

Abstract
Previous studies have established that pro-oxidative stressors suppress host immunity due to their ability to generate oxidized lipids with PAF-receptor (PAF-R) agonist activity. Although exposure to the pro-oxidative stressor cigarette smoke (CS) is known to exert immunomodulatory effects, little is known regarding the role of platelet-activating factor (PAF) in these events. The current studies sought to determine the role of PAF-R signaling in CS-mediated immunomodulatory effects. We demonstrate that CS exposure induces the generation of a transient PAF-R agonistic activity in the blood of mice. CS exposure inhibits contact hypersensitivity in a PAF-R-dependent manner as PAF-R-deficient mice were resistant to these effects. Blocking PAF-R agonist production either by systemic antioxidants or treatment with serum PAF-acetyl hydrolase enzyme blocked both the CS-mediated generation of PAF-R-agonists and PAF-R dependent inhibition of CHS reactions, indicating a role for oxidized glycerophosphocholines with PAF-R agonistic activity in this process. In addition, cyclooxygenase-2 (COX-2) inhibition did not block PAF-R agonist production but prevented CS-induced inhibition of CHS. This suggests that COX-2 acts downstream of the PAF-R in mediating CS-induced systemic immunosuppression. Moreover, CS-exposure induced a significant increase in the expression of the regulatory T cell reporter gene in FoxP3EGFP mice but not in FoxP3EGFP mice on a PAF-R-deficient background. Finally, Treg depletion via anti-CD25 antibodies blocked CS-mediated inhibition of CHS, indicating the potential involvement of Tregs in CS-mediated systemic immunosuppression. These studies provide the first evidence that the pro-oxidative stressor CS can modulate cutaneous immunity via the generation of PAF-R agonists produced through lipid oxidation.
Keywords: Cigarette smoke, Oxidized glycerophosphocholines, Platelet-activating factor, antioxidants
INTRODUCTION
It has been estimated that half of the world’s population are exposed to environmental or tobacco smoke [1–2]. Among various environmental pollutants, cigarette smoke (CS) exposure (active and passive) has been a leading cause of morbidity and mortality associated with many disorders ranging from chronic lung and vascular diseases to oral and lung cancers [1,3]. In addition to containing various bioactive compounds including nicotine and carcinogens, CS exerts immunomodulatory effects resulting in alterations of both innate & adaptive systemic immunity [4–11]. These effects on host immunity are multifaceted involving both pro-inflammatory and suppressive effects. The absolute effect of CS on the immune system depends on several variables, including the dose and type of tobacco, the route and chronicity of exposure, and the specific inflammatory mediators present at the time of immune cell stimulation. In addition, several studies have demonstrated that CS exposure leads to imbalances in Th1/Th2 responses resulting in T cell anergy [10–13]. The mechanism underlying these events has remained largely undetermined. Several studies have claimed nicotine to be a causative factor in CS-mediated suppression of immune cells and increased risk of human cancers [5–7,13]. Other studies have shown contrasting results, indicating that CS-induced effects are independent of nicotine [10,14]. Most importantly, multiple lines of evidence have suggested that the immunomodulatory effects of CS exposure in part are attributed to the ability of CS to induce the production of reactive oxygen species (ROS), which in turn act as an initiating event in modulating host immunity [10,15–17].
Several groups including ours have characterized the importance of various pro-oxidative stressors including UVB to suppress host immunity through a mechanism involving platelet-activating factor (1-alkyl-2-acetyl-glycerophosphocholine; PAF)(reviewed in [18,19]). PAF is produced enzymatically in a tightly-controlled process [20]. In addition, pro-oxidative stressors can act directly on glycerophosphocholines (GPC) to produce oxidized GPC (Ox-GPC) which are potent PAF-receptor (PAF-R) agonists [21–23]. The ability of various pro-oxidative stressors to suppress host immunity is classically measured by their ability to inhibit contact hypersensitivity (CHS) responses to either chemical antigens such as 2, 4-dinitrofluorobenzene (DNFB) or delayed type hypersensitivity responses to antigens such as Candida albicans (24–28). Employing CHS responses to DNFB in PAF-R expressing C57BL/6 wild type (WT) and gene-deficient (Ptafr−/−) mice, it has been previously demonstrated that the ability of Ox-GPCs to mediate systemic immunosuppression is dependent on the presence of the PAF-R and can be blocked by systemic administration of antioxidants, cyclooxygenase type 2 (COX-2) inhibitors, PAF-metabolizing enzymes, PAF-acetyl hydrolase (PAF-AH) or neutralizing antibodies against the immunosuppressive cytokine interleukin 10 (IL-10) [24–28]. In addition, we have shown that these Ox-GPCs/PAF-R agonists augment experimental murine melanoma tumor growth in a process involving IL-10 and regulatory T cells (Tregs) [29]. Since CS exposure acts as a pro-oxidative stressor [15–17], and has been shown to generate PAF-like mediators in hamsters [30], we hypothesized that CS-induced suppression of the host immune system is mediated through PAF/PAF-R.
Delineation of the specific mechanisms by which CS affects host immunity is important to identify potentially novel therapeutic approaches for the management of CS-mediated diseases. Thus, the present studies sought to determine the role of the PAF/PAF-R system in CS-mediated immunomodulatory effects. For these studies, we utilized several model systems including PAF-receptor (PAFR)-expressing (KBP) and deficient (KBM) cells, wild-type C57BL/6 (WT) and PAF-R-deficient (Ptafr−/−) mice, and FoxP3 reporter mice, and a well characterized DNFB allergen CHS model. These studies provide the first evidence that PAF-R signaling is involved in the immunosuppressive effects of CS.
MATERIAL AND METHODS
Reagents and CS exposure
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise. As previously reported, CS exposure was performed using a total body exposure method [31]. Briefly, mice were exposed for the indicated number of days, for 5h/day, 5 days/week in a Teague-10E exposure chamber (Teague Enterprises, Woodland, California) to a mixture of 90% sidestream and 10% mainstream cigarette smoke. The exposure chamber atmosphere was monitored for total suspended particulates (average 90 μg/m3) and carbon monoxide (average 350 ppm). The cigarettes used were research-grade cigarettes (1R3F) or low nicotine cigarettes (1R5F) from the Kentucky Tobacco Research and Development Center (University of Kentucky, Lexington, KY).
Mice
Female C57BL/6-wild type mice (PAF-R expressing; age 6–8 week) were purchased from The Charles River Laboratories. Age-matched female Ptafr−/− mice on a C57BL/6 background, generated as described previously (32), were a kind gift of Professor Takao Shimizu (University of Tokyo Department of Biochemistry). FoxP3EGFP knock-in transgenic mice on the C57BL/6 background (age 8–12 wk) were procured from the Jackson Laboratories (33). FoxP3EGFP-Ptafr−/− mice were generated to determine the involvement of PAF-R in Treg-mediated inhibition of contact hypersensitivity reactions by CS. In brief, FoxP3EGFP-WT female mice were crossed with Ptafr−/− males and offspring from each generation were genotyped and crossed to finally obtain FoxP3EGFP-Ptafr−/− mice. In some experiments mice were placed on vitamin C-enriched (10g/kg; Research Diets, Inc., New Brunswick, NJ) and 5 mM N-acetyl cysteine (NAC) in water for 10 days prior to CS exposure and for the duration of the study as per our previous studies (28,29). All mice were housed under specific pathogen-free conditions at the Indiana University School of Medicine. All procedures were approved by the Animal Care and Use Committee of Indiana University School of Medicine.
Measurement of PAF-R agonists by calcium mobilization and IL-8 production
The presence of systemic PAF-R agonists in lipid extracts derived from the blood of treated mice was measured by the ability of the lipid extracts to induce an intracellular Ca2+ mobilization response in PAF-R expressing KBP cells, but not in KBM cells lacking the PAF-R, as previously described (28). In brief, KBP and KBM cells were preloaded with the Ca2+-sensitive indicator, fura-2-AM (4 μM in Hanks’ balanced salt solution) at 37°C for 90 min, washed and resuspended in Hanks’ balanced salt solution at room temperature before use. Lipid extracts from whole blood obtained from groups of CS- vs untreated (sham) exposed mice were added to an aliquot of these cells (1.0–1.5 × 106 cells/2 ml) in a cuvette at 37°C with constant stirring. CPAF and endothelin-1 (ET-1) dissolved in ethanol (adjusted to 1μM) were used as positive controls. Fura-2-AM fluorescence was monitored in a Hitachi F-4010 spectrophotometer with excitation and emission wavelengths of 331 and 410 nm, respectively. The Ca2+ influx in suspensions was calculated as described [28] and shown as percentage of maximal peak calcium flux induced by either CPAF or ET-1. In separate experiments, WT mice were exposed to low nicotine cigarettes (0.16 mg/cig [<10% of standard amounts found in standard reference cigarette]; obtained from University of Kentucky Reference Labs) and its effect on PAF agonists production was similarly determined. In some experiments KBM and KBP cells were exposed to lipid extracts and supernatants were collected to measure IL-8 protein by ELISA as previously described [34].
Contact hypersensitivity (CHS) reactions
CHS to DNFB was conducted as previously described [22–23]. In brief, to evaluate the effect of CS on sensitization reactions, WT and Ptafr−/− mice were exposed to CS for 5h/day for 5 days according to the published protocol [31]. Three days following the CS exposure, a 2.5 × 2.5 cm area of distal back skin was shaved under anesthesia and sensitized with the application 25μl of 0.5% DNFB in acetone: olive oil (4:1, v/v). Nine days later, ear thickness was measured and then 10μl 0.5% DNFB was painted on the dorsal sides of one ear while the other ear was painted with vehicle. After 24 hours, ear thickness was measured using a digital caliper as a final read out. Intraperitoneal injection of PAF agonist, carbamoyl-PAF (CPAF; 250ng/mouse) and intradermal injection of histamine (200ng/mouse) were used as a positive control. In separate experiments, the mice were pretreated with antioxidants, COX-2 inhibitors SC-236 and NS-398, PAF-AH and neutralizing antibodies against CD25 as described in the appropriate figure legends. In CHS experiments, DNFB was added to ears of naive mice for 24h, which consistently did not elicit an inflammatory response.
Real time RT-PCR
Total RNA was extracted from lymph nodes of sham versus CS-exposed mice using the RNAeasy kit (QIAGEN). In brief, tissue was homogenized in RLT buffer containing β-mercaptoethanol by bullet blender (Next Advance., Inc, Averill Park, NY) using carbide beads following the manufacturer’s protocol (QIAGEN). Purified RNA was quantitated with the Nano Drop 2000 (Thermo Fisher Scientific, Lafayette, CO). Reverse transcription of whole RNA was done using Super Script cDNA synthesis kit (Invitrogen) with random hexamers. Quantitative RT-PCR was performed for EGFP and TGF-β expression and normalized against CD3ε using the ΔΔCt method on a Step One Real time PCR machine (BioRad Laboratories, Hercules, CA). Each assay was performed in triplicate in a 20-μl reaction volume with 2× SYBR green mastermix (SA Biosciences, Frederick, MD), 2μl of cDNA, and primers at 10μM. The following PCR conditions were used: 95°C for 15 minutes, followed by 45 cycles of 94°C for 15 seconds, 50°C for 30 seconds, and 72°C for 30 seconds, followed by 72°C for a final 10 minutes. Each PCR reaction was also tested to assure a single product of the predicted size.
Flow cytometry analysis
Tissue samples (draining lymph nodes) were harvested from isotype control antibody or anti-CD25 antibody treated FoxP3EGFP mice at day 17 (considering 15 day period for CHS experiment including −2 days of antibodies injection) and processed for the flow analysis. In brief, these tissues were dissociated into single cell suspension in CM media using gentleMACS™ (Miltenyl Biotech, Auburn, CA). Tissue samples were filtered with cell strainer (40μm) and centrifuged at 12,000 rpm for 5 minutes. Cells were washed twice with FACS buffer (PBS containing 2% FBS) and counted. 1×106 cells from each organ were incubated with Fc blocking (CD16/32) antibody for 10 minutes after which cells were washed twice with FACS buffer. Foxp3 positive cell populations were quantitated based on EGFP as surrogate by FACS. Data files were analyzed using FlowJo software (Tree Star, Inc, Ashland, OR).
Statistical analysis
In the present study, at least five mice/group was used in all murine experiments. All statistical calculations were performed using GraphPad Prism 5.0. Nonparametric one-way analysis of variance (ANOVA) followed by Bonferroni post hoc multiple comparison tests were used to test the statistical significance difference between multiple groups. The data represent mean values with SE. Differences were considered statistically significant when the P value was less than 0.05.
RESULTS
CS-exposure generates systemic PAF-R agonists
Previously, Lehr et al demonstrated that CS-exposure generates PAF-like oxidized lipids in hamsters [30]. Our first studies verified that CS induces oxidized lipids with PAF-R activity in mice Although some of the structures of Ox-GPC PAF-R agonists have been elucidated [19,22,23,35,36], it appears that numerous as yet undefined bioactive Ox-GPC PAF-R agonistic species exist [36]. Hence, our studies measured total PAF-R biochemical activity [28,35]. To test whether CS exposure can stimulate the production of PAF agonists, WT mice were first exposed to CS for 5 hours [31]. Control mice were not exposed to CS. Groups of mice were then sacrificed at various times post CS exposure and blood was drawn by cardiac puncture and underwent lipid extraction [28,35]. These lipids were tested for the presence of total PAF-R agonistic activity by their ability to induce intracellular calcium ion (Ca2+) mobilization in PAF-R expressing KBP cells, but not PAF-R negative KBM cells. We observed that lipid extracts derived from the blood of CS-treated WT mice, but not from sham treated mice, resulted in an immediate intracellular Ca2+ mobilization response in KBP cells (Figure 1A). However, no Ca2+ mobilization response was detected with lipid extracts from sham or CS-treated mice in KBM cells (Figure 1B). We then assessed the time course for CS-mediated generation of PAF-R agonists. As shown in figure 1C, the maximum amounts of PAF-R agonistic activity (as a percentage of 1 μM CPAF response) were generated when the blood was drawn immediately after completing 5 hours of CS (Time 0) and were substantially reduced after 60 minutes and were at baseline levels by 240 minutes following CS exposure. Of interest, approximately similar levels of PAF agonists were measured in WT and PAF-R-deficient mice indicating that the PAF-R was not necessary for CS-mediated generation of PAF-R agonists (data not shown). In addition to the receptor-mediated intracellular calcium mobilization studies discussed above, we also utilized a second biochemical assay to verify that CS induces the production of PAF-R agonists. Several studies including ours have demonstrated that activation of the epidermal PAF-R via PAF-R agonists results in the release of interleukin 8 (IL-8) [34,37]. Consistent with these findings, we demonstrate that lipid extracts derived from CS-treated induced a greater increase in IL-8 secretion in KBP cells in comparison to sham-treated mice. However, blood derived from CS-treated mice did not result in IL-8 release in PAF-R-negative KBM cells (Fig 1E). Taken together, our data indicates that PAF-R agonists are indeed produced in response to CS exposure.
Figure 1. Identification of PAF-R agonistic activity in blood from CS-treated mice.
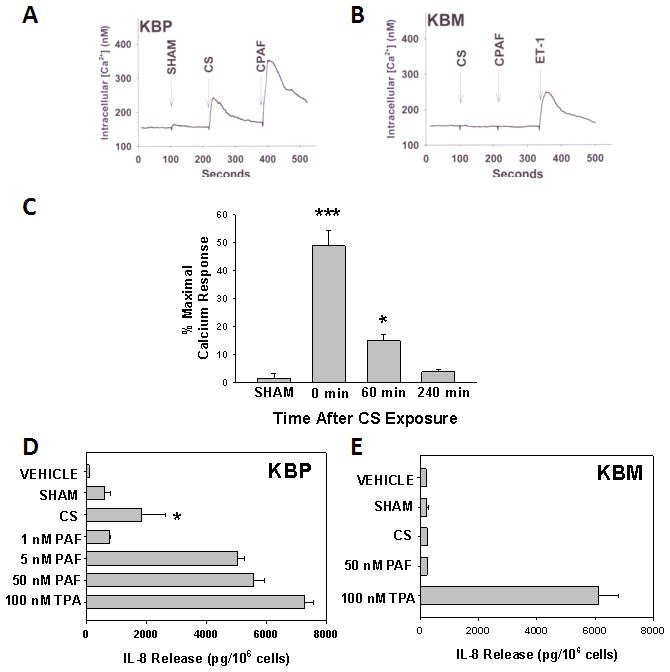
Lipid extracts from pooled whole blood from groups of 3–4 female mice (approx 2 ml total for each sample) were taken immediately (time 0) or various times following 5h of cigarette exposure, versus sham-treated mice. 25% of the sample (0.5 ml blood) from time = 0 was added to A) KBP cells or B) KBM cells loaded with FURA-2 dye and fluorescence monitored with spectrophotofluorimeter, and converted to intracellular Ca2+ levels. Excess (1 μM) CPAF or ET- 1 was added at the end of the incubation as positive control. C). Time course of PAF-R agonist measurement. Lipid extract-induced Ca2+ mobilization responses (from 0.5 ml of pooled blood) are expressed as a percentage of the maximal response elicited by CPAF (1μM) in KBP cells. The data are mean ± SE % maximal (CPAF) response at time = 0 from at least 4 pooled samples at each time point. D) KBP or E) KBM cells were incubated with various concentrations of PAF, 100 nM of the phorbol ester TPA or lipid extracts from blood taken 1h following sham- versus CS-exposed mice (as in Figure 1C). After 6h, the supernatants were removed and IL-8 measured by ELISA. The data are mean ± SD IL-8 protein from at least 3 pooled samples. Statistically significant (* = p < 0.05 & *** = p <0.0001) differences from vehicle-treated CS PAF-R agonist formation measurements.
Pharmacologic manipulation of CS-generated systemic PAF-R agonist formation
Our previous studies demonstrated that the generation of Ox-GPC PAF-R agonists by the pro-oxidative stressor ultraviolet B radiation can be blocked by antioxidants or the PAF-metabolizing enzyme serum PAF-AH [28,35]. Moreover, PAF-induced systemic immunosuppression is mediated via downstream COX-2/prostaglandin signaling [24,25]. Finally, several studies have implicated nicotine, a major constituent of cigarettes, as a mediator in CS-induced modulation of T-cells responsiveness and increase risk of human cancers [5–7,13]. We therefore determined the effects of antioxidants, PAF-AH, the COX-2 inhibitor NS-398, and low nicotine cigarettes on CS-mediated generation of PAF-R agonists. To that end, WT mice were either fed with a vitamin C enriched diet (10g/kg) and supplemented with NAC (5 mM) in drinking water for 10 days or regular diet prior to CS exposure. This antioxidant regimen has been shown to successfully block UVB-mediated augmentation of experimental melanoma growth, a process that is dependent upon Ox-GPC PAF-R agonists [29]. In separate experiments, WT mice were pretreated either with PAF-AH (5mg/kg) or with the COX-2 antagonist, NS-398 (200ng) injected i.p. followed by exposing them to cigarette smoke for 5 hours. Again, blood was drawn immediately following CS exposure and lipid extracts were tested for the presence of PAF-R agonists by the KBP Ca2+ mobilization assay. As shown in Figure 2, pretreatment with the PAF and Ox-GPC-metabolizing enzyme PAF-AH blocked CS-mediated generation of PAF-R agonists. Moreover, the antioxidant-enriched diet also ameliorated CS generation of PAF-R agonists, suggesting the importance of oxidized PAF-R agonists in this process (Figure 2). These data are in agreement with studies by Lehr et al demonstrating that antioxidants can block lipid PAF-R agonists generated by CS exposure in hamsters [30]. In contrast, pre-treatment with the COX-2 inhibitor NS-398 had no effect on CS-mediated generation of PAF-R agonists. To define the role of nicotine in CS generation of PAF-R agonistic activity, WT mice were exposed to low nicotine (<10% of usual content) instead of standard reference cigarettes. As shown in Figure 2, exposure to low nicotine cigarettes generated similar levels of PAF-R agonists as standard cigarettes. Collectively, these data suggest the importance of oxidative stress rather than nicotine in mediating the CS generation of PAF-R agonists.
Figure 2. Antioxidants and PAF-AH but not COX-2 inhibitor block CS-exposure generated systemic PAF-R agonists; lack of role for nicotine.

Female mice were placed on 10g/kg vitamin C-enriched chow + 5 mM NAC in water for 10 days vs standard chow/water, or i.p. injections of 5 mg/kg PAF-AH (24 h before CS) or 200 ng of the COX-2 inhibitor NS-398 or vehicle control (2h before CS), or were treated with low-nicotine CS. Groups of 3–4 mice were exsanguinated immediately following 5 h of CS exposure and PAF-R agonistic activity measured as in Fig 1. The data are mean ± SE % maximal (CPAF) response from at least 4 pooled samples. ***Statistically significant (p < 0.001) differences from control CS-treated.
Effect of Cigarette smoke-mediated generation of PAF-R agonists on CHS in WT and Ptafr−/− mice
Given our findings demonstrating that CS exposure generates systemic PAF-R agonists as well as previous studies demonstrating that these potent lipid mediators can suppress host immunity (24–27), our next experiments assessed the effect of CS-generated PAF-R agonists on CHS reactions. To that end, mice were exposed to CS for 5h/day for 5 days, followed by sensitization of the back skin with DNFB 3 days later, and finally the elicitation of the CHS response by reapplying DNFB to one ear 9 days after the sensitizing the mice to DNFB and vehicle to the contralateral ear. The elicitation reactions were assessed by measuring the differences in ear thickness between DNFB and vehicle treated ears 24 hours after the application of the eliciting agent (as a measure of inflammation). In other groups, treatment with metabolically stable PAF-R agonist CPAF (250ng i.p.), histamine (200 ug s.c.) or 0.1% DMSO vehicle (i.p.) were given 5 days before sensitization. As shown in Figure 3, there was no difference in the CHS reactions to DNFB between PAF-R positive WT and Ptafr−/−mice in sham control groups. However, CS-exposure inhibited CHS reactions to DNFB selectively in WT mice and not in Ptafr−/− mice. Similarly, intraperitoneal injection of the CPAF, used as a positive control, resulted in an inhibition of CHS reactions only in WT mice. Intradermal injections of histamine, which suppresses systemic CHS responses independently of the PAF-R [38,39], induces systemic immunosuppression in both WT and Ptafr−/− mice. These findings correlate with previous studies demonstrating that PAF-R agonists generated via UVB mediate systemic immunosuppression only in WT and not in Ptafr−/− mice [26,27]. These data also implicate the importance of PAF-R agonists generated via CS-exposure in mediating systemic immunosuppression.
Figure 3. Smoking exposure inhibits CHS to DNFB in WT but not PAF-R−/− mice.
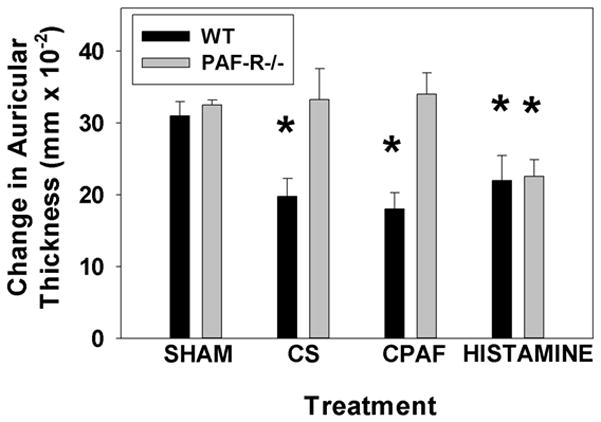
Groups of 6–8 WT or PAF-R-deficient female mice were exposed to 5d of 5h/day cigarette smoke followed 3 d later by treating back skin with DNFB (sensitization). 9 d later ear thickness measured, then one ear was treated with DNFB, the other with vehicle and ear thickness (as a measure of inflammation) measured 24h later. In other groups, treatment with CPAF (250ng i.p.), histamine (200 ug s.c.) or 0.1% DMSO vehicle given 5 d before sensitization. The data are mean ± SE differences in ear thickness measurements. *Denotes statistically significant (p < 0.05) differences from vehicle-treated mice.
Effect of anti-oxidants on CS-mediated inhibition of CHS in WT mice
The next studies were designed to assess whether CS-generated PAF-R agonistic activity could be suppressed by systemic antioxidants in vivo using the CHS model. To achieve this, mice were fed either standard diet or a diet enriched with antioxidants (vitamin C-and NAC) for 10 days before CS exposure and then were maintained on this diet during the experimental period. As above, mice were exposed to CS for 5h/day for 5 days or were exposed to ambient air as sham controls and CHS to DNFB was assessed by measuring the elicitation reaction (ear thickness). We observed that CS-mediated inhibition of CHS reactions was observed in WT mice on a regular diet, but was blocked in mice treated with systemic antioxidants (Figure 4). These studies indicate that CS-exposure generates PAF-R agonists via a process involving ROS. These findings are consistent with our previous studies demonstrating that vitamin C blocks the generation of UVB-mediated PAF-R agonists and restored the inhibition of CHS reactions in WT mice (28).
Figure 4. Treatment with antioxidants blocks CS-mediated inhibition of CHS to DNFB.
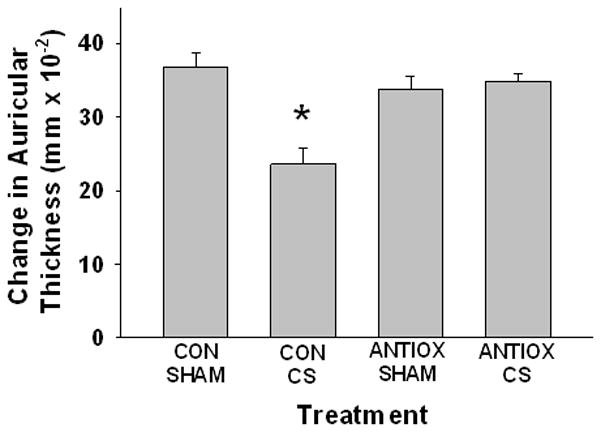
Groups of 6–8 WT female mice were treated with standard chow or antioxidant diet (10g/kg vitamin C-enriched chow and 5 mM NAC in water) for 10 days before being exposed to 5d of 5h/day cigarette smoke or sham followed 3 d later by treating back skin with DNFB (sensitization). 9 d later ear thickness measured, then one ear was treated with DNFB, the other with vehicle and ear thickness (as a measure of inflammation) measured 24h later. The data are mean ± SE differences in ear thickness measurements. *Denotes statistically significant (p < 0.05) differences from sham- or vehicle-treated mice.
Effect of PAF-AH and COX-2 antagonists on CS-mediated inhibition of CHS in WT mice
Given our findings that PAF-AH treatment blocks CS-mediated generation of PAF-R agonists and supporting evidence that PAF-AH prevented the inhibition of CHS by UVB [28], we next planned to determine whether exogenous PAF-AH can block the inhibition of CHS mediated by CS-exposure. To that end, WT mice were pretreated with recombinant PAF-AH two days prior to CS-exposure and the effect of CS on CHS reactions was assessed. We observed that PAF-AH treatment did not affect CHS responses in sham-treated mice, yet blocked the CS-induced inhibition of CHS (Figure 5). Of interest, previous studies have demonstrated that PAF-AH does not block the effects of the metabolically stable PAF-R agonist CPAF on CHS reactions [28].
Figure 5. Treatment with PAF-AH, and COX-2 inhibitors block CS-mediated inhibition of CHS to DNFB.
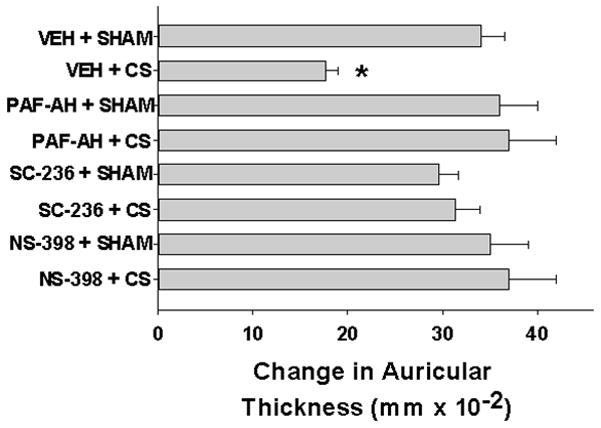
Groups of 6–8 WT female mice were treated with either 5 mg/kg PAF-AH (24 h before) or 200ng of COX-2 antagonists SC-236 or NS-398 or vehicle 1h before being exposed to 5d of 5h/day cigarette smoke or sham followed 3 d later by treating back skin with DNFB (sensitization). 9 d later ear thickness measured, then one ear was treated with DNFB, the other with vehicle and ear thickness (as a measure of inflammation) measured 24h later. The data are mean ± SE differences in ear thickness measurements. * Denotes statistically significant (p < 0.05) differences from sham- or vehicle-treated mice.
Previous studies have shown that COX-2 metabolites are downstream mediators of PAF in UVB-mediated inhibition of CHS [24]. Hence, we tested the hypothesis that COX-2 could be involved in CS-mediated inhibition of CHS reactions. For this, WT mice were pretreated with COX-2 antagonists, NS-398 and SC-236 two days prior to 5h/day for 5 days of CS-exposure and the effect of CS on CHS reactions was similarly assessed. As shown in Figure 5, blocking COX-2 by two pharmacologically distinct COX-2 enzymatic antagonists prevented CS-mediated inhibition of CHS to DNFB.
Involvement of regulatory T cells in CS-mediated inhibition of CHS
Several studies have implicated CD4+CD25+ regulatory T cells (Tregs) as mediators of UVB-induced local and systemic immunosuppression [40–42]. In addition, our studies have demonstrated that Tregs are important in mediating UVB/PAF-R agonist-induced systemic immunosuppression and the augmentation of the growth of experimental murine melanoma [29]. We have also shown that the depletion of Tregs by neutralizing antibodies against CD25 abrogates UVB-mediated increase in Foxp3 expression, the master regulator of Tregs, in lymph nodes as measured by quantitative PCR, and these findings correlated very well with flow cytometry analysis [29]. This depletion of Tregs attenuated UVB-mediated enhanced melanoma tumor growth [29]. Based on these observations, we hypothesized that Tregs could be mediating CS-induced systemic immunosuppression. To answer this question we utilized a Foxp3 reporter mouse model (FoxP3EGFP mice) that expresses EGFP under the control of the FoxP3 promoter [33]. We first exposed FoxP3EGFP mice to 5h/day of CS for two alternate days or left them unsmoked for sham control and isolated lymph nodes and quantified the expression of Foxp3 by performing qPCR for EGFP, a surrogate marker for Tregs, after normalization against the T cell gene Cd3. We observed a significant increase in the expression of EGFP by CS-exposure compared to their sham controls (Figure 6A). We next defined whether this increase in the expression of EGFP by CS is dependent on PAF-R signaling. For this, we crossed FoxP3EGFP mice with Ptafr−/− mice to generate FoxP3EGFP mice on PAF-R-deficient background (FoxP3EGFP-Ptafr−/−) and exposed them to CS and quantified Foxp3 expression from the lymph nodes as described. Unlike the wild type FoxP3EGFP mice, we observed that CS-exposure did not result in the enhanced expression of EGFP in FoxP3EGFP-Ptafr−/− mice (Figure 6A). These studies indicate a systemic increase in Tregs following CS-exposure occurs in a PAF-R dependent manner. Interestingly, we also observed statistically (p < 0.05) increased expression of the Treg-associated immunosuppressive cytokine TGFβ in lymph nodes by CS in a PAF-R dependent manner (data not shown).
Figure 6. Evidence for Treg-mediation of CS- and CPAF-inhibition of CHS to DNFB. A) CS induces Tregs.
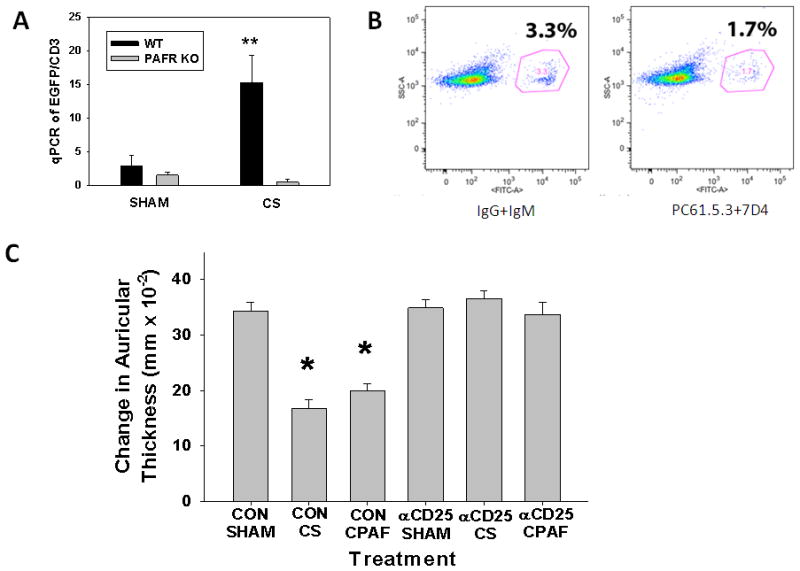
A group of 4–5 FoxP3EGFP transgenic mice on WT or PAF-R-deficient backgrounds were either sham- or CS-exposed for 5h/day for three days before removal of lymph nodes for RNA extraction and cDNA synthesis. The expression of EGFP was analyzed by qRT-PCR and normalized against CD3. The data represent mean ± SD. ** Denotes statistically significant (p< 0.01) difference from sham-treated mice. B) Depleting antibodies against CD25 block expression of EGFP-positive Tregs. FoxP3 EGFP mice were injected with either isotype control antibodeis (IgG and IgM) or depleting antibodies against CD25 (PC61.5.3 and 7D4). The expression of EGFP (marker for FoxP3-regulatory T cells) in lymph nodes of these mice were then evaluated on the 10th day using EGFP as surrogate by FACS. C) Treatment with anti-CD25 inhibits CS- and CPAF-mediated inhibition of CHS. WT mice (groups of 6–8) were injected either with isotype control antibodies against IgG and IgM or depleting antibodies against two different clones of anti-CD25 (PC61.5.3 and 7D4) (1 mg each) two days before beginning standard sham or CS (5h/day × 5 days) exposure. 3 days later mice were then sensitized to DNFB followed 9 d later by elicitation as outlined in Fig 3. The data are mean ± SE differences in ear thickness measurements. * Denotes statistically significant (p < 0.05) differences from sham-treated mice.
Given our findings that CS exposure increased LN Foxp3 expression in a PAF-R-dependent fashion, coupled with our previous studies demonstrating by flow cytometry increased numbers of FoxP3-expressing T cells in lymph nodes following systemic CPAF treatment [29], we next tested whether depletion of Tregs by anti-CD25 antibodies could abrogate CS-mediated inhibition of CHS reactions. To that end, FoxP3EGFP mice were injected either with anti-CD25 or isotype control antibodies at −2 days followed by treatment with or without CS for 5 days and CHS to DNFB was measured. We included i.p. injections of CPAF and vehicle as positive controls. As shown in figure 6B, CS exposure or CPAF treatment inhibited CHS to DNFB in isotype control-injected mice compared to sham control mice. Depleting anti-CD25 antibodies inhibited FoxP3 expression in lymph nodes, and ablated the decrease in CHS reactions to DNFB both in CS exposed and CPAF-treated mice (Figure 6B, C), suggesting the contribution of Tregs in CS/CPAF-induced systemic immunosuppression.
Discussion
Because cigarette smoke is one of the most important immunosuppressive environmental exposures and is a well-known risk factor for lung cancer induction, it is important to understand the mechanisms involved. The present studies describe a previously unappreciated mechanism by which CS exposure can result in systemic immunosuppression. These data support the model that Ox-GPC PAF-R agonists produced in response to ROS from CS can exert systemic immunosuppressive effects in a process involving COX-2 metabolites and Tregs. Moreover, we present evidence that this novel immunomodulatory pathway is susceptible to pharmacologic inhibition.
Oxidation of esterified fatty acyl residues introduces oxy functions, rearranges bonds and fragments carbon-carbon bonds by β-scission that generate a myriad of phospholipid reaction products including PAF-R agonists [19–22,35,36]. In contrast to the tightly controlled enzymatic pathways for PAF biosynthesis, high levels of Ox-GPC PAF-R agonists can be produced non-enzymatically. In this regard, cellular membranes serve as the source of oxidized phospholipids and are thus the source of CS-mediated PAF formation. Though these studies did not address the exact cellular source for the CS-generated PAF-R agonists, this is likely to be the lung epithelium, in a process similar to skin keratinocytes and UVB.
Previous findings indicated that CS treatment of both hamsters and humans resulted in the generation of a PAF-R agonistic activity, in a process inhibited by the antioxidant vitamin C [30,40]. The present studies confirm that CS exposure generates PAF-R agonists in murine blood. That antioxidants ameliorated the CS-mediated production of PAF agonists, and that low nicotine CS exposure generated equal levels of these novel lipids as the standard cigarettes, all fit with the notion that ROS, not nicotine, mediates this response.
Exogenous pro-oxidative stressors ranging from aromatic hydrocarbons to UVB have been shown to inhibit contact hypersensitivity via PAF-R signaling [18,19]. Consistent with the ability of CS exposure to generate PAF-R agonists, CS induces systemic immunosuppression in a PAF-R-dependent process blocked by antioxidants. Production of PAF-R agonists begins a cascade of events leading to systemic immunosuppression. The cytokines which appear to be critical for the immunosuppression include IL-10 and COX-2-generated eicosanoids [24–27]. Regulatory T cells also are implicated in PAF-R-dependent systemic immunosuppression [29]. The ability of a Treg-depleting strategy successfully used to block UVB-mediated immunosuppression [29] to similarly block CS-mediated inhibition of CHS confirms Treg involvement. Of interest, use of a Treg depleting strategy has been reported to modestly enhance CHS reactions [41,42]. As depicted in Figure 6, our neutralizing antibody protocol did not result in an enhancement of baseline CHS reactions, though this strategy did deplete Tregs by approximately 50%. It is possible that the level of Treg depletion in our study was not enough to exert this enhancement.
The model that has emerged indicates that mast cells are also crucial mediators of PAF-induced systemic immunosuppression. Mast cell-deficient mice do not undergo systemic immunosuppression in response to PAF agonist generators such as UVB or JP-8 jet fuel [38,39,43]. In response to these agents, mast cells traffic to lymph nodes in a CXCR4-dependent manner in that a CXCR4 antagonist blocks not only UVB/PAF-R-mediated mast cell migration from skin to lymph nodes, but also the systemic immunosuppression [39]. Elegant studies transplating mast cells into mast cell-deficient mice have indicated that mast cells appear to be the source for eicosanoids which mediate the systemic immunosuppression [39].
PAF-R agonists have been reported to be measured in the blood from human volunteers exposed to CS [40], suggesting that this process could be generalized to humans. Indeed, CS-mediated systemic immunosuppression has been documented in humans [7,10,44]. Of interest, a large study of subjects undergoing hepatitis B vaccination indicates that smokers were more likely to experience vaccine failure than non-smokers [45]. Though the current studies focused upon the ability of CS to inhibit the sensitization of a neoantigen in a PAF-R-dependent manner, our previous studies have demonstrated that systemic CPAF exposure can also inhibit elicitation resactions to a sensitized antigen [25]. Thus, the CS generation of PAF agonists could have a profound effect on immunity. The findings from the present studies implicating Ox-GPC PAF-R agonist involvement in CS-mediated systemic immunosuppression suggests that strategies such as antioxidants and COX-2 inhibitors could potentially have clinical use in augmenting vaccine efficiency in smokers.
The major enzyme for PAF/Ox-GPC degradation is serum PAF-AH (PLA2G7); reviewed in [46]. PAF-AH deficiencies have been described, including a genetic loss of function mutation found to be homozygous in approximately 4% of Japanese individuals [47]. In addition, acquired PAF-AH deficiencies have been reported in disorders such as lupus erythematosus [46–48]. Acute CS exposure has also been reported to inactivate PAF-AH [49]. However, CS has also been demonstrated to increase serum PAF-AH levels [50]. The present studies indicate that PAF-AH could play an important role in protecting against CS-mediated systemic immunosuppression via the inactivation of PAF-R agonists. Hence, there appears to be considerable variability in the ability of individuals to metabolize PAF/Ox-GPC generated by CS due to genetic/environmental influences, which could have clinical significance.
In summary, the present studies provide the first evidence that PAF-R signaling plays an important role in CS-mediated immunosuppression in a murine model of CHS. Inasmuch as this process involves the pro-oxidative qualities of CS rather than nicotine, it is possible that other smoke exposures including pollution could exert similar effects on the immune system. That these CS-mediated effects are neutralized by relatively simple and safe measures (e.g., antioxidants) could provide the impetus for future studies to define the clinical significance of this novel pathway in humans.
Acknowledgments
This research was supported in part by grants from the Riley Memorial Association, and the National Institutes of Health grant R01 HL062996 (JBT&RLK) & R21 ES020965 (RLK), R01 HL077328 (IP), and Veteran’s Administration Merit Award (JBT) and a Postdoctoral Fellowship grant from American Institute for Cancer Research 09A062 (RPS).
The authors wish to acknowledge the technical assistance of Drs. Mohammed Al-hassani and Kelly S. Schweitzer and Ms. Sonia DaSilva. We also wish to acknowledge the helpful suggestions from Dr. Mark Kaplan.
Abbreviations usedd in this paper
- CHS
contact hypersensitivity
- COX-2
cyclooxygenase type 2
- CPAF
1-hexadecyl-2-N-methylcarbamoyl glycerophosphocholine
- CS
cigarette smoke
- DNFB
dinitrofluorobenzene
- NAC
N-acetylcysteine
- PAF
platelet-activating factor
- GPC
glycerophosphocholine
- Ox-GPC
oxidized GPC
- PAF-R
PAF receptor
- PAF-AH
PAF acetylhydrolase
- ROS
reactive oxygen species
References
- 1.Wipfli H, Samet JM. Global economic and health benefits of tobacco control: part 1. Clin Pharmacol Therapeu. 2009;86:263–271. doi: 10.1038/clpt.2009.93. [DOI] [PubMed] [Google Scholar]
- 2.Hatsukami DK, Stead LF, Gupta PC. Tobacco addiction. Lancet. 2008;371:2027–2038. doi: 10.1016/S0140-6736(08)60871-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Eng J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esber HJ, Menninger FF, Jr, Bogden AE, Mason MM. Immunological deficiency associated with cigarette smoke inhalation by mice. Primary and secondary hemagglutinin response. Arc Environ Health. 1973;27:99–104. doi: 10.1080/00039896.1973.10666328. [DOI] [PubMed] [Google Scholar]
- 5.SucIu-Foca N, Molinaro A, Buda J, Reemtsma K. Cellular immune responsiveness in cigarette smokers. Lancet. 1974;1:1062–1064. doi: 10.1016/s0140-6736(74)90474-7. [DOI] [PubMed] [Google Scholar]
- 6.Geng Y, Savage SM, Razani-Boroujerdi S, Sopori ML. Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. J Immunol. 1996;156:2384–2390. [PubMed] [Google Scholar]
- 7.Onari K, Sadamoto K, Takaishi M, Inamizu T, Ikuta T, Yorioka N, Ishioka S, Yamakido M, Nishimoto Y. Immunological studies on cigarette smokers. Part II: cell mediated immunity in cigarette smokers and the influence of the water-soluble fraction of cigarette smoke (WSF) on the immunity of mice. Hirosh J Med Sci. 1980;29:29–34. [PubMed] [Google Scholar]
- 8.Lu LM, Zavitz CC, Chen B, Kianpour S, Wan Y, Stampfli MR. Cigarette smoke impairs NK cell-dependent tumor immune surveillance. J Immunol. 2007;178:936–943. doi: 10.4049/jimmunol.178.2.936. [DOI] [PubMed] [Google Scholar]
- 9.Mian M, Lauzon NM, Stampfli MR, Mossman KL, Ashkar AA. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J Leukoc Biol. 2008;83:774–784. doi: 10.1189/jlb.0707481. [DOI] [PubMed] [Google Scholar]
- 10.Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. 2009;9:377–384. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 11.Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91:142–149. doi: 10.1177/0022034511421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smyth LJ, Starkey C, Vestbo J, Singh D. CD4-regulatory cells in COPD patients. Chest. 2007;132:156–163. doi: 10.1378/chest.07-0083. [DOI] [PubMed] [Google Scholar]
- 13.Kaifra R, Singh SP, Savage SM, Finch CL, Sopori ML. Effect of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca2+ stores. J Pharmacol Exp Ther. 2000;293:166–171. [PubMed] [Google Scholar]
- 14.Imaizumi T, Satoh K, Yoshida H, Kawamura Y, Hiramoto M, Takamatsu S. Effect of cigarette smoking on the levels of platelet-activating factor-like lipid(s) in plasma lipoproteins. Atherosclerosis. 1991;87:47–55. doi: 10.1016/0021-9150(91)90231-q. [DOI] [PubMed] [Google Scholar]
- 15.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pryor WA, Stone K. Oxidants in cigarette smoke. Ann NY Acad Sci. 1993;686:12–28. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- 17.Seet RC, Lee CY, Loke WM, Huang SH, Huang H, Looi WF, Chew ES, Quek AM, Lim EC, Halliwell B. Biomarkers of oxidative damage in cigarette smokers: which biomarkers might reflect acute versus chronic oxidative stress? Free Rad Biol Med. 2011;50:1787–1793. doi: 10.1016/j.freeradbiomed.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res. 2005;571:185–205. doi: 10.1016/j.mrfmmm.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 19.Konger RL, Marathe GK, Yao Y, Zhang Q, Travers JB. Oxidized glycerophosphocholines as biologically active mediators for ultraviolet radiation-mediated effects. Prost Lipid Med. 2008;87:1–8. doi: 10.1016/j.prostaglandins.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braquet P, Touqui L, Shen TY, Vargaftig BB. Platelet-activating Factor. Pharmacol Rev. 1987;39:97–145. [PubMed] [Google Scholar]
- 21.Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, Murphy RC, Zimmerman GA, McIntyre TM, Travers JB. Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J Biol Chem. 2005;280:35448–35457. doi: 10.1074/jbc.M503811200. [DOI] [PubMed] [Google Scholar]
- 22.Murphy RC. Oxidized lipids with platelet-activating factor activity. Adv Exp Med Biol. 1996;416:51–58. [Google Scholar]
- 23.Patel KD, Zimmerman GA, Prescott SM, McIntyre TM. Oxidized phospholipids. J Biol Chem. 1992;267:15168–15175. [PubMed] [Google Scholar]
- 24.Walterscheid JP, Ullrich SE, Nghiem DX. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J Exp Med. 2002;195:171–179. doi: 10.1084/jem.20011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Mousdicas N, Yi Q, Al-Hassani M, Billings S, Howard KM, Ishii S, Shimizu T, Travers JB. Staphylococcal Lipoteichoic Acid Inhibits Delayed Type Hypersensitivity Reactions via the Platelet-activating Factor Receptor. J Clin Invest. 2005;115:2855–2861. doi: 10.1172/JCI25429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf P, Nghiem DX, Walterscheid JP, Byrne S, Matsumura Y, Bucana C, Ananthaswamy HN, Ullrich SE. Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol. 2006;169:795–805. doi: 10.2353/ajpath.2006.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, Yao Y, Konger RL, Sinn AL, Cai S, Pollok KE, Travers JB. UVB radiation-mediated inhibition of contact hypersensitivity reactions is dependent on the platelet-activating factor system. J Invest Dermatol. 2008;128:1780–1787. doi: 10.1038/sj.jid.5701251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, Travers JB. Ultraviolet B Radiation Generated Platelet-activating Factor Receptor Agonist Formation Involves EGF-R-mediated Reactive Oxygen Species. J Immunol. 2009;182:2842–2848. doi: 10.4049/jimmunol.0802689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahu RP, Turner MJ, Dasilva SC, Rashid BM, Ocana JA, Perkins SM, Konger RL, Touloukian CE, Kaplan MH, Travers JB. The environmental stressor ultraviolet B radiation inhibits murine antitumor immunity through its ability to generate platelet-activating factor agonists. Carcinogenesis. 2012;33:1360–1367. doi: 10.1093/carcin/bgs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehr HA, Weyrich AS, Saetzler RK, Jurek A, Arfors KE, Zimmerman GA, Prescott SM, McIntyre TM. Vitamin C blocks inflammatory platelet-activating factor mimetics created by cigarette smoking. J Clin Invest. 1997;99:2358–2364. doi: 10.1172/JCI119417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clauss M, Voswinckel R, Rajashekhar G, Sigua NL, Fehrenbach H, Rush NI, Schweitzer KS, Yildirim AAO, Kamocki K, Fisher AJ, Gu Y, Safadi B, Nikam S, Hubbard WC, Tuder RM, Twigg HL, 3rd, Presson RG, Sethi S, Petrache I. Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J Clin Invest. 2011;121:2470–2479. doi: 10.1172/JCI43881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishii S, Kuwaki T, Nagase T, Maki K, Tashiro F, Sunaga S, Cao WH, Kume K, Fukuchi Y, Ikuta K, Miyazaki J, Kumada M, Shimizu T. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J Exp Med. 1998;187:1779–1788. doi: 10.1084/jem.187.11.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 34.Pei Y, Barber LA, Murphy RC, Johnson CA, Kelley SW, Dy LC, Fertel RH, Nguyen TM, Williams DA, Travers JB. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol. 1998;161:1954–1961. [PubMed] [Google Scholar]
- 35.Yao Y, Harrison KA, Al-Hassani M, Murphy RC, Rezania S, Konger RL, Travers JB. Platelet-activating factor agonists mediate Xeroderma Pigmentosum A photosensitivity. J Biol Chem. 2012;287:9311–9321. doi: 10.1074/jbc.M111.332395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gruber F, Bicker W, Oskolkova OV, Tschachler E, Bochkov VN. A simplified procedure for semi-targeted lipidomic analysis of oxidized phosphatidylcholines induced by UVA irradiation. J Lipid Res. 2012;53:1232–1242. doi: 10.1194/jlr.D025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Countryman NB, Pei Y, Yi Q, Spandau D, Travers JB. Evidence for involvement of the platelet-activating factor receptor in ultraviolet B radiation-induced interleukin-8 production. J Invest Dermatol. 2000;115:267–272. doi: 10.1046/j.1523-1747.2000.00058.x. [DOI] [PubMed] [Google Scholar]
- 38.Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay-Jones JJ. Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J Exp Med. 1998;187:2045–2053. doi: 10.1084/jem.187.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byrne SN, Limon-Flores AY, Ullrich SE. Mast cell migration from the skin to the draining lymph nodes upon ultraviolet irradiation represents a key step in the induction of immune suppression. J Immunol. 2008;180:4648–4655. doi: 10.4049/jimmunol.180.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imaizumi T. Intravascular release of a platelet-activating factor-like lipid (PAF-LL) induced by cigarette smoking. Lipids. 1991;26:1269–1273. doi: 10.1007/BF02536545. [DOI] [PubMed] [Google Scholar]
- 41.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamimura Y, Iwai H, Piao J, Hashiguchi M, Azuma M. The glucocorticoid-induced TNF receptor-related protein (GITR)-GITR ligand pathway acts as a mediator of cutaneous dendritic cell migration and promotes T cell-mediated acquired immunity. J Immunol. 2009;182:2708–2716. doi: 10.4049/jimmunol.0803704. [DOI] [PubMed] [Google Scholar]
- 43.Limon-Flores AY, Chacon-Salinas R, Ramos G, Ullrich SE. Mast cells mediate the immune suppression induced by dermal exposure to JP-8 jet fuel. Toxicol Sci. 2009;112:144–152. doi: 10.1093/toxsci/kfp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaieb AM, Berman B, Smith B, Krumpe P. Epidermal Langerhans cell density in patients with pulmonary malignancies and chronic obstructive pulmonary disease. J Dermatol Surg Oncol. 1987;13:991–996. doi: 10.1111/j.1524-4725.1987.tb00575.x. [DOI] [PubMed] [Google Scholar]
- 45.Winter AP, Follett EA, McIntyre J, Stewart J, Symington IS. Influence of smoking on immunological responses to hepatitis B vaccine. Vaccine. 1994;12:771–772. doi: 10.1016/0264-410x(94)90283-6. [DOI] [PubMed] [Google Scholar]
- 46.McIntyre TM, Prescott SM, Stafforini DM. The emerging roles of PAF acetylhydrolase. J Lipid Res. 2009;50(Suppl):S255–259. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miwa M, Miyake T, Yamanaka T, Sugatani J, Suzuki Y, Sakata S, Araki Y, Matsumoto M. Characterization of serum platelet-activating factor (PAF) acetylhydrolase. Correlation between deficiency of serum PAF acetylhydrolase and respiratory symptoms in asthmatic children. J Clin Invest. 1988;82:1983–1991. doi: 10.1172/JCI113818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stafforini DM. PAF acetylhydrolase gene polymorphisms and asthma severity. Pharmacogenomics. 2001;2:163–175. doi: 10.1517/14622416.2.3.163. [DOI] [PubMed] [Google Scholar]
- 49.Miyaura S, Eguchi H, Johnston JM. Effect of a cigarette smoke extract on the metabolism of the proinflammatory autacoid, platelet-activating factor. Circ Res. 1992;70:341–347. doi: 10.1161/01.res.70.2.341. [DOI] [PubMed] [Google Scholar]
- 50.Ichimaru T, Tai HH. Alteration of platelet activating factor (PAF) metabolism in rat pulmonary alveolar macrophages and plasma by cigarette smoking. Prost Leukot Essent Fatty Acids. 1992;47:123–128. doi: 10.1016/0952-3278(92)90148-c. [DOI] [PubMed] [Google Scholar]