

Abstract
In this work, we establish a methodology for comparing the efficiencies of different hydrazide labels for detecting protein carbonyls. We have chosen acrolein- modified human serum albumin as a model. This system provides a convenient means of reproducibly generating carbonylated protein. Five hydrazide-based labels were tested. Three carry a biotin affinity tag and the others are simple fatty acid hydrazides. For the biotin-based labels, the yield of the labeling reaction varies considerably and the most commonly used label, biotin hydrazide, gives the lowest yield. The total MS/MS spectrum counts of modified peptides are similar for all of the biotin-based tags indicating that factors beyond the labeling efficiency are important in determining the effectiveness of the label. In addition, there is a large variation in the number of spectra obtained for specific, modified peptides depending on the nature of the labeling group. This variation implies that the relative dectability of a particular modification site is highly dependent on the tagging reagent, and more importantly, titration schemes aimed at identifying the most reactive site based on its threshold concentration will be biased by the choice of tagging reagent. The fatty acid hydrazides are somewhat more effective than the biotin-based hydrazides in generating identifiable MS/MS spectra, but offer no opportunity for enrichment. For the biotin-based tags, avidin affinity chromatography was used with the tryptic digests and each tag led to similar enrichment levels.
Keywords: Acrolein, Oxidative Stress, Protein Carbonylation, Mass Spectrometry
INTRODUCTION
Among the posttranslational protein modifications, carbonylation has attracted a great deal of attention due to its correlation with oxidative stress, aging and age-related diseases [1,2]. Protein carbonyls involve the introduction of an aldehyde group on the side-chain of an amino acid and have been identified as important biomarkers for disease and aging [3]. These protein carbonyls are formed on amino acid side chains either via direct oxidation with reactive oxygen species (ROS), such as hydroxyl radicals, or by the addition of α,β–unsaturated aldehydes, which are the result of lipid peroxidation processes [4–6]. A number of studies have reported that protein carbonylation is selective and only certain proteins are carbonylated in the systems under oxidative stress [5–13]. Identifying carbonylation sites has proven to be challenging with in vivo experiments and only a limited number of studies have reported specific modification sites [16–26]. A key problem is the low concentration of modification sites in samples. For example, if one has a sample of roughly 200 proteins, each of which yields roughly 50 tryptic peptides, the concentration ratio of a specific modified peptide to the background in a bottom-up LC/MS/MS experiment is at best 1:10,000 (50 * 200), but likely much lower because only a fraction of any particular protein is expected to be carbonylated at any specific site. As a result, samples must generally be fractionated in some way before analysis to concentrate the modifications. The typical approach has been to take advantage of the unique reactivity of protein carbonyls (aldehydes) with hydrazine and hydrazide reagents as an entryway to an affinity tag. The most common has been biotin and a variety of strategies have been employed for immobilizing and isolating the biotin-tagged protein carbonyls. Despite this activity, there has not been a systematic evaluation of the efficiency of hydrazide-based tags in isolating and identifying protein carbonyls. However, Guo and Prokai have recently published a paper focused on the ability of a series of hydrazides to produce useable CID spectra from a collection of six 4-hydroxynonenal-labeled peptides. The work indicates that biotin hydrazide labeling can increase the quality of CID spectra (suppress neutral loss), but in 4 of 6 cases, labeling with biotin hydrazide failed to produce identifiable MS/MS spectra, potentially due to inefficient labeling [24].
In the present study, we test the effectiveness of five hydrazide-labeling agents in identifying protein carbonyls (Scheme 1). The first three are commercially-available biotin hydrazides with varying spacer groups between the two components. The last two are hydrazides of simple fatty acids. They have been included because Muddiman and co-workers reported that hydrophobic tagging groups can substantially increase the electrospray ionization efficiency of some peptides.[27] To stabilize the resulting hydrazones, they have been reduced with sodium cyanoborohydride to give hydrazine linkages as described by Yoo and Regnier[28]. Scheme 2 outlines the process with biotin hydrazide. As a model protein, we have chosen human serum albumin (HSA). It is readily available and been the subject of several studies of in vitro oxidative stress.[22,29–31] We have found that the treatment of HSA with acrolein provides a convenient and reproducible source of protein carbonyls that can be used for testing various isolation strategies. Acrolein is an α,β-unsaturated aldehyde and can undergo Michael additions with histidine, lysine, and cysteine to produce protein carbonyls (Scheme 3) – it is also capable of a variety of other reactions including Schiff base formation with lysine as well as cross-linking processes. This reaction is akin to the condensation of proteins with α,β-unsaturated aldehydes derived from lipid peroxidation processes – a key source of protein carbonyls for systems under oxidative stress. In the present study, we simply use acrolein as a convenient means of generating protein carbonyls – no effort was made to assess the relative reactivity of the residues in HSA with acrolein (we have shown that more sophisticated quantitation schemes are needed for that task).[32] Using MS/MS spectrum counts of modified peptides as a measure of a label’s effectiveness, we have done experiments with and without affinity chromatography of the tagged peptides. The results of this study indicate that the choice of label has a significant impact on the tagging efficiency and relative detectability of the modification sites. As a result, care must be taken in the choice of a tagging group and in the interpretation of data from pseudo-quantitative experiments such as titrations aimed at identifying the relative reactivity of sites.
Scheme 1.

Hydrazide reagents used in the study.
Scheme 2.

Labeling oxidized protein with biotin hydrazide.
Scheme 3.

Michael addition of acrolein to the side chains of cysteine, lysine, and histidine.
MATERIALS AND METHODS
Human serum albumin (HSA), acrolein, biotin hydrazide, extended chain biotin hydrazide, PEG4 linked biotin hydrazide, and sodium cyanoborohydride were purchased from Sigma (St. Louis, MO). Palmityl hydrazide and lauryl hydrazide were purchased from Alfa Aesar ( Ward Hill, MA). Sequencing grade, modified trypsin used for the enzymatic digestion was obtained from Promega (Madison, WI). Immobilized monomeric avidin kits and BCA protein assay kits were purchased from Pierce (Rockford, IL). MCX columns were purchased from Waters (Milford, MA). All other chemicals and solvents were obtained from commercial sources and were of the highest purity available.
MODIFICATION OF HSA
HSA (1 mg/mL) was treated with acrolein in a 1:10 (HSA:acrolein) final molar ratio and then incubated at 25°C for 1 hour according to a method described by Dalle-Donne[33–34]. Following the modification, excess acrolein was removed by filtering the solution through a Microcon YM-30 filter (Millipore) with PBS (phosphate-buffered saline) buffer (pH 7.4). Filtration was accomplished by centrifugation of the Microcon assembly at 8000 rpm for about 4 min. The filtration/centrifugation process was repeated three times and subsequently the modified HSA sample was labeled.
LABELING MODIFIED PROTEIN
Labeling of the carbonyl groups of oxidized HSA was performed by reaction with the appropriate hydrazide using an approach based on the method of Yoo and Regnier [28]. In summary, the oxidized protein samples were dried using a Speed-Vac concentrator, then dissolved at a concentration of 2 mg/mL in PBS buffer (pH 7.4) containing 5 mM of the hydrazide. The samples were incubated at room temperature for 2 h with shaking and then cooled to 0 °C. An equal volume of 30 mM sodium cyanoborohydride in PBS buffer was subsequently added to stabilize the reaction products. Following the labeling, the HSA samples were precipitated with 20% trichloroacetic acid in an ice bath for 1 h, and the excess reagents were removed as a part of the supernatant. The pellets were washed three times with ethanol/ethyl acetate (1:1) and then dissolved in ammonium bicarbonate buffer in preparation for enzymatic digestion.
ENZYMATIC DIGESTION
The labeled HSA samples in 50 mM ammonium bicarbonate buffer at a concentration of 1 mg/mL were reduced with 30 mM dithiothreitol at 50 °C for 20 min and alkylated with 55 mM iodoacetamide at room temperature for 30 min in the dark. The excess of dithiothreitol and iodoacetamide were removed using a Microcon YM-30 filter at 8000 g for 15 min. The proteins were redissolved in 50 mM ammonium bicarbonate, and subsequently digested with trypsin (substrate/enzyme ratio of 40/1, wt/wt) at 37 °C for 24 h. The digestion was terminated by adding 0.1% formic acid (120 μL).
ENRICHING MODIFIED PEPTIDES
Modified peptides were enriched with a monomeric avidin kit as described by the manufacturer. After preconditioning the column, samples were loaded. Non-bound peptides were washed away with PBS buffer and bound ones were eluted with the biotin blocking and elution buffer. The resulting peptide solutions were pre-concentrated with an MCX column as described by the manufacturer. The peptide solutions were dried with a Speed Vac concentrator and peptide concentrations were determined using a BCA assay kit as described by the manufacturer.
QUANTIFICATION
Quantification of carbonylated proteins was done as described by Levine[35]. In this protocol, reactive carbonyl groups were converted to the corresponding hydrazone by reaction with 10 mM 2,4-dinitrophenyl hydrazine (2,4-DNPH) in 2 M HCl (1:500 protein to 2,4-DNPH ratio). The reaction mixture was incubated for 15 min with shaking and the protein mixtures were precipitated with trichloroacetic acid (final concentration 15% vol/vol ratio). After incubation in an ice bath for 45 min, the precipitate was centrifuged at 13400 rpm for 5 min. The supernatant was discarded and the precipitate was washed 3 times with an ethanol-ethyl acetate mixture (1:1 vol/vol ratio). The precipitate was resuspended in 6 M guanidine-HCl solution (pH 2.3). The absorbance was measured at 370 nm and 276 nm. The former corresponds to the hydrazone maximum with an estimated molar absorptivity coefficient of 22,000 M−1 cm−1 [35]. We have used commercial HSA as a control. It shows a significant background signal and there was concern that it may be partially carbonylated. To test this hypothesis, we pre-reduced the commercial HSA with sodium borohydride (NaBH4) and then used it as a control in the DNPH analysis. There was only a negligible difference in absorbances between the pre-reduced and native control samples. With DNPH assay, the ratio of active carbonyls per protein molecule, corrected for background signal, is 2.7, and suggests 2–3 carbonyl modifications per HSA molecule. Given the 10:1 ratio of acrolein to HSA, it also suggests that 70 % of the acrolein did not result in protein carbonyl modifications under our reaction conditions.
BIOTIN QUANTIFICATION ASSAY
The extent of HSA biotinylation was measured with a HABA-biotin quantitation assay kit as described by the manufacturer (Pierce Biotechnology). In short, 100 μl of biotinylated HSA was added to 900 μl of the HABA-avidin solution and then the absorbance of the mixture was measured at 500 nm. The ratio of biotin to protein was calculated based on the difference in absorbance before and after addition of biotinylated HSA using a HABA-avidin extinction coefficient of 34,000 M−1cm−1. The assay was repeated three times, each with three technical replicates, for the biotin labels, 1, 2 and 3. As a positive control, commercial, biotinylated BSA (Sigma, St. Louis, MO) was used. The manufacturer reports an average of 9 biotinylation sites per protein for this sample.
LC/ESI-MS/MS ANALYSIS
Liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS) analysis of digested HSA samples (300 pmol/injection) was performed using a capillary HPLC system (ThermoFinnigan, San Jose, CA) connected to an LTQ ion trap mass spectrometer (ThermoFinnigan, San Jose, CA) with a nanospray source. Tryptic peptides were separated by reverse-phase chromatography on a C18 column (100*0.18 mm; Thermo, 5 um particle size) with a split flow system. The split is approximately 100:1 with a master flow rate of 50 μl/min ( ~0.5 μl/min in the analytical column). The digested peptides were eluted from the column using two mobile phases, A (0.1% formic acid in water) and B (0.1% formic acid in methanol). The gradient started from 2% B, then increased to 15% B over 15min, then increased to 80% B over 70 min, and finally increased to 95% B over 25 min. The eluted peptides (singly, doubly, or triply charged ions) were analyzed using a data-dependent scan procedure. To exclude the redundant processing of a few dominant ions in the MS/MS analyses, a dynamic exclusion feature was used that only allows a particular m/z value to be processed twice in a 2 min time window. The acquired MS/MS spectra were then searched against the NCBI non-redundant protein database (GI: 4502027) using the Finnigan Bioworks v.3.2 (Sequest) software. The database searching was set to only consider peptides within the mass range 500–4000 Da. Up to two missed cleavage sites were allowed, and cysteines were defined as modified with carbamidomethylation. To search for labeled peptides, differential modifications of cysteine, histidine, lysine, and arginine were set to, and 298.4, 411.5, 545.3, 254.3, and 310.4 Da, respectively for 1 – 5. In order to filter the search results, the Yates Rule[36] was applied and the Xcorr threshold was set to 1.9, 2.2 and 3.75 for +1, +2 and +3 charged peptides, respectively. ΔC was set to 0.1 and the number of top matches was set to 1.
RESULTS AND DISCUSSION
HSA was modified with acrolein and labeled with five different hydrazide compounds to compare their effectiveness in LC/MS/MS analysis. The sample preparations were repeated at least three times to establish reproducibility and the data are derived from multiple LC/MS/MS runs on each sample. Using tryptic digests, average sequence coverage was over 85% with our experimental arrangement.
MS/MS Spectra
A key issue with a label’s efficiency in LC/MS/MS experiments is its impact on the production of useful fragments, b and y ions, with our CID protocol. If the label diverts signal intensity into unproductive side-chain losses (i.e., breakdown of the label group), the quality of the MS/MS spectra will be degraded and the identification efficiency will potentially be reduced. For comparison, sample MS/MS spectra for peptides with and without modifications at His170 are shown in Figures 1 through 6 (+2 charge state). The spectra for the biotin-containing tags (1 – 3) exhibit a similar number of identified b and y ions, and share the pattern of strong y12, but weak y8 ions for the expected proline-directed cleavages. There is a bias towards cleavage near the N-terminus (modification site) with strong signals for b2, b3, y11, y12 and y13 ions. A dehydrated b3 ion is particularly intense in the parent as well as the biotinylated spectra, but the b2 ion is only intense in the biotinylated system. This bias is charge state dependent and more evenly distributed cleavages are seen in the corresponding triply-charged peptides (Supporting Information). In these spectra, no intense peaks were identified for fragmentation on the biotin label groups themselves (some unlabeled peaks in the spectra are for multiply-charged b and y ions with either H2O or NH3 loss). The lack of label breakdown with the biotin hydrazides is consistent with Prokai’s recent observations [24]. With the fatty acid derived tags, typical fragmentation spectra are observed, but there are some differences compared to those seen for the biotin hydrazides. A key difference is that the fatty acid tags at His170 suppress tryptic cleavage at Arg169 and lead to peptides extended by one residue at the N-terminus – tryptic cleavage occurs at Arg168. This is an unexpected effect of the labeling group. Although modification at the cleavage site (i.e., acrolein addition at a lysine) does suppress tryptic cleavage, this is the first case we have seen of modification of a neighboring residue suppressing tryptic cleavage. One does not expect the fatty acid to interact with the guanidine group of the arginine, so a disruptive, hydrophobic interaction with trypsin is a more likely explanation. The added arginine at the N-terminus provides a minor bias towards b ions with the most intense b ion being about 2-fold more intense than the most intense y ion. In addition, it appears that there are fewer ions from dehydration and ammonia loss with the fatty acid labels. Finally, each of them gives an intense peak for the loss of the label with the remnants of the acrolein group – this leads to the full-length peptide, but with no modifications (doubly-charged, m/z 950.2). Some label group breakdown is also evident in spectra for other peptides with the fatty acid hydrazides (Supporting Information). Overall, each of the tagging agents leads to useful data with spectra that contain extended spans of b and y ions.
Figure 1.

MS/MS spectrum of peptide containing residues 170–183 in the +2 charge state, m/z 872.21. Blue peaks are y ions and red peaks are b ions. Orange ones represent either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
Figure 6.

MS/MS spectrum of peptide containing residues 169–183 in the +2 charge state, m/z 1104.85. Modification of His-170 with acrolein and label 5. Blue peaks are y ions and red peaks are b ions. Orange ones represent either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
MS/MS Spectrum Counts
In bottom-up proteomics approaches, a reasonable measure of a labeling scheme’s effectiveness is the number of times a peptide with the label is targeted for an MS/MS scan and identified with acceptable confidence by a cross-correlation approach such as that found in SEQUEST. It is important to note that we are not correlating spectrum counts with peptide concentration, but instead, using them as a practical measure of the likelihood that a peptide would be detected in an LC/MS/MS experiment. The spectrum count total is based on multiple factors. (1) To be selected for an MS/MS scan, the labeled peptide must present sufficient signal intensity to be competitive with other signals in the full scan. The intensity relates to the efficiency of the labeling reaction as well as to the ionization/detection efficiency of the labeled peptide. (2) To be identified, the labeled peptide must give an MS/MS spectrum with an adequate set of characteristic fragment ions. (3) The number of competing species in the sample with relatively similar retention times affects the probability of a peptide being chosen for an MS/MS scan. Because the impacts of these individual factors can be constant across samples, particularly in complex mixtures, it has been reported that peptide hits and spectrum counts can be used as measures of relative protein abundance and detectability [37–39]. In the present context, MS/MS spectrum counts will be used to assess the ability of the labels to produce useful proteomic data. In these systems, each label is applied to the same protein sample and should produce analogous collections of modified peptides. Because the majority of peptides are not modified in these samples, the background matrix is similar for each of the labeled mixtures. In Table 1, the effectiveness of each label can be assessed in terms of the fraction of MS/MS identifications that include the label. The fractions are roughly similar for the biotin-based labels (1 – 3), but rise for the fatty acid based labels (4 and 5). This is consistent with Muddiman’s observation that hydrophobic tags enhance the ionization properties of some peptides [27]. As noted in the Methods section, a standard 2,4-dinitrophenylhydrazine assay for the level of protein carbonylation in the acrolein-modified HSA indicates approximately 2.7 active carbonyls per protein molecule. Given that HSA yields roughly 75 tryptic peptides with identifiable MS/MS spectra, one expects that about 3.5% of the peptides in the tryptic digest would be labeled (2.7/75). Based on this crude estimate, it appears that the labels are generally enhancing the detectability of the peptides. The effect is significant in label 4 where over a 2-fold enhancement is seen.
Table 1.
Fraction of MS/MS Spectra Identified for Labeled Peptides from Modified/Labeled HSA.
| Label | Fraction Labeled |
|---|---|
| 1 | 0.041 ± 17% |
| 2 | 0.044 ± 33% |
| 3 | 0.037 ± 14% |
| 4 | 0.095 ± 12% |
| 5 | 0.068 ± 7% |
Number of MS/MS spectra for labeled peptides divided by total number of MS/MS spectra obtained for HSA samples (labeled + unlabeled). For each of three preparations, the sum of spectra from 3 LC/MS runs was used. Uncertainties are projected from standard deviations in the log of the ratios from three preparations.
An important question in interpreting the data is whether the labels were equally successful in modifying the protein carbonyls in the sample. To address this issue, we have turned to an established biotinylation assay. As noted in the Methods section, a DNPH analysis of the acrolein-treated HSA indicated that the protein, on average, carried ~2.7 active carbonyl groups. This is a measure of the number of DNPH molecules captured by the acrolein-treated HSA and therefore is sensitive to the efficiency of the reaction of the DNPH with the protein carbonyls (the reported value assumes 100% efficiency). Using a HABA-avidin assay for the biotin-based labels, we obtained the data in Figure 7. As a control, we have included commercial, biotinylated BSA, which is reported to carry 9 biotinylation sites per protein. This value is reproduced within experimental uncertainty in the HABA-avidin assay. The data for labels 1, 2, and 3 suggest a marked variation in the efficiency of the hydrazide labeling reactions. Although there are significant uncertainties in the experimental values, the results indicate the following order of efficiency in the reactions with protein carbonyls: 3 > 2 > 1. The absolute values are in the general range suggested by the DNPH results and it appears that under our specific reaction conditions, 3 is about as effective as DNPH in labeling protein carbonyls. The low labeling efficiency, particularly with label 1, may partially explain the lack of MS/MS spectra for biotin hydrazides in Prokai’s study [24]. The chemical basis for the variation in reactivity is likely linked to two factors, (1) interference, possibly steric, by the biotin/linker in the hydrazide reaction and (2) the hydrophobicity of the labeling reagent (which also impacts its solubility). Label 3 benefits from having the most flexible linker and the greatest aqueous solubility. These data offer a contrasting picture to the spectral counts in Table 1 and suggest that the labeling groups impact the ionizability/detectability of the peptides in such a way that the concentration variations indicated in Figure 7 are nearly canceled in the spectral counts (i.e., ionizability/detectability follows the opposite trend, 1 > 2 > 3). This is an intriguing result and indicates that label 3 would be superior in situations where detection of biotinylated peptides is not the primary goal (e.g., identification/isolation of proteins with carbonyl modifications).
Figure 7.
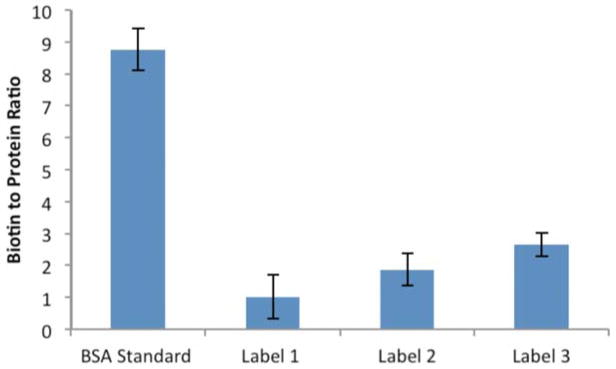
Quantification of biotin labels using HABA-avidin assay. BSA standard is a commercially-available, biotinylated BSA sample.
Data presented in Figure 8 show the number of MS/MS identifications at a set of common modification sites. These are the sum of three separate preparations, each subjected to 3 LC/MS/MS runs. The modified peptides give a limited number of spectra (generally less than 40 per peptide per preparation), so there is significant statistical variation between preparations, but the summed data in Figure 8 should provide a good qualitative picture of label performance. The results clearly indicate that the effectiveness of a labeling reagent (on the basis of spectral counts) is dependent on the structure/nature of the labeled peptide. Despite the fact that all of the biotin labels give roughly similar totals of labeled MS/MS spectra, there are multi-fold variations in relative effectiveness at a given modification site. For example, although Lys186 and Lys375 exhibit relatively similar detectability with the three biotin hydrazides (a modest advantage for 2), Lys549 exhibits a nearly 4-fold preference for label 1 relative to label 3. Variability is also seen in the response of the labeled histidines with the biotin hydrazides, particularly in His312, where label 3 provides no MS/MS spectra with our search parameters, whereas the response is fairly similar with His170 for all the biotins. With the fatty acid hydrazides, there is greater variability in the response. For example, label 5 gives over five times more spectra than label 1 for His312, but nearly 5 times fewer than label 1 for His170 – a 25 fold shift in response. There are many variables that impact these data and the number of spectral counts is low in some cases, so one can make only general rationalizations of the preferences. His170 is an interesting outlier in that it gives very few spectra with the hydrophobic alkyl labels (4 and 5). The sequence is HPYFYAPELLFFAK and contains the highest percentage of hydrophobic residues of the set shown in Figure 8. In addition, Lys549 (KQTALVELVK – modification leads to a missed cleavage) contains a high fraction of hydrophobic residues and the hydrophobic alkyl labels are also less effective relative to the biotin labels (specifically 1 and 2) with this peptide. In contrast, His312 exhibits the strongest bias in favor of the hydrophobic alkyl labels and the peptide, SHCIAEVENDEMPADLPSLAADFVESK, is one of the richest in hydrophilic residues in the set. Therefore, it appears that the alkyl labels can enhance the hydrophobicity of a relatively hydrophilic peptide giving a beneficial effect, but in some cases, such as His312, push the peptide to be too hydrophobic for efficient analysis. The differences exhibited between the individual biotin hydrazides are too subtle to analyze with the limited data. Overall, the variability is somewhat surprising and indicates that label choice will have a significant effect on the modifications that are detected. This will critically impact titration experiments where minimum detection level is used as a measure of the concentration of a modified peptide. Finally the data in the figure suggest that labels 1 and 2 are the least sensitive to peptide structure and give the most even response.
Figure 8.

Spectrum counts for HSA peptides modified by acrolein and labeled with different hydrazide tags.
Avidin Column Enrichment Of Biotinylated Peptides
The three biotin-based labels were also assessed in assays involving enrichment with a monomeric avidin column. Samples were prepared using the same protocol as above and then split. One fraction was analyzed directly and the other subjected to an affinity chromatography cycle before analysis. Table 2 shows the fraction of biotinylated peptides that were detected before and after enrichment. Combining the data from three runs, each of the labels leads to an enrichment of a factor of about 8–9 fold for the biotinylated peptides. Avidin enrichment leads to a reduction in the total number of identified MS/MS spectra of about 75%, but generally an increase in the MS/MS spectra for biotinylated peptides (Supporting Information). The behavior of all of the tags is roughly equivalent suggesting that there is no significant advantage between the tags in the enrichment phase – this is an expected result. The level of enrichment in this system is potentially limited by the sample’s complexity because the small number of unique biotinylated peptides leaves large gaps in the chromatographic run and allows even low abundance non-biotinylated peptides to be identified in those gaps. Future work will concentrate on more complex biological samples where the potential level of enrichment can be more accurately assessed.
Table 2.
Fraction of Labeled Peptides Identified in MS/MS Spectra from Modified/Labeled HSA Before and After Enrichment.
| Label | Before | After |
|---|---|---|
| 1 | 0.041 ± 17% | 0.39 ± 8% |
| 2 | 0.044 ± 33% | 0.34 ± 27% |
| 3 | 0.037 ± 14% | 0.33 ± 33% |
Uncertainties are projected from standard deviations in the log of the ratios from three experiments.
CONCLUSIONS
Carbonylation of human serum albumin with acrolein offers the opportunity to conveniently assess the labeling efficiency of various hydrazide compounds. Based on a HABA-avidin assay, we find that the labeling efficiencies vary considerably among the biotin hydrazides, but this variation is effectively canceled by the impact of the label on the ionizability/detectability of the peptides in this data set. For each of the modification sites tested, the pattern of relative responses from the labeling groups, as measured by MS/MS spectrum counts, varied considerably, suggesting that features of the individual peptides had an impact on the label’s effectiveness in producing identifiable MS/MS spectra. As a result, care must be taken in titration experiments that attempt to link the onset of identifiable MS/MS spectra with the reactivity of a particular modification site because the results will vary considerably with the choice of labeling group. Although none of the labeling groups is markedly superior in terms of spectrum counts, label 3 is the most efficient of the biotin hydrazides in reacting with protein carbonyls. At the peptide level, these data indicate that labels 1 and 2 give the most consistent response pattern and are best suited for identifying the maximum number of modification sites; however, other labeling groups can give much better response for particular modification sites. The three tested biotin hydrazides appeared to perform equally well during affinity chromatography and in our system, gave multi-fold enrichments in MS/MS counts (relative to background) for the biotinylated peptides.
Supplementary Material
Figure 2.

MS/MS spectrum of peptide containing residues 170–183 in the +2 charge state, m/z 1022.02. Modification of His-170 with acrolein and label 1. Blue peaks are y ions and red peaks are b ions. Orange ones show either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
Figure 3.

MS/MS spectrum of peptide containing residues 170–183 in the +2 charge state, m/z 1077.94. Modification of His-170 with acrolein and label 2. Blue peaks are y ions and red peaks are b ions. Orange ones show either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
Figure 4.

MS/MS spectrum of peptide containing residues 170–183 in the +2 charge state, 1145.35. Modification of His-170 with acrolein and label 3. Blue peaks are y ions and red peaks are b ions. Orange ones show either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
Figure 5.

MS/MS spectrum of peptide containing residues 169–183 in the +2 charge state, m/z 1078.01. Modification of His-170 with acrolein and label 4. Blue peaks are y ions and red peaks are b ions. Orange ones show either loss of water or loss of ammonia from b and y ions. b° and y° represent loss of water and b* or y* represent loss of ammonia from b and y ions (doubly charged ammonia and water loss peaks are not assigned). Black peaks were not identified as b or as y ions.
Acknowledgments
We would like to thank Dr. Simpson for his assistance in the implementation of the DNPH protocol. We acknowledge the support of the Virginia Commonwealth University and the National Institute of Health (1R01AG034167).
References
- 1.Eal MFLB. Oxidatively Modified Proteins in Aging and Disease. Science. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 2.Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 4.Madian AG, Regnier FE. Proteomic Identification of Carbonylated Proteins and Their Oxidation Sites. J Proteome Res. 2010;9:3766–3780. doi: 10.1021/pr1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki YJ, Carini M, Butterfield DA. Protein carbonylation. Antioxid Redox Signaling. 2010;12:323–325. doi: 10.1089/ars.2009.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grimsrud Pa, Xie H, Griffin TJ, Bernlohr Da. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.England K, O3Driscoll C, Cotter TG. Carbonylation of glycolytic proteins is a key response to drug-induced oxidative stress and apoptosis. Cell Death Differ. 2004;11:252–260. doi: 10.1038/sj.cdd.4401338. [DOI] [PubMed] [Google Scholar]
- 8.Linton S, Davies MJ, Dean RT. Protein oxidation and ageing. Exp Gerontol. 2001;36:1503–18. doi: 10.1016/s0531-5565(01)00136-x. [DOI] [PubMed] [Google Scholar]
- 9.Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem. 2000;275:27393–27398. doi: 10.1074/jbc.M003140200. [DOI] [PubMed] [Google Scholar]
- 10.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci U S A. 1998;95:12896–901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan LJ, Levine RL, Sohal RS. Effects of aging and hyperoxia on oxidative damage to cytochrome c in the housefly, Musca domestica. Free Radical Biol Med. 2000;29:90–97. doi: 10.1016/s0891-5849(00)00323-3. [DOI] [PubMed] [Google Scholar]
- 12.Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci U S A. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol. 2004;142:231–55. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johansson E, Olsson O, Nyström T. Progression and specificity of protein oxidation in the life cycle of Arabidopsis thaliana. J Biol Chem. 2004;279:22204–22208. doi: 10.1074/jbc.M402652200. [DOI] [PubMed] [Google Scholar]
- 15.Maisonneuve E, Ducret A, Khoueiry P, Lignon S, Longhi S, Talla E, Dukan S. Rules governing selective protein carbonylation. PLoS ONE. 2009 doi: 10.1371/journal.pone.0007269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzaei H, Regnier F. Creation of Allotypic Active Sites during Oxidative Stress. J Proteome Res. 2006;5:2159–2168. doi: 10.1021/pr060021d. [DOI] [PubMed] [Google Scholar]
- 17.Mirzaei H, Regnier F. Identification and quantification of protein carbonylation using light and heavy isotope labeled Girard3s P reagent. J Chromatogr, A. 2006;1134:122–133. doi: 10.1016/j.chroma.2006.08.096. [DOI] [PubMed] [Google Scholar]
- 18.Mirzaei H, Regnier F. Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal Chem. 2005;77:2386–2392. doi: 10.1021/ac0484373. [DOI] [PubMed] [Google Scholar]
- 19.Palmese A, De Rosa C, Marino G, Amoresano A. Dansyl labeling and bidimensional mass spectrometry to investigate protein carbonylation. Rapid Commun Mass Spectrom. 2011;25:223–231. doi: 10.1002/rcm.4863. [DOI] [PubMed] [Google Scholar]
- 20.Chavez J, Chung WG, Miranda CL, Singhal M, Stevens JF, Maier CS. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: protein carbonylation is diminished by ascorbic acid. Chem Res Toxicol. 2010;23:37–47. doi: 10.1021/tx9002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chavez JD, Bisson WH, Maier CS. A targeted mass spectrometry-based approach for the identification and characterization of proteins containing α-aminoadipic and γ-glutamic semialdehyde residues. Anal Bioanal Chem. 2010;398:2905–2914. doi: 10.1007/s00216-010-4289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aldini G, Gamberoni L, Orioli M, Beretta G, Regazzoni L, Facino RM, Carini M. Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. J Mass Spectrom. 2006;41:1149–1161. doi: 10.1002/jms.1067. [DOI] [PubMed] [Google Scholar]
- 23.Roe MR, Xie H, Bandhakavi S, Griffin TJ. Proteomic Mapping of 4-Hydroxynonenal Protein Modification Sites by Solid-Phase Hydrazide Chemistry and Mass Spectrometry. Peptides. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- 24.Guo J, Prokai L. To tag or not to tag: a comparative evaluation of immunoaffinity-labeling and tandem mass spectrometry for the identification and localization of posttranslational protein carbonylation by 4-hydroxy-2-nonenal, an end-product of lipid peroxidation. J Proteomics. 2011;74:2360–2369. doi: 10.1016/j.jprot.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo J, Prokai-Tatrai K, Nguyen V, Rauniyar N, Ughy B, Prokai L. Protein targets for carbonylation by 4-hydroxy-2-nonenal in rat liver mitochondria. J Proteomics. 2011;74:2370–2379. doi: 10.1016/j.jprot.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rauniyar N, Prokai-Tatrai K, Prokai L. Identification of carbonylation sites in apomyoglobin after exposure to 4-hydroxy-2-nonenal by solid-phase enrichment and liquid chromatography-electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2010;45:398–410. doi: 10.1002/jms.1725. [DOI] [PubMed] [Google Scholar]
- 27.Bereman MS, Comins DL, Muddiman DC. Increasing the hydrophobicity and electrospray response of glycans through derivatization with novel cationic hydrazides. Chem Commun. 2010;46:237–239. doi: 10.1039/b915589a. [DOI] [PubMed] [Google Scholar]
- 28.Yoo BS, Regnier FE. Proteomic analysis of carbonylated proteins in two-dimensional gel electrophoresis using avidin-fluorescein affinity staining. Electrophoresis. 2004;25:1334–1341. doi: 10.1002/elps.200405890. [DOI] [PubMed] [Google Scholar]
- 29.Temple A, Yen T-Y, Gronert S. Identification of specific protein carbonylation sites in model oxidations of human serum albumin. J Am Soc Mass Spectrom. 2006;17:1172–1180. doi: 10.1016/j.jasms.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 30.Mörtstedt H, Jeppsson MC, Ferrari G, Jönsson BaG, Kåredal MH, Lindh CH. Strategy for identification and detection of multiple oxidative modifications within proteins applied on persulfate-oxidized hemoglobin and human serum albumin. Rapid Commun Mass Spectrom. 2011;25:327–340. doi: 10.1002/rcm.4867. [DOI] [PubMed] [Google Scholar]
- 31.Funk WE, Li H, Iavarone AT, Williams ER, Riby J, Rappaport SM. Enrichment of cysteinyl adducts of human serum albumin. Anal Biochem. 2010;400:61–68. doi: 10.1016/j.ab.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, Simpson DC, Gronert S. The Reactivity of human serum albumin towards trans-4-hydroxy-2-nonenal. J Mass Spectrom. 2012;411:411–424. doi: 10.1002/jms.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossi R, Colombo R, Carini M, Milzani A, Dalle-Donne I. Water-Soluble α,β,-Unsaturated Aldehydes of Cigarette Smoke Induce Carbonylation of Human Serum Albumin. Antioxid Redox Signaling. 2010;12:349–364. doi: 10.1089/ars.2009.2806. [DOI] [PubMed] [Google Scholar]
- 34.Dalle-Donne I, Carini M, Vistoli G, Gamberoni L, Giustarini D, Colombo R, Maffei Facino R, Rossi R, Milzani A, Aldini G. Actin Cys374 as a nucleophilic target of alpha,beta-unsaturated aldehydes. Free Radical Biol Med. 2007;42:583–598. doi: 10.1016/j.freeradbiomed.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Levine RL, Wehr N, Williams Ja, Stadtman ER, Shacter E. Determination of carbonyl groups in oxidized proteins. Methods Mol Biol. 2000;99:15–24. doi: 10.1385/1-59259-054-3:15. [DOI] [PubMed] [Google Scholar]
- 36.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 37.Pang JX, Ginanni N, Dongre AR, Hefta SA, Opiteck GJ. Biomarker Discovery in Urine by Proteomics. J Proteome Res. 2002;1:161–169. doi: 10.1021/pr015518w. [DOI] [PubMed] [Google Scholar]
- 38.Gao J, Opiteck GJ, Friedrichs MS, Dongre AR, Hefta SA. Changes in the Protein Expression of Yeast as a Function of Carbon Source. J Proteome Res. 2003;2:643–649. doi: 10.1021/pr034038x. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Sadygov RG, Yates JR. A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.