to be autolytically activated over the same Ca2+ range in the samples (Fig. 1), in agreement with our previous studies (Murphy et al. 2006a,b). It was further shown that JP1 in skeletal muscle fibres could be proteolysed by raising the intracellular [Ca2+] by supraphysiological stimulation of muscles in vitro (Fig. 3). Proteolysis of JP1 was seen in rat, mouse and human skeletal muscle and seemingly occurs at a single site near the C-terminal end, leading to formation of ∼75 kDa N-terminal and ∼15 kDa C-terminal fragments, with the latter remaining embedded in the SR membrane. The N-terminal end of JP1 is thought to bind via MORN motifs to the T-tubule membrane (Takeshima et al. 2000), but following proteolysis the 75 kDa fragment evidently becomes diffusible to some extent (Fig. 3E), and in such a situation JP1 could no longer be functioning as a physical link between the T-tubule and SR membranes (see Fig. 2D).
Thus, the loss of EC coupling that occurs in skinned fibres exposed to raised [Ca2+] (Lamb et al. 1995) can be accounted for at least in part by the apparent Ca2+-dependent proteolysis of JP1 occurring in such fibres (Fig. 2). This is also consistent with the following previous observations: (i) the Ca2+ treatment activates μ-calpain in the skinned fibres, and activated μ-calpain both disrupts EC coupling (Verburg et al. 2009) and cleaves triad junctions (Kim et al. 1990); and (ii) supraphysiological stimulation of isolated muscles or fibres activates μ-calpain (Murphy et al. 2006b) and disrupts EC coupling (Lamb et al. 1995; Chin & Allen, 1996) without proteolysis of the DHPRs or RyRs themselves. Ryanodine receptors in skinned fibres can be proteolysed by application of exogenous m-calpain in 2 mm Ca2+ for 60 min (Iino et al. 1992), but the findings here (Fig. 3) indicate that activation of the endogenous calpains in intact muscle fibres appears to have a proportionately greater effect on JP1 than on the RyRs. Disruption of coupling may nevertheless be determined by the summed effect of proteolysis of a number of different junctional proteins. The postulated change at the triad junction occurring with JP1 proteolysis (Fig 2D) may also account for the increased SR Ca2+ leakage seen after Ca2+ treatment (Lamb & Cellini, 1999), brought about by the loss of the normal inhibitory actions of the DHPRs on the RyRs.
The Ca2+-dependent proteolysis of JP1 is also likely to contribute to the disruption of EC coupling that occurs after eccentric contractions (see Introduction). Corona et al. (2010) found that levels of full-length JP1 and JP2 were reduced following eccentric contractions in mouse muscle, although they attributed this to mechanical damage to the proteins rather than to Ca2+-dependent proteolysis, because their previous experiments had found that the force loss was not prevented by calpain inhibitors or removal of extracellular Ca2+. However, using a similar preparation another group has observed that μ-calpain becomes activated with eccentric contractions (Zhang et al. 2012), and that the force loss can be reduced both by Ca2+ removal and by the calpain inhibitor, leupeptin (Zhang et al. 2008). We have previously shown that Ca2+ disruption of EC coupling in skinned fibres can be reduced with calpain inhibitors, but only if the uncoupling occurs at a low rate (Lamb et al. 1995), probably owing to the difficulty of controlling Ca2+-dependent processes within the triad junction, particularly given that the inhibitors act on calpain only after it is activated.
The Ca2+-dependent proteolysis of JP1 may also underlie reductions in Ca2+ release seen in dystrophic muscle (Woods et al. 2004; Yeung et al. 2005). In mdx mice, limb muscles display a transient period of degeneration and weakness at 3–4 weeks of age, at which time there is calpain activation and major proteolytic loss of both JP1 and JP2 (Fig. 4). In contrast to the limb muscles, the diaphragm shows progressively greater dystrophy and weakness into and throughout adulthood (Dupont-Versteegden & McCarter, 1992), and marked calpain activation and junctophilin proteolysis can be seen in mdx mice at 7 months (Fig. 4). Stretch readily induces elevated intracellular [Ca2+] in mdx muscle fibres (Yeung et al. 2005), probably due to stretch-induced production of reactive oxygen species (Khairallah et al. 2012), leading to both increased influx of extracellular Ca2+ (Whitehead et al. 2010) and increased release of SR Ca2+ (Bellinger et al. 2009). The findings here suggest that increases in intracellular [Ca2+] by either mechanism would readily lead to calpain activation and proteolysis of the junctophilins.
Finally, our results suggest that Ca2+-dependent proteolysis of JP2 may contribute to the impaired SR Ca2+ release and contractility occurring in heart muscle after ischaemia–reperfusion. Ischaemia–reperfusion in rat hearts sufficient to induce marked deterioration of left ventricular developed pressure is accompanied by increased calpain activity (Singh et al. 2012), and calpain inhibitors reduce both calpain activity and the decrease in left ventricular developed pressure (Singh & Dhalla, 2010). In the present study, a loss of JP2 following ischaemia–reperfusion was observed (Fig. 5), which might be expected to have had a deleterious effect on SR Ca2+ release, given that cardiac-specific knockdown of JP2 has been shown to result in disrupted junctional membrane complexes and reduced EC coupling gain (van Oort et al. 2011).
Concluding remarks
Our findings show that both JP1 and JP2 undergo Ca2+-dependent proteolysis in skeletal and cardiac muscle and suggest that such proteolysis may be a significant factor in reduced Ca2+ release and consequent muscle weakness and cardiac dysfunction in a range of circumstances.
Acknowledgments
We thank Maria Cellini and Heidy Latchman for technical assistance, Professor Kathy North for facilitating access to the Neurogenetic Biospeciman Bank, and the National Health & Medical Research Council of Australia for financial support (grant number 602538). The monoclonal antibody directed against RyR1 was developed by Drs J. Airey and J. Sutko and obtained from the Development Studies Hybridoma Bank, under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA.
Glossary
- CrP
creatine phosphate
- DHPR
dihydropyridine receptor
- EC coupling
excitation–contraction coupling
- EDL
extensor digitorum longus
- HDTA
hexa-methylene-diamine-tetraacetate
- JP1
junctophilin-1
- JP2
junctophilin-2
- MHC
myosin heavy chain
- MORN
membrane occupation and recognition nexus
- RT
room temperature
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- TA
tibialis anterior
- T-system
transverse tubular system
- wt
wild-type
Author contributions
R.M.M. and G.D.L. conceived and designed most of the experiments and drafted the manuscript. All authors contributed to collection, analysis and interpretation of data. All authors have approved the final version of the submitted manuscript. Biochemical analysis of all tissue and physiological measurements on skinned muscle fibres were performed at La Trobe University, and the whole heart preparation was performed at University of Melbourne.
References
- Allen DG. Skeletal muscle function: role of ionic changes in fatigue, damage and disease. Clin Exp Pharmacol Physiol. 2004;31:485–493. doi: 10.1111/j.1440-1681.2004.04032.x. [DOI] [PubMed] [Google Scholar]
- Balnave CD, Allen DG. Intracellular calcium and force in single mouse muscle fibres following repeated contractions with stretch. J Physiol. 1995;488:25–36. doi: 10.1113/jphysiol.1995.sp020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin ER, Allen DG. The role of elevations in intracellular [Ca2+] in the development of low frequency fatigue in mouse single muscle fibres. J Physiol. 1996;491:813–824. doi: 10.1113/jphysiol.1996.sp021259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona BT, Balog EM, Doyle JA, Rupp JC, Luke RC, Ingalls CP. Junctophilin damage contributes to early strength deficits and EC coupling failure after eccentric contractions. Am J Physiol Cell Physiol. 2010;298:C365–C376. doi: 10.1152/ajpcell.00365.2009. [DOI] [PubMed] [Google Scholar]
- Dupont-Versteegden EE, McCarter RJ. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve. 1992;15:1105–1110. doi: 10.1002/mus.880151008. [DOI] [PubMed] [Google Scholar]
- Gilchrist JS, Wang KK, Katz S, Belcastro AN. Calcium-activated neutral protease effects upon skeletal muscle sarcoplasmic reticulum protein structure and calcium release. J Biol Chem. 1992;267:20857–20865. [PubMed] [Google Scholar]
- Golini L, Chouabe C, Berthier C, Cusimano V, Fornaro M, Bonvallet R, Formoso L, Giacomello E, Jacquemond V, Sorrentino V. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J Biol Chem. 2011;286:43717–43725. doi: 10.1074/jbc.M111.292755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, Zhao X, Thornton A, Komazaki S, Takeshima H, Ma J, Pan Z. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Takano-Ohmuro H, Kawana Y, Endo M. Enhancement of Ca2+-induced Ca2+ release in calpain treated rabbit skinned muscle fibers. Biochem Biophys Res Commun. 1992;185:713–718. doi: 10.1016/0006-291x(92)91684-i. [DOI] [PubMed] [Google Scholar]
- Ingalls CP, Warren GL, Williams JH, Ward CW, Armstrong RB. E-C coupling failure in mouse EDL muscle after in vivo eccentric contractions. J Appl Physiol. 1998;85:58–67. doi: 10.1152/jappl.1998.85.1.58. [DOI] [PubMed] [Google Scholar]
- Inomata M, Nomoto M, Hayashi M, Nakamura M, Imahori K, Kawashima S. Comparison of low and high calcium requiring forms of the calcium-activated neutral protease (CANP) from rabbit skeletal muscle. J Biochem. 1984;95:1661–1670. doi: 10.1093/oxfordjournals.jbchem.a134779. [DOI] [PubMed] [Google Scholar]
- Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154:1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen YW, Raiteri R, Lederer WJ, Dorsey SG, Ward CW. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal. 2012;5:ra56. doi: 10.1126/scisignal.2002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KC, Caswell AH, Brunschwig JP, Brandt NR. Identification of a new subpopulation of triad junctions isolated from skeletal muscle; morphological correlations with intact muscle. J Membr Biol. 1990;113:221–235. doi: 10.1007/BF01870074. [DOI] [PubMed] [Google Scholar]
- Komazaki S, Ito K, Takeshima H, Nakamura H. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett. 2002;524:225–229. doi: 10.1016/s0014-5793(02)03042-9. [DOI] [PubMed] [Google Scholar]
- Lamb GD, Cellini MA. High intracellular [Ca2+] alters sarcoplasmic reticulum function in skinned skeletal muscle fibres of the rat. J Physiol. 1999;519:815–827. doi: 10.1111/j.1469-7793.1999.0815n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb GD, Junankar PR, Stephenson DG. Raised intracellular [Ca2+] abolishes excitation–contraction coupling in skeletal muscle fibres of rat and toad. J Physiol. 1995;489:349–362. doi: 10.1113/jphysiol.1995.sp021056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Snow RJ, Lamb GD. μ-Calpain and calpain-3 are not autolyzed with exhaustive exercise in humans. Am J Physiol Cell Physiol. 2006a;290:C116–C122. doi: 10.1152/ajpcell.00291.2005. [DOI] [PubMed] [Google Scholar]
- Murphy RM, Verburg E, Lamb GD. Ca2+ activation of diffusible and bound pools of μ-calpain in rat skeletal muscle. J Physiol. 2006b;576:595–612. doi: 10.1113/jphysiol.2006.114090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Vissing K, Latchman H, Lamboley C, McKenna MJ, Overgaard K, Lamb GD. Activation of skeletal muscle calpain-3 by eccentric exercise in humans does not result in its translocation to the nucleus or cytosol. J Appl Physiol. 2011;111:1448–1458. doi: 10.1152/japplphysiol.00441.2011. [DOI] [PubMed] [Google Scholar]
- Ono Y, Kakinuma K, Torii F, Irie A, Nakagawa K, Labeit S, Abe K, Suzuki K, Sorimachi H. Possible regulation of the conventional calpain system by skeletal muscle-specific calpain, p94/calpain 3. J Biol Chem. 2004;279:2761–2771. doi: 10.1074/jbc.M308789200. [DOI] [PubMed] [Google Scholar]
- Phimister AJ, Lango J, Lee EH, Ernst-Russell MA, Takeshima H, Ma J, Allen PD, Pessah IN. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J Biol Chem. 2007;282:8667–8677. doi: 10.1074/jbc.M609936200. [DOI] [PubMed] [Google Scholar]
- Singh RB, Dhalla NS. Ischemia–reperfusion-induced changes in sarcolemmal Na+/K+-ATPase are due to the activation of calpain in the heart. Can J Physiol Pharmacol. 2010;88:388–397. doi: 10.1139/Y10-012. [DOI] [PubMed] [Google Scholar]
- Singh RB, Hryshko L, Freed D, Dhalla NS. Activation of proteolytic enzymes and depression of the sarcolemmal Na+/K+-ATPase in ischemia–reperfused heart may be mediated through oxidative stress. Can J Physiol Pharmacol. 2012;90:249–260. doi: 10.1139/y11-128. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg E, Dutka TL, Lamb GD. Long-lasting muscle fatigue: partial disruption of excitation-contraction coupling by elevated cytosolic Ca2+ concentration during contractions. Am J Physiol Cell Physiol. 2006;290:C1199–C1208. doi: 10.1152/ajpcell.00469.2005. [DOI] [PubMed] [Google Scholar]
- Verburg E, Murphy RM, Richard I, Lamb GD. Involvement of calpains in Ca2+-induced disruption of excitation-contraction coupling in mammalian skeletal muscle fibers. Am J Physiol Cell Physiol. 2009;296:C1115–C1122. doi: 10.1152/ajpcell.00008.2009. [DOI] [PubMed] [Google Scholar]
- Warren GL, Lowe DA, Hayes DA, Karwoski CJ, Prior BM, Armstrong RB. Excitation failure in eccentric contraction-induced injury of mouse soleus muscle. J Physiol. 1993;468:487–499. doi: 10.1113/jphysiol.1993.sp019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PloS One. 2010;5:e15354. doi: 10.1371/journal.pone.0015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CE, Novo D, DiFranco M, Vergara JL. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J Physiol. 2004;557:59–75. doi: 10.1113/jphysiol.2004.061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BT, Whitehead NP, Gervasio OL, Reardon TF, Vale M, Fatkin D, Dietrich A, Yeung EW, Allen DG. Pathways of Ca2+ entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol. 2012;112:2077–2086. doi: 10.1152/japplphysiol.00770.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BT, Yeung SS, Allen DG, Qin L, Yeung EW. Role of the calcium-calpain pathway in cytoskeletal damage after eccentric contractions. J Appl Physiol. 2008;105:352–357. doi: 10.1152/japplphysiol.90320.2008. [DOI] [PubMed] [Google Scholar]