

Abstract
Intermedin (IMD) is a cardiac peptide synthesized in a prepro form, which undergoes a series of proteolytic cleavages and amidations to yield the active forms of 47 (IMD1−47) and 40 amino acids (IMD8−47). There are several lines of evidence of increased IMD expression in rat models of cardiac pathologies, including congestive heart failure and ischaemia; however, its myocardial effects upon cardiac disease remain unexplored. With this in mind, we investigated the direct effects of increasing concentrations of IMD1−47 (10−10 to10−6 m) on contraction and relaxation of left ventricular (LV) papillary muscles from two rat models of chronic pressure overload, one induced by transverse aortic constriction (TAC), the other by nitric oxide (NO) deficiency due to chronic NO synthase inhibition (NG-nitro-l-arginine, l-NAME), and respective controls (Sham and Ctrl). In TAC and l-NAME rats, exogenous administration of IMD1−47 elicited concentration-dependent positive inotropic and lusitropic effects. By contrast, in Sham and Ctrl rats, IMD1−47 induced a negative inotropic response without a significant effect on relaxation. Both TAC and l-NAME rats presented LV hypertrophy, elevated LV systolic pressures, preserved systolic function and elevated peroxynitrite levels. In the normal myocardium (Ctrl and Sham), IMD1−47 induced a 3-fold increase of endothelial nitric oxide synthase (eNOS) phosphorylation at Ser1177, indicating enhanced eNOS activity. In TAC and l-NAME rats, eNOS phosphorylation was increased at baseline, and its response to IMD1−47 was blunted. In addition, the distinct myocardial response to IMD1−47 was accompanied by distinct subcellular mechanisms. While in Sham rats the addition of IMD1−47 induced the phosphorylation of cardiac troponin I due to NO/cGMP activation, in TAC rats IMD1−47 induced phospholamban phosphorylation possibly associated with cAMP/protein kinase A activation. Therefore, we demonstrated for the first time a reversed myocardial response to IMD1−47 neurohumoral stimulation due to impairment of eNOS activation in TAC and l-NAME rats. These results not only reveal the distinct myocardial effects and subcellular mechanisms for IMD1−47 in normal and hypertrophic hearts, but also highlight the potential pathophysiological relevance of cardiac endothelial dysfunction in neurohumoral myocardial action.
Key points
Intermedin (IMD) is a cardiac endogenous peptide upregulated in several models of heart disease.
We show a depressant effect of IMD1−47 on contractility of the normal myocardium, mediated by increased nitric oxide (NO) production due to endothelial nitric oxide synthase (eNOS) phosphorylation.
In rat models of cardiac hypertrophy or NO deficiency, IMD1−47 enhances contractility and hastens relaxation associated with phospholamban phosphorylation.
This opposing response in normal and diseased myocardium is due to impairment of IMD1−47-induced eNOS phosphorylation.
The results reveal distinct myocardial effects and subcellular mechanisms for IMD1−47 in the hypertrophic heart, suggesting a key role of cardiac endothelial dysfunction with potential pathophysiological relevance.
Introduction
The last decade has witnessed an upsurge in the identification of novel neurohormones with a putative role in heart failure (HF). Among these, the newest member of the calcitonin gene-related peptide (CGRP) family, named intermedin (IMD) or adrenomedullin-II, is gaining increasing interest (Roh et al. 2004; Takei et al. 2004). Similar to other peptidic hormones, IMD is synthesized in a prepro form (prepro-IMD101−147), which undergoes a series of proteolytic cleavages and amidation to produce the active forms of a 47 amino-acid peptide (IMD1−47) and a shorter 40 amino-acid peptide (IMD8−47) (Roh et al. 2004). IMD and its receptors are widely expressed, especially in the pituitary gland and gastrointestinal tract. In the heart, its expression is scarce in the normal myocardium, while there are several reports of increased expression in rat models of congestive HF (Hirose et al. 2008), arterial hypertension (Zhao et al. 2006; Bell et al. 2008) and myocardial ischaemia (Zhang et al. 2009), suggesting a pathophysiological role of IMD in heart disease. Furthermore, there is in vitro evidence of anti-hypertrophic (Pan et al. 2005) and anti-fibrotic effects (Yang et al. 2009) of IMD, as well as attenuation of cardiac ischaemia/reperfusion injury (Yang et al. 2005), indicating a cardioprotective action of IMD similar to adrenomedullin (AM) (Kato et al. 2003; Bell et al. 2010).
Regarding the cardiovascular function of IMD, this peptide has similar biological actions to AM in the vasculature (e.g. vasodilator, vasodepressor) (Takei et al. 2004) and in the heart both were shown to modulate myocardial function (Pan et al. 2005; Fontes-Sousa et al. 2009; Pires et al. 2012). However, in contrast to the normal heart where its action has been comprehensively studied (Roh et al. 2004; Takei et al. 2004; Munzel et al. 2011; Pires et al. 2012) the myocardial effects of IMD in the diseased heart remain unexplored. With this in mind, we investigated in the present study the myocardial action of IMD1−47 and the underlying mechanisms in well-known rat models of left ventricular (LV) hypertrophy due to pressure overload (transverse aortic constriction, TAC) or nitric oxide (NO) deficiency associated with chronic NO synthase inhibition
Methods
Ethical approval
The animal handling and experiments were subjected to the Portuguese law on animal welfare, were carried out according to the guidelines laid down by the Faculty of Medicine of the University of Porto (Porto, Portugal) animal welfare committee, and conform to the principles of UK regulations, as described in Drummond (2009).
Animals
Transverse aortic constriction
Male Wistar–Han rats (Rattus norvegicus) of 7 weeks of age were anaesthetized with sevoflurane (2%) and connected to a rodent ventilator (Topo, small animal ventilator, Kent Scientific, Torrington, CT, USA) after tracheal intubation. The different muscle layers were spread apart and cut through a small incision at the level of the second intercostal space. After isolation of the aortic arch, a 3–0 non-absorbable silk suture was placed around the aorta and tied together with a 25G needle (TAC, n = 9). The needle was only used to standardize the constriction and therefore was immediately removed after tying the suture. The chest cavity was then closed with 4–0 polypropylene suture and all layers of muscle repositioned and skin closed with 4–0 silk suture. Sham animals underwent a similar surgery, but without the final tightening of the silk suture (Sham, n = 12). At 28 weeks of age the rats underwent either haemodynamic evaluation or papillary muscle experiments as illustrated in Fig. 1. After haemodynamic evaluation they were killed via an injection of sodium pentobarbital (200 mg kg−1, i.p.). The heart was then excised and weighed, and myocardial samples were collected for molecular and histological studies. Similarly, the rats undergoing papillary experiments were killed with sodium pentobarbital and the LV papillary muscles isolated for functional studies as explained below.
Figure 1. Diagram of the experimental protocol.
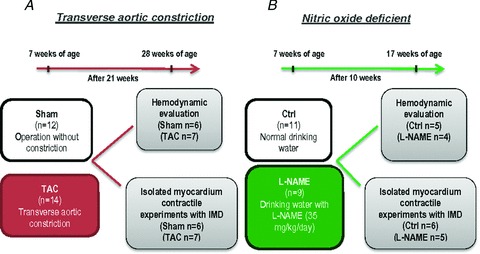
A, transverse aortic constriction model (TAC) protocol. B, nitric oxide deficient (l-NAME) rat protocol.
NO-deficient rats (l-NAME)
Seven-week-old male Wistar–Han rats were assigned to receive: (1) drinking water (controls; n = 11) or (2) NO synthase inhibitor NG-nitro-l-arginine (l-NAME, 35 mg kg−1 day−1, n = 9) in drinking water for 10 weeks. At 17 weeks of age the rats were submitted to either haemodynamic evaluation or papillary muscle experiments after which the animals were killed as described above. The heart was then excised and weighed, and myocardial samples were collected for molecular and histological studies and the LV papillary muscles isolated for functional studies as explained below (Fig. 1).
LV haemodynamics
A 1.4Fr microtip pressure–volume catheter (SPR-838; Millar Instruments, Houston, TX, USA), externally calibrated and balanced with an MPV-300 Millar control unit, was inserted through an apical puncture wound into the LV cavity. Prior to insertion in the LV, the catheter was soaked in a distilled water bath at 37°C for 30 min. The catheter was connected to an MVPS-300 conductance system (Millar Instruments), coupled to a PowerLab16/30 converter (AD Instruments, Colorado Springs, CO, USA) and a personal computer for data acquisition. After 10 min of stabilization, data were recorded continuously, in mmHg and units of conductance (RVU, relative volume units).
Isolated papillary muscle experiments
After cardiectomy, the LV was opened, the papillary muscles were dissected free from the LV wall using a dissecting microscope in a bath with Krebs–Ringer (KR) solution (composition in mm: 98 NaCl, 4.7 KCl, 2.4 MgSO4, 1.2 KH2PO4, 4.5 glucose, 1.8 CaCl2, 17 NaHCO3, 15 sodium pyruvate, 5 sodium acetate), cardioplegic 2,3-butanedione monoxime (BDM, 3%) and 5% newborn calf serum. After dissection, papillary muscles were mounted vertically in a 10 ml plexi glass organ bath containing KR solution, BDM and the serum at 30°C. The lower muscular end was fixed in a phosphorbronze clip and the upper tendinous end was attached to an electromagnetic length-tension transducer (University of Antwerp, Belgium). Preload was initially set between 3 and 4 mN according to the muscle dimensions. The preparations were stimulated at 0.6 Hz with a voltage of 10% above threshold (typically 30–60 mV) by rectangular pulses of 5 ms duration through two platinum electrodes arranged longitudinally alongside the entire muscle. After 10 min, bathing solutions were replaced by corresponding serum-KR solution without BDM at which time the muscle started to contract. Fifteen minutes later, bathing solution was replaced by corresponding serum-free KR solution. During the next 15–30 min the muscles stabilized. Finally, the muscles were stretched to a length at which active force development was maximal (Lmax). Protocols were initiated after obtaining two similar isotonic and isometric control twitches separated by a 5 min interval. At the end of the experiment the muscles were removed, lightly blotted and weighed. Muscle cross-sectional area was calculated by dividing the weight of the muscle by its length at Lmax. A cylindrical shape and a specific density of 1.0 were assumed. Muscle tension was then expressed as force normalized per cross-sectional area (mN mm−2).
Experimental protocols
The effects of increasing concentrations of intermedin (IMD1−47; 10−10 to 10−6 m) on myocardial contraction and relaxation parameters were studied in rat papillary muscles from the different animal groups (TAC, Sham, l-NAME and Ctrl). Muscle twitches were recorded after a stable response was obtained. Isotonic and isometric twitches were recorded and analysed. Selected parameters include active tension (AT, mN mm−2) and maximal velocities of tension rise (dT/dtmax, mN mm−2 s−1) and decline (dT/dtmin, mN mm−2 s−1). At the end of the experiments papillary muscles were immediately frozen in liquid nitrogen and stored in a freezer at −80°C until use for protein quantification by Western blot analysis.
Western blot analysis
Western blot analysis was applied to evaluate levels of total cardiac troponin I (t-cTnI), phosphorylated cTnI at Ser23/24 (p-cTnI), phospholamban (PLB), phosphorylated PLB at Ser16 (p-PLB), total endothelial nitric oxide synthase (eNOS) and phosphorylated eNOS at Ser1177 (p-eNOS) in LV papillary muscles: (1) under control conditions without IMD (n = 4, experiments performed two to three times), and (2) after treatment with increasing concentrations of IMD (n = 4, experiments performed two to three times). Briefly, tissues were homogenized on ice in 1 ml RIPA lysis buffer containing phenylmethylsulfonyl fluoride (1 mm), aprotonin (10 g ml−1), leupeptin (10 μg ml−1) and pepstatin (10 μg ml−1) all from Sigma Chemicals (St Louis, MO, USA) as the protease inhibitors. Tissue was then centrifuged at 14,000 g for 20 min at 4°C. The supernatants were collected and total protein concentration was determined. Samples containing 40 μg of protein were loaded on a 12% SDS-PAGE gel for cTnI, 15% for PLB or 6% for eNOS, run and electroblotted onto polyvinylidene difluoride membrane. Prestained molecular weight marker proteins were used as standards for the SDS-PAGE. Ponceau staining was performed to verify the quality of the transfer and to ensure equal protein loading. Blots were blocked in 5% non-fat skimmed milk in PBS for 1 h, treated overnight with antibody against the different proteins (eNOS, Cell Signaling Technology, Inc., Danvers, MA, USA, ref. 9572; PLB, Abcam Inc., Cambridge, MA, USA, ref. ab15000; cTnI, Cell Signaling Technology, ref. 4002S; β-actin, Cell Signaling Technology, ref. 4967; p-eNOS, Cell Signaling Technology, ref. 9571; p-PLB, Abcam, ref. ab85146; p-cTnI, Cell Signaling Technology, ref. 4004S) followed by incubation with alkaline phosphatase secondary antibodies for 1 h. Immunoblots were developed with an ECF Western blotting detection system (GE Healthcare, Little Chalfont, UK). Protein content was determined using a Bio-Rad protein assay kit.
Real-time PCR
Total RNA was extracted from tissue samples using Tripure (Roche, Mannheim, Germany). RT-PCR was performed with total RNA, followed by real-time PCR analyses using the SYBR Green method in a LightCycler 2.0 (Roche). Specific primers for GTP cyclohydrolase 1 (GTPCH-1) are given in Table 1. Normalization of samples was performed against GAPDH.
Table 1.
Real-time primer sequences (all 5′ to 3′)
| Forward | Reverse | |
|---|---|---|
| GAPDH | GAAACCCATCACCATCTTCCA | ACCCCATTTGATGTTAGCGG |
| gTPch-1 | TGGAGAAGCCGCGGGGTGTA | AGTAAGCGGCCGCCAGGTTG |
NOS activity
For determination of eNOS activity (Stratagene, La Jolla, CA, USA, ref. 204500) l-arginine to l-citrulline conversion was assayed in LV myocardial homogenates as described previously (Moens et al. 2008). Briefly, LV samples were homogenized in a solution of Tris-HCl (250 mM, pH 7.4), EDTA (10 mM) and EGTA (10 mM) and spun at full speed for 5 min at 4°C. The supernatant was incubated in NADPH (10 M), l-[3H]arginine (ref. NET1123250UC, from Perkin Elmer, Waltham, MA, USA; 1 mCi ml−1), CaCl2 (6 mM), Tris-HCl (50 mM, pH 7.4), tetrahydrobiopterin (6 mM), flavin adenine dinucleotide (2 mM) and flavin mononucleotide (2 mM) for 60 min at 24°C. As instructed by the kit manufacturer the reaction was stopped with Hepes (50 mM, pH 5.5), EDTA (5 mM) and added equilibrated resin, which binds to arginine. This solution was then transferred into a spin cup and spun at full speed for 30 s. The supernatant radioactivity, which represented the l-citrulline, was measured by liquid scintillation counting. To determine the ratio of unreacted arginine to l-citrulline, 400 μl of elution buffer was added to the spin cup, the spin cup was placed in a microcentrifuge tube and spun at full speed for 30 s. Enzyme activity was expressed as l-citrulline production in pmol (mg of protein)−1 h−1.
Quantification of nitrotyrosine
Nitrotyrosine, a biomarker of peroxynitrite formation, was determined in the LV myocardium by estimating the levels of protein nitration using immunochemical methods. For immunostaining, sections (6 μm) of LV were washed with PBS and fixed in ice-cold acetone, for 10 min. Sections were then permeabilized for 10 min in 1% Triton X-100 in PBS, pH 7.4, and blocked with 10% goat serum, for 30 min. Primary antibodies were diluted in PBS containing 0.02% bovine serum albumin (PBS/BSA). The primary antibody anti-nitrotyrosine was added and the sections were incubated overnight at 4°C. After incubation, the sections were extensively washed with PBS/BSA solution. Sections were then incubated with the secondary antibodies, diluted in PBS/BSA for 1 h. The coverslips were washed before mounting with Glycergel Dako mounting medium (Dako, Carpinteria, CA, USA). Immunostained sections were visualized with a Leica DMIRE200 fluorescence microscope. Sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole), examined and photographed. Microscope and camera settings were kept constant for all preparations. Fluorescence was quantified using ImageJ (1.40g; NIH, http://rsb.info.nih.gov/ij/).
Chemicals and solutions
All chemicals were obtained from Sigma, with the exception of IMD, which corresponds to the active form of the 47 amino-acid peptide (IMD1−47) that was obtained from Bachem (Bubendorf, Switzerland), eNOS activity kit from Stratagene (La Jolla, CA, USA) and [3H]arginine which was obtained from Perkin Elmer. The antibodies were obtained from Cell Signaling Technology Inc. (eNOS, cTnI) and Abcam (PLB). The stock solutions, including IMD, were prepared in distilled water and stored as frozen aliquots at −20°C until use.
Statistical analysis
GraphPad Prism 5.0 software was used for data analysis. Values are presented as means ± standard error of the mean (SEM) and n represents the number of animals or papillary muscles. Differences in treatment effects among the different animal groups were evaluated using a two-way analysis of variance (ANOVA). When significant differences were detected with any of the ANOVA tests, Dunnett's test was selected to perform pairwise multiple comparisons, with P < 0.05 being considered significant. In all other experiments, statistical significance was assessed using a two-tailed Student's unpaired t test with P < 0.05 being considered significant.
Results
Characterization of animal models
Both TAC and l-NAME rats developed LV hypertrophy as indicated by an 18 ± 2 and 12 ± 3% increase in heart weight/body weight (HW/BW) versus respective controls (Table 2). In addition, LV haemodynamic evaluation showed that, in comparison with respective controls, TAC and l-NAME rats presented a significant increase in LV peak systolic pressure, but only TAC showed elevated end-diastolic pressure and slower LV relaxation (prolonged tau). Conversely, both pressure overload groups presented no significant changes in LV contractility (Table 2), suggesting normal systolic function.
Table 2.
Morphometric and haemodynamic parameters of the different groups
| Sham | TAC | Ctrl | l-NAME | |
|---|---|---|---|---|
| HW/BW (g kg−1) | 2.9 ± 0.1 | 3.4 ± 0.2* | 2.3 ± 0.1 | 2.7 ± 0.2** |
| Heart rate (beats min−1) | 403 ± 11 | 386 ± 17 | 410 ± 19 | 378 ± 21 |
| LVSP (mmHg) | 120 ± 5 | 158 ± 9* | 110 ± 3 | 147 ± 12** |
| LVEDP (mmHg) | 4.0 ± 0.4 | 10.0 ± 2.0* | 3.7 ± 0.4 | 5.6 ± 1.0 |
| dP/dtmax (mmHg s−1) | 7559 ± 143 | 7234 ± 629 | 6612 ± 430 | 7131 ± 900 |
| dP/dtmin (mmHg s−1) | −9436 ± 474 | −8065 ± 948 | −8714 ± 2 | −9382 ± 1583 |
| tau | 8 ± 0.3 | 12 ± 1.0* | 9 ± 2.0 | 10 ± 1.0 |
All data are means ± SEM. The number of animals for haemodynamic data were 4–7, and for morphometric data 7–8 animals. HW, heart weight; BW, body weight; LVSP, left ventricle systolic pressure; LVEDP, left ventricle diastolic pressure; dP/dtmax, maximum velocity of pressure rise; dP/dtmin, maximum velocity of pressure decline; tau, time constant index of LV relaxation. *P < 0.05 vs. Sham, **P < 0.05 vs. Ctrl.
IMD1−47 concentration response in LV papillary muscles
Mean values of contractile and relaxation parameters in papillary muscles from the different animal groups were globally similar in all experimental protocols and are summarized in Table 3. Only tHR was higher in l-NAME animals, as expected by the lack of relaxation hastening by NO in this experimental group.
Table 3.
Summary of contractile parameters at baseline in all groups
| Sham (n = 6) | TAC (n = 7) | Ctrl (n = 6) | l-NAME (n = 5) | |
|---|---|---|---|---|
| AT (mN mm−2) | 15 ± 2 | 16 ± 4 | 18 ± 3 | 23 ± 8 |
| dT/dtmax (mN mm−2 s−1) | 141 ± 35 | 198 ± 53 | 199 ± 20 | 230 ± 81 |
| dT/dtmin (mN mm−2 s−1) | −71 ± 10 | −99 ± 20 | −112 ± 9 | −115 ± 39 |
| PS (%Lmax) | 0.1 ± 0.01 | 0.08 ± 0.01 | 0.08 ± 0.01 | 0.1 ± 0.01 |
| dL/dtmax (Lmax s−1) | 1.5 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 |
| dL/dtmin (Lmax s−1) | −1.7 ± 0.3 | −1.3 ± 0.2 | −1.2 ± 0.2 | −1.2 ± 0.2 |
| tHR (ms) | 292 ± 34 | 257 ± 16 | 265 ± 21 | 318 ± 19* |
All data are means ± SEM. AT, active tension; dT/dtmax, maximal velocity of tension rise; dT/dtmin, maximal velocity of tension decline; PS, peak isotonic shortening; dL/dtmax, maximum velocity of shortening; dL/dtmin, maximum velocity of lengthening; tHR, time to half relaxation; *P < 0.05 vs. Ctrl.
Upon IMD1−47 addition to isolated LV papillary muscles from Sham rats we observed a decrease in AT of 11 ± 5% and in the rate of force generation (dT/dtmax) of 5 ± 4% and no significant effect in relaxation parameters (Fig. 2). Similar results were observed in Ctrl rats (AT −12 ± 4%, dT/dtmax−7 ± 4%). By contrast, in LV papillary muscles from TAC rats IMD1−47 induced a rise in contractility characterized by an increase of 11 ± 3% in AT and of 20 ± 3% in dT/dtmax (Fig. 2A and B), as well as a hastening effect on relaxation evident by an increase of 23 ± 4% in the rate of relaxation (dT/dtmin; Fig. 2D). In l-NAME rats the response to IMD1−47 was similar to that of TAC rats (AT 13 ± 3%, dT/dtmax 20 ± 3%, dT/dtmin 31 ± 6%; Fig. 2A–C). Therefore, these results demonstrate reversal of IMD1−47 myocardial action in TAC and l-NAME rats into positive inotropic and lusitropic effects when compared with Sham and Ctrl, respectively, in which IMD1−47 had a negative inotropic effect.
Figure 2. Distinct effect of increasing concentrations of intermedin in LV papillary muscles.
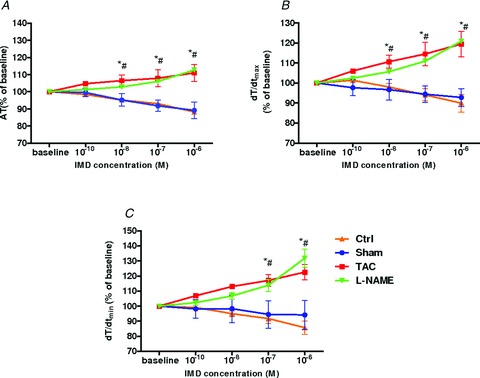
Increasing concentrations of intermedin (IMD, 10−10 to 10−6 m) in LV papillary muscles from: Sham (n = 6), TAC rats (n = 7), Ctrl (n = 6) and l-NAME (n = 5) rats. A–C, effect on: active tension (AT) (A), peak rate of tension rise (dT/dtmax) (B) and peak rate of tension decline (dT/dtmin) (C). *P < 0.05 vs. Sham; #P < 0.05 vs. Ctrl.
Cardiac endothelial dysfunction in TAC rats
As we have previously shown that IMD1−47 myocardial action depends of endothelial NO production, we examined whether rats with LV hypertrophy presented endothelial dysfunction. In comparison with Sham, TAC animals showed no significant differences in eNOS protein expression (Fig. 3A). However, nitrotyrosine levels were increased (Fig. 3C), indicating elevation of peroxynitrite in LV myocardium of TAC and l-NAME rats. In addition to oxidative stress, eNOS uncoupling can also be due to a decrease of BH4 ((6R-)5,6,7,8-tetrahydro-l-biopterin). We observed no significant differences in GTPCH-1 levels between groups (Fig. 3B); as GTPCH-1 is the rate-limiting enzyme in de novo synthesis of BH4 this result suggests no alterations in BH4 production. However, analysis of eNOS activity at baseline by quantification of conversion of l-arginine to l-citruline revealed a significant reduction of eNOS activity in TAC rats relative to Sham rats (Fig. 3D).
Figure 3.
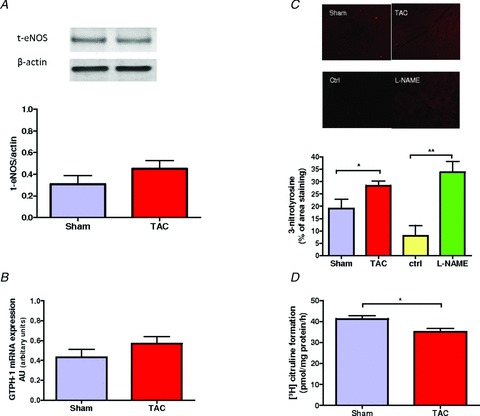
A, Western blot analyses of total eNOS protein (total-eNOS) expression in LV papillary muscles from Sham and TAC rats. B, immunohistochemical staining of cross-sections of LV myocardium from Sham, Ctrl, TAC and l-NAME rats (n = 5–6); the last two groups presented increased nitrotyrosine levels when compared to respective controls. C, levels of GTPCH-1 mRNA expression were unchanged in TAC compared to Sham. D, eNOS activity based on l-citruline formation is decreased in TAC rats. *P < 0.05 vs. Sham.
Activation of eNOS by IMD1−47
Papillary muscles from Sham rats treated with IMD1−47 and analysed by immunoblots presented a 3-fold increase in eNOS phosphorylation at Ser1177. In TAC rats, eNOS phosphorylation levels were increased at baseline and IMD1−47 failed to further increase it (Fig. 4A). The immunoblots from the l-NAME rats showed a similar trend (Fig. 4B). These results suggest impairment of IMD1−47-induced eNOS activation through phosphorylation in both TAC and l-NAME rats.
Figure 4.
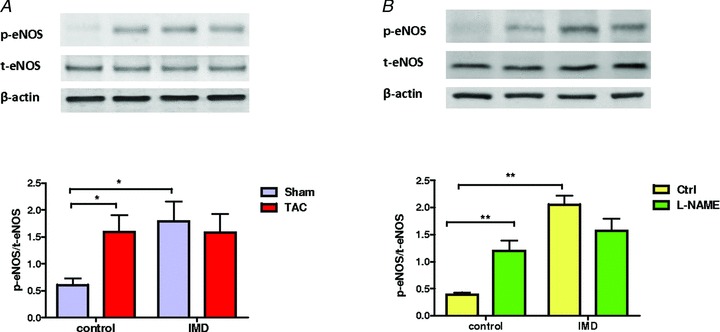
A and B, Western blot analyses of eNOS phosphorylation at Ser1177 (p-eNOS) and total-eNOS (t-eNOS) in LV papillary muscles from (A) Sham and TAC, (B) Ctrl and l-NAME rats, after isolated papillary muscle experiments, without IMD (control) and upon stimulation with IMD (IMD). In both control groups (Sham and Ctrl) IMD induced an increase in the p-eNOS/total-eNOS ratio in LV papillary muscles, and this was blunted in both pressure-overloaded groups. *P < 0.05 vs. Sham, #P < 0.05 vs. Ctrl.
Phosphorylation of regulatory proteins by IMD1−47
Papillary muscles treated with increasing concentrations of IMD1−47 from Sham rats and analysed by immunoblots presented, in accordance with previous reports (Pires et al. 2012), a 3-fold increase in p-cTnI, when compared with untreated muscles (Fig. 5A). Conversely, in LV papillary muscles from TAC and l-NAME rats, p-cTnI levels were not significantly altered by exposure to IMD1−47 (Fig. 5A). However, in TAC animals, but not the Sham group, IMD1−47 significantly increased p-PLB (Fig. 5B). This suggests differential signalling pathway activation, accompanied by distinct protein phosphorylation. However, in the l-NAME rats although there was a trend for an increase in PLB phosphorylation upon IMD1−47, this was not statistically significant, suggesting the possible involvement of other regulatory proteins.
Figure 5.
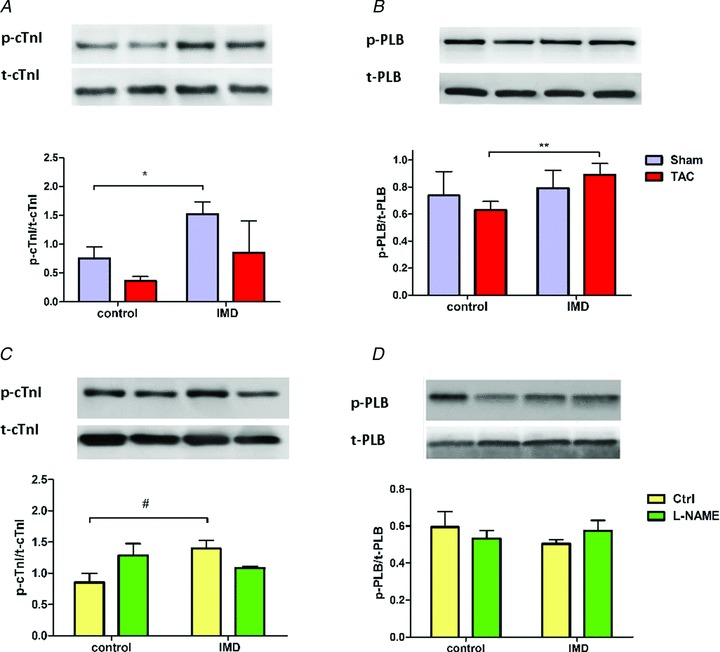
A–D, myofilament phosphorylation in LV papillary muscles from (A and B) Sham and TAC, (C and D) Ctrl and l-NAME rats, after isolated papillary muscle experiments, without IMD (control) and upon stimulation with IMD (IMD). Upon IMD there was an increase in cardiac troponin I (cTnI) phosphorylation in both Sham and Ctrl, but this was blunted in TAC (A) and l-NAME rats (C). There was an increase in phospholamban (PLB) phosphorylation upon IMD treatment in TAC rats but not in Sham (B); l-NAME rats demonstrated a similar tendency but this was not statistically significant (D). *P < 0.05 vs. Sham, #P < 0.05 vs. Ctrl, **P < 0.05 vs. TAC + IMD.
Discussion
The present study demonstrated striking differences in the myocardial response to direct neurohumoral stimulation with IMD1−47 between normal hearts and those that were subject to chronic LV pressure overload, due either to TAC banding or to chronic inhibition of NO synthase. Whereas in the normal myocardium IMD1−47 had an inhibitory effect on contractility, in the overloaded myocardium IMD1−47 induced a significant increase in contractility and a powerful hastening effect on relaxation. As underlying mechanisms, our findings indicate that reversed IMD myocardial actions in TAC rats are due to cardiac endothelial dysfunction, characterized by blunted IMD-induced eNOS activation. At the subcellular level, the overloaded myocardium presented, in response to IMD1−47, impaired cTnI phosphorylation and enhanced PLB phosphorylation.
Role of eNOS in IMD1−47 myocardial effects
The role of NO in the modulation of cardiac function has been established, based mainly on studies that used exogenous NO donors to evaluate its effects. In these circumstances, a negative inotropic effect has been reported for high doses of NO donors and a positive inotropic action for low doses (Mohan et al. 1995; Shah & MacCarthy, 2000). In cardiac tissue, as in the vasculature, one of the major NO producers is eNOS. The normal function of this enzyme is dependent on its homodimer form, which conversely may be altered into a monomeric uncoupled format under pathological conditions, such as in HF (Moens et al. 2008). In fact, increasing evidence points to a key function of eNOS uncoupling in the pathophysiology of HF, namely in pressure overload models in which it has been associated with myocardial remodelling and fibrosis (Takimoto et al. 2005; Moens et al. 2008). However, the action of endogenous NO and eNOS activation in the myocardial function in disease settings remains poorly understood. In accordance with previous reports, we observed in rats with pressure overload induced by TAC a reduction of eNOS enzyme activity, as indicated by the reduced conversion of l-arginine to l-citruline. In addition, LV myocardium from TAC rats presented increased levels of nitrotyrosine, an indicator of increased peroxynitrite, despite unchanged protein levels of eNOS and unaltered mRNA expression of the rate-limiting enzyme for BH4 production, namely GTPCH-1 (Thony et al. 2000). These observations suggest reduced eNOS activity in TAC rats. In addition, eNOS enzyme activity and subcellular localization are also intimately controlled by post-translational modifications including phosphorylation, nitrosylation and acylation, especially upon stimulation by extracellular stimuli. For instance, eNOS phosphorylation at Ser1177, Ser635 or Ser617 is stimulatory while at Thr495 and Ser116 is inhibitory (Bauer et al. 2003; Mount et al. 2007). In the present study, we observed in normal hearts a 3-fold increase in eNOS phosphorylation at Ser1177 upon IMD1−47 treatment, thus suggesting IMD stimulation of eNOS activity. Interestingly, TAC rats presented elevated eNOS phosphorylation at baseline, while IMD1−47 failed to further increase it. At first glance elevated levels of phosphorylated eNOS before IMD1−47 addition may seem contradictory with the reduced eNOS activity in the LV of TAC rats; however, its worth mentioning that similar observations were previously reported in TAC mice (Moens et al. 2009). These observations, allied to our results, raise the possibility that elevated eNOS phosphorylation due to neurohumoral activation could be a characteristic of HF, and a potential mechanisms of cardiac endothelial dysfunction in these conditions.
Further evidence supporting the role of decreased eNOS activity in the reversal of IMD1−47 myocardial action comes from the similar responses to IMD1−47 in both TAC and NO chronic deficient rat models observed herein.
Signalling pathways and regulatory proteins
Several neurohumoral agents act on the myocardium inducing a negative inotropic effect by activation of NO release, for example substance P (Mohan et al. 1995) and AM (Fontes-Sousa et al. 2009). Similarly, there are several reports of IMD induction of NO release from the endothelium (Yang et al. 2005; Kandilci et al. 2008). Such NO release was previously shown by us to play a critical role in mediating the myocardial effects of IMD1−47 in healthy animals (Pires et al. 2012). The myocardial signal transduction pathways activated by NO can be divided into cGMP-mediated (e.g. reduction of myofilament desensitization and modulation of sarcolemmal Ca2+) and cGMP-independent (e.g. direct reaction of NO with thiol and other protein residues). Concerning cGMP-mediated NO actions, the main myocardial targets are cGMP-dependent protein kinases, which are responsible for phosphorylation of various proteins (Shah & MacCarthy, 2000). In fact, there are several reports of NO-induced decrease in contractility due to cGMP-mediated reduction of Ca2+ myofilament responsiveness, as a result of cTnI phosphorylation (Kojda & Kottenberg, 1999; Shah & MacCarthy, 2000). In the normal myocardium and in accordance with our previous report (Pires et al. 2012), IMD1−47 induced a negative inotropic effect accompanied by an increment in cTnI phosphorylation. Conversely, in TAC rats, IMD1−47 induced an increase in contractility and relaxation rate, associated with enhanced phosphorylation of PLB at Ser16. These distinct effects of IMD1−47 on myocardial function and phosphorylation of sarcomeric proteins suggests IMD1−47 activation of an alternative pathway in the pressure-overloaded myocardium. The most probable is activation of cAMP/protein kinase A (PKA) pathway, as there are several reports of PLB phosphorylation by PKA at Ser16 following, for example, adrenergic activation. This post-translational modification is sufficient to remove the inhibitory effect of PLB on SERCA (sarcoplasmic/endoplasmic reticulum calcium ATPase), which ultimately results in increased sarcoplasmic reticulum Ca+2 uptake with enhanced cardiomyocyte contractility and relaxation (Chu et al. 2000). Consonant with this is the previous observation of a blunted IMD1−47-positive contractile effect in l-NAME-treated muscles upon PKA inhibition (Pires et al. 2012). In addition, the prior report from Dong et al. (2006) of a PKA-mediated positive inotropic effect of IMD1−47 in isolated murine cardiomyocytes also fits in our findings. However, we cannot exclude the possible involvement of other regulatory proteins, as in the l-NAME rats there was a tendency for an increase in PLB phosphorylation upon IMD1−47, but which did not reach statistical significance.
Conclusion
This is the first report to demonstrate opposite myocardial action of IMD1−47 between normal and pressure-overloaded myocardium, suggesting a potential stimulatory effect of IMD1−47 on cardiac function during heart disease.
In addition, these results add novel details to the pathophysiological importance of eNOS function in myocardial response to neurohumoral activation and suggest impairment of eNOS activation by phosphorylation as a potential new mechanism of cardiac endothelial dysfunction in heart disease.
Acknowledgments
This work was supported by the Portuguese Foundation for Science and Technology Grants PEst-C/SAU/UI0051/2011 through the Cardiovascular R&D Unit and by European Commission Grant FP7-Health-2010; MEDIA-261409. A.L.P. received financial support from Fundação para a Ciência e a Tecnologia Grant SFRH/BD/19544/2004.
Glossary
- AM
adrenomedullin
- AT
active tension
- BDM
cardioplegic 2,3-butanedione monoxime
- BH4
(6R-)5,6,7,8-tetrahydro-l-biopterin
- BSA
bovine serum albumin
- BW
body weight
- cTnI
cardiac troponin I
- eNOS
endothelial nitric oxide synthase
- GTPCH-1
guanosine triphosphate cyclohydrolase-1
- HF
heart failure
- HW
heart weight
- IMD1−47
intermedin
- KR solution
Krebs–Ringer solution
- l-NAME
NG-nitro-l-arginine
- LV
left ventricle
- NO
nitric oxide
- NOS
nitric oxide synthase
- PLB
phospholamban
- TAC
transverse aortic constriction
Author contributions
Animal models, real-time PCR, papillary muscles and haemodynamic experiments were undertaken at the Department of Physiology and Cardiovascular Surgery of the Faculty of Medicine of the University of Porto. Western blot, immunohistochemistry and NO synthase measurement were performed in the Department of Physiology of the Faculty of Medicine of the University of Coimbra. A.L.P. led in the collection and analysis of data, conception and design of the project, and wrote the manuscript. She performed haemodynamic LV evaluation, TAC surgery, and papillary muscles experiments with help from M.P. and B.S.A. The real-time experiments were performed by S.P. Western blot, immunohistochemistry and NO synthase measurement were performed by M.P., C.S. and A.L.P. Finally, A.F.L.-M. critically reviewed the conception and design of the project, analysis and interpretation of the data, and manuscript elaboration. R.M.S. and C.S. critically reviewed previous drafts of the manuscript. All authors gave final approval of the version to be published.
References
- Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, Sessa WC. Compensatory phosphorylation and protein–protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278:14841–14849. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- Bell D, Campbell M, Wang X, Earle JA, Cosby SL, McDermott BJ. Adrenomedullin gene delivery is cardio-protective in a model of chronic nitric oxide deficiency combining pressure overload, oxidative stress and cardiomyocyte hypertrophy. Cell Physiol Biochem. 2010;26:383–394. doi: 10.1159/000320562. [DOI] [PubMed] [Google Scholar]
- Bell D, Zhao Y, McCoy FP, Devine A, McDermott BJ. Expression of the counter-regulatory peptide intermedin is augmented in the presence of oxidative stress in hypertrophied cardiomyocytes. Cell Physiol Biochem. 2008;21:409–420. doi: 10.1159/000129633. [DOI] [PubMed] [Google Scholar]
- Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J Biol Chem. 2000;275:38938–38943. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- Dong F, Taylor MM, Samson WK, Ren J. Intermedin (adrenomedullin-2) enhances cardiac contractile function via a protein kinase C- and protein kinase A-dependent pathway in murine ventricular myocytes. J Appl Physiol. 2006;101:778–784. doi: 10.1152/japplphysiol.01631.2005. [DOI] [PubMed] [Google Scholar]
- Fontes-Sousa AP, Pires AL, Carneiro CS, Bras-Silva C, Leite-Moreira AF. Effects of adrenomedullin on systolic and diastolic myocardial function. Peptides. 2009;30:796–802. doi: 10.1016/j.peptides.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Hirose T, Totsune K, Mori N, Morimoto R, Hashimoto M, Nakashige Y, Metoki H, Asayama K, Kikuya M, Ohkubo T, Hashimoto J, Sasano H, Kohzuki M, Takahashi K, Imai Y. Increased expression of adrenomedullin 2/intermedin in rat hearts with congestive heart failure. Eur J Heart Fail. 2008;10:840–849. doi: 10.1016/j.ejheart.2008.06.020. [DOI] [PubMed] [Google Scholar]
- Kandilci HB, Gumusel B, Lippton H. Intermedin/adrenomedullin-2 (IMD/AM2) relaxes rat main pulmonary arterial rings via cGMP-dependent pathway: role of nitric oxide and large conductance calcium-activated potassium channels (BK(Ca)) Peptides. 2008;29:1321–1328. doi: 10.1016/j.peptides.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Kato K, Yin H, Agata J, Yoshida H, Chao L, Chao J. Adrenomedullin gene delivery attenuates myocardial infarction and apoptosis after ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;285:H1506–1514. doi: 10.1152/ajpheart.00270.2003. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovasc Res. 1999;41:514–523. doi: 10.1016/s0008-6363(98)00314-9. [DOI] [PubMed] [Google Scholar]
- Moens AL, Leyton-Mange JS, Niu X, Yang R, Cingolani O, Arkenbout EK, Champion HC, Bedja D, Gabrielson KL, Chen J, Xia Y, Hale AB, Channon KM, Halushka MK, Barker N, Wuyts FL, Kaminski PM, Wolin MS, Kass DA, Barouch LA. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the β3-adrenoreceptor. J Mol Cell Cardiol. 2009;47:576–585. doi: 10.1016/j.yjmcc.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. [Google Scholar]
- Mohan P, Brutsaert DL, Sys SU. Myocardial performance is modulated by interaction of cardiac endothelium derived nitric oxide and prostaglandins. Cardiovasc Res. 1995;29:637–640. [PubMed] [Google Scholar]
- Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Munzel G, Schlier A, Schreckenberg R, Abdallah Y, Schluter KD. Rat intermedin1−47 does not improve functional recovery in postischemic hearts. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:535–542. doi: 10.1007/s00210-011-0680-4. [DOI] [PubMed] [Google Scholar]
- Pan CS, Yang JH, Cai DY, Zhao J, Gerns H, Yang J, Chang JK, Tang CS, Qi YF. Cardiovascular effects of newly discovered peptide intermedin/adrenomedullin 2. Peptides. 2005;26:1640–1646. doi: 10.1016/j.peptides.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Pires AL, Pinho M, Sena CM, Seica RM, Leite-Moreira AF. Intermedin elicits a negative inotropic effect in rat papillary muscles mediated by endothelial derived nitric oxide. Am J Physiol Heart Circ Physiol. 2012;302:H1131–1137. doi: 10.1152/ajpheart.00877.2011. [DOI] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279:7264–7274. doi: 10.1074/jbc.M305332200. [DOI] [PubMed] [Google Scholar]
- Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther. 2000;86:49–86. doi: 10.1016/s0163-7258(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: a potent cardiovascular and renal regulator. FEBS Lett. 2004;556:53–58. doi: 10.1016/s0014-5793(03)01368-1. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Cai Y, Duan XH, Ma CG, Wang X, Tang CS, Qi YF. Intermedin1–53 inhibits rat cardiac fibroblast activation induced by angiotensin II. Regul Pept. 2009;158:19–25. doi: 10.1016/j.regpep.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Yang JH, Qi YF, Jia YX, Pan CS, Zhao J, Yang J, Chang JK, Tang CS. Protective effects of intermedin/adrenomedullin2 on ischemia/reperfusion injury in isolated rat hearts. Peptides. 2005;26:501–507. doi: 10.1016/j.peptides.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Jiang W, Liu JY, Li Y, Chen CL, Xin HB, Huang DJ. Intermedin is upregulated and has protective roles in a mouse ischemia/reperfusion model. Hypertens Res. 2009;32:861–868. doi: 10.1038/hr.2009.120. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Bell D, Smith LR, Zhao L, Devine AB, McHenry EM, Nicholls DP, McDermott BJ. Differential expression of components of the cardiomyocyte adrenomedullin/ intermedin receptor system following blood pressure reduction in nitric oxide-deficient hypertension. J Pharmacol Exp Ther. 2006;316:1269–1281. doi: 10.1124/jpet.105.092783. [DOI] [PubMed] [Google Scholar]