

Abstract
Trastuzumab (Herceptin®) is an effective targeted therapy in HER2 overexpressing human breast carcinoma. However, many HER2-positive patients initially or eventually become resistant to this treatment, so elucidating mechanisms of trastuzumab resistance that emerge in breast carcinoma cells is clinically important. Here we show that autocrine motility factor (AMF) binds to HER2 and induces cleavage to the ectodomain-deleted and constitutively active form p95HER2. Mechanistic investigations indicated that interaction of AMF with HER2 triggers HER2 phosphorylation and metalloprotease-mediated ectodomain shedding, activating PI3K and MAPK signaling and ablating the ability of trastuzumab to inhibit breast carcinoma cell growth. Further, we found that HER2 expression and AMF secretion were inversely related in breast carcinoma cells. Based on this evidence that AMF may contribute to HER2-mediated breast cancer progression, our findings suggest that AMF-HER2 interaction might be a novel target for therapeutic management of breast cancer patients whose disease is resistant to trastuzumab.
Introduction
HER2 (ERBB2/Neu), a family member of epidermal growth factor receptors (HERs) is overexpressed in ~ 25% of invasive breast carcinomas (1, 2, 3) and is a major approved target for breast cancer therapy. The crystal structure of HER2 suggests that its extracellular domain (ECD) exists in a constitutively active conformation resembling the ligand-bound state of the other HERs (4, 5), while, HER2-ECD targeting antibodies that are antagonistic or agonistic at the levels of HER2 phosphorylation and cell growth, suggest the presence of binding partner(s) necessary for complete activation of HER2 (1, 6, 7). Herceptin/Trastuzumab has improved the outcome in HER2 overexpressing breast carcinoma patients (8, 9). However, a substantial proportion of HER2-positive breast cancer patients is intrinsically resistant to Trastuzumab or acquires resistance following initial treatment (10). The mechanisms of resistance to Herceptin/Trastuzumab are mainly involved in the restoration of the phosphoinositide-3-kinase (PI3K)/AKT signaling pathways either via an epitope masking (Mucin) and escaping (truncated p95HER2), alternative compensation of receptor tyrosine kinases, or the constitutive mutations of PI3K pathways (10, 11, 12). Retrospective studies suggest that the oncogenic p95HER2 variant is most likely responsible for clinical resistance to Herceptin/Trastuzumab treatment (13, 14).
Phosphoglucose isomerase (EC: 5.3.1.9) (PGI) is a housekeeping dimeric enzyme that catalyzes the reversible isomerization of glucose-6-phosphate and fructose-6-phosphate in glycolysis/gluconeogenesis (15). PGI belongs to the moonlighting family of proteins having multiple functions/activities within a single polypeptide chain, not resulting from multiple domains of a protein, alternative RNA splicing, gene fusions, and/or post-translational processing (16). Secreted form of PGI in the extracellular milieu of transformed cells and several tissues was identified as neuroleukin (NLK), a neurotrophic factor that mediates the differentiation of neurons and autocrine motility factor (AMF), a tumor-secreted C-X-X-C cytokine that is involved in cell motility (17, 18). Aberrant secretion of AMF was observed in the blood and urine of cancer patients, suggesting a prognostic value (15, 19). Functionally, AMF was shown to induce cell proliferation, differentiation, and survival of various cancer and immune cells (15). Independent reports have shown that AMF activates mitogenic MAPK/ERK or pro-survival PI3K/AKT pathways, similarly to the signaling mode of growth factors as emphasized in the resistance to HER2-targeted therapy (20, 21). The receptor of AMF i.e., gp78/AMFR was identified as a seven transmembrane domain containing protein. However, gp78/AMFR-null cells still respond to AMF, suggesting the presence of yet another unidentified receptor (22, 23).
Here, we show that in human breast carcinoma cells AMF binds to HER2, induces its phosphorylation, ectodomain shedding, activates its downstream signaling pathways and overcomes Heceptin/Trastuzumab effect. The data suggest that AMF may be a novel therapeutic target for breast cancer patients in conjunction with Heceptin/Trastuzumab therapy.
Materials and Methods
Antibodies and Chemicals
Purified rabbit phosphoglucose isomerase (PGI/AMF) was purchased from Sigma for AMF stimulation. Monoclonal anti-PGI (12F9A6, Pfizer) and rabbit anti-PGI (H300, Santa Cruz) antibodies were used for Western blot and immunoprecipitation. p-ERK (E-4), ERK1/2(MK1), p-Tyr (PY20), anti-HER2-ICD (Neu, C-18), anti-HER2-ECD (9G6), p-HER2 antibodies and Lapatinib were purchased from Santa Cruz. Anti-p-AKT (Ser473) and AKT antibodies were from Cell Signaling. Anti-rabbit IgG-TRITC and anti-IgG-FITC antibodies, Marimastat (BB2516), lysophophatidic acid, pertussis toxin (P2980) were purchased from Sigma. Wortmannin and U0126 were obtained from Calbiochem. 3, 3 ′-Dithiobis(sulfosuccinimidylpropionate) (DTSSP) was purchased from Pierce. Trastuzumab was a kind gift from Dr. Wei-Zen Wei of Wayne State University. Anti-V5, anti-HER2-ECD antibodies (poly-2 and CB11 clone), siRNAs against gp78, HER2 and AMF were purchased from Invitrogen. MTT [3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] was purchased from Sigma.
Cell culture and treatments
T47D and EBNA 293 cells obtained from American Type Culture Collection (ATCC) were grown in DMEM supplemented with 10% FBS and antibiotics. SkBr3, BT474 were kindly gifted by Dr. Arun Rishi of Wayne State University. SkBr3 cells were cultured in complete McCoy’s Modified 5A Medium. Before pretreatment with inhibitors or addition of stimulators (EGF, AMF), 50% confluent cells were rinsed two times with 1X phosphate saline buffer (PBS) and then serum-starved for 16 hr.
Cross-linking with DTSSP was performed to identify interaction of AMF (AMF-V5) and HER2. T47D cells were washed with 1X PBS and then exposed to AMF (AMF-V5) along with DTSSP for 1hr at 4°C. Reactions were terminated by the addition of 20 mM Tris–HCl (pH 7.5) for 15 min at room temperature. Cells were extracted with lysis buffer and insoluble material was removed by centrifugation and supernatants were processed for immunoprecipitation.
Transfection and RNA interference
Expression plasmid of AMF-V5 was constructed in pCDNA3.1/V5 (Invitrogen) harboring human full-length PGI/AMF and confirmed by sequence analysis. CMV-HER2 (p185HER2) was purchased from Addgene and transiently transfected into EBNA293 cells using Fugene HD (Roche). Breast cancer cells were seeded at 50% confluence per well in six well plates overnight and transfected for 48 hr with 20 nM siRNAs using Lipofectamine™ RNAiMAX Reagent (Invitrogen) according to the manufacturer’s instructions.
Western blotting and Immunoprecipitation
Western blotting and immunoprecipitation was performed according to routine protocol. Briefly, the cells were extracted in lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 2.5 mM Sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml Leupeptin]. After BCA protein assay (Pierce), immunoprecipitation with appropriate antibodies (1 μg) was carried out overnight. Each experiment was repeated at least, twice.
Immunofluorescence and Immunohistochemistry
For immunofluorescence studies, SkBr3 cells grown on 12 mm round coverslips (Fisher) were processed after AMF-V5 treatment for 15 min. The cells were washed and fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 5 min. For double immunofluorescence labelling, the cells were incubated with primary antibodies (anti-HER2 and anti-V5) for 1hr separately to minimize the cross reactivity. After three washes, appropriate secondary antibodies (FITC-conjugated, TRITC-conjugated) were also incubated separately. Immunofluorescence images were obtained using a Zeiss Confocal Laser Microscope LSM 510. 4 micron sections obtained from formalin fixed, paraffin embedded tissue blocks were deparaffinized, rehydrated and microwaved on high 2X for five minutes in 1mM sodium citrate buffer, pH 6.0. The sections were washed 3X in PBS and blocked with Super Block (Skytek Laboratories) for 10 min. Sequential sections were then incubated in PBS and linked with biotinylated mouse anti-AMF antibodies at 4°C overnight. The sections were then washed 3X for 10 min each in PBS and linked with biotinylated anti-mouse secondary antibodies (Vector Laboratories) followed by Texas Red conjugated Avidin. After washing, the sections were incubated at 4°C overnight with rabbit anti-HER2 antibodies (Invitrogen) and with fluorescein conjugated anti-rabbit secondary antibodies. Images were documented with an OLYMPUS BX40 microscope supported by a DP72 CCD Camera, and CellSens Dimension Imaging Software (Olympus).
Cell growth assay
BT474 cells were seeded at a density of 5×104 cells per well in 24 well plates. After 24 hr, cells were treated with either 40 μg/ml Trastuzumab or a combination of AMF (90 nM) with Trastuzumab. Cell growth was monitored at 550 nm after MTT assay. All experiments were performed in quadruplicates and repeated twice. Statistical analysis was done using Student t test. P < 0.005 was regarded as significant.
Cell Migration Assay
Transwell (Corning Costar) migration assays were conducted on SkBr3 cells following pre-treatment with Trastuzumab for 16 hr. 5×104 SkBr3 cells in serum-free medium were introduced into the upper compartment of transwell chambers (8 μM pore) and were allowed to migrate in lower chambers with 10% FBS or AMF (0.1 μg/ml) for 16 hr. Migrated cells were fixed and stained with Hema-3 stain kit (Fisher). Each condition was assayed in triplicate and each experiment was repeated three times.
Results
AMF induces PI3K/MAPK signaling pathways in a HER family dependent manner
To study whether AMF acts independently of HER2 signaling, we initially established whether AMF induces PI3K/MAPK signaling pathways through gp78/AMFR in low HER2 expressing breast cancer cell line T47D. Treatment with purified AMF showed that it activates both PI3K/AKT and MAPK/ERK (Fig. 1A–C). Furthermore, gp78/AMFR knockdown study revealed that gp78/AMFR mediates AMF-induced p-ERK but not p-AKT, suggesting involvement of another AMF receptor for AKT activation (Supplementary Fig. S1). Since participation of a pertussis-toxin sensitive G protein in the signal transduction of AMF-stimulated cell motility was previously reported (24), we ventured that presence of yet another AMF receptor might be a G protein coupled receptor (GPCR), which transactivates the HER family through ectodomain shedding of membrane-bound HER ligand precursors by metalloproteases (MMPs), resulting in the activation of PI3K/MAPK pathways (25). Therefore, we tested whether AMF is involved in the transactivation of the HER family for AKT activation. Interestingly, the results show that AMF-induced p-AKT/p-ERK was partially suppressed by Lapatinib (dual kinase inhibitor of EGFR and HER2) compared with control and EGF, indicating that AMF-induced p-AKT/p-ERK is mediated by HER receptors and implying the responsibility of gp78/AMFR for partial suppression by Lapatinib (Fig. 1D). To study whether a GPCR mediates AMF-induced p-AKT/p-ERK, we performed suppression studies using pertussis-toxin (G protein inhibitor) or Marimastat (MMPs inhibitor) for blocking GPCR signaling. We found that these inhibitors did not suppress AMF-induced p-AKT/p-ERK (Fig. 1E), signifying that AMF signaling is not linked to GPCR but independently activates EGFR and/or HER2.
Figure 1. AMF induces PI3K/MAPK signaling pathways in a HER family dependent manner in T47D cells.
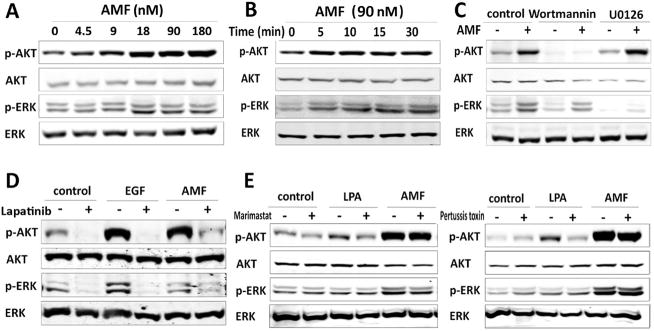
A, Cells serum-starved for 16 hr were stimulated with purified AMF, p-AKT and p-ERK levels were monitored in dose-dependent treatment for 15 min and B, for 5 to 30 min with AMF (90 nM). C, Suppression of AMF-induced p-AKT/p-ERK by 100 nM Wortmannin (PI3K inhibitor) and 20 μM U0126 (MAPK inhibitor). Serum-starved cells were pretreated with inhibitors for 1hr before AMF stimulation for 15 min. D, Effect of Lapatinib, dual tyrosine kinase inhibitor (10 μM) on p-AKT/p-ERK expression. Serum starved cells pretreated with Lapatinib for 1hr and stimulated by AMF (90 nM) or EGF (16 nM) for 15 min. Control indicates bovine serum albumin. E, After pretreatment with broad spectrum MMPs inhibitor (50 μM, Marimastat) for 1hr or 0.2 ng/ml pertussis toxin (G protein transduction inhibitor) for 16 hr, the cells were stimulated with AMF or lysophosphatidic acid (LPA, 10 μM), a GPCR stimulator for 15 min.
AMF induces phosphorylation of HER2 and enhances its cleavage
As HERs phosphorylation is induced by homo- and hetero-dimerization following the engagement of HER ligand and a cognate receptor (2), we addressed whether AMF induces the phosphorylation of EGFR and/or HER2. As shown in Fig. 2A, phosphorylation of both EGFR and HER2 were induced by exogenous EGF, which does not bind to HER2 (2, 3), implying EGFR/HER2 hetero-dimerization. While AMF did not induce phosphorylation of EGFR and kinetic studies indicated that AMF induces the phosphorylation of HER2 within a very short time frame of 2 min (Fig. 2B). Moreover, we found that both background p-HER2 and unphosphorylated p185HER2 vanished concomitantly with the emergence of cleaved p95HER2. As ectodomain shedding of p185HER2 was previously reported to generate membrane-anchored p95HER2 (95 to 100 kDa) by sheddases such as metalloproteases (MMPs) at the juxtamembrane region of HER2 (26), we addressed whether HER2 cleavage is directly affected by AMF or is mediated by MMPs. MMPs inhibitor suppressed AMF-induced cleavage of p185HER2 and also stabilized phosphorylated HER2 by AMF (Fig. 2C), indicating that AMF is sufficient for the phosphorylation of HER2 and AKT activation and also cooperates with MMPs for HER2 cleavage.
Figure 2. AMF induces phosphorylation of HER2 and its cleavage.

A, T47D cells starved and stimulated by AMF or EGF were immunoprecipitated with anti-phosphotyrosine antibodies (p-Y). Input lysate indicates lysate used for immunoprecipitation. B, HER2 phosphorylation and cleavage were monitored following AMF treatment in T47D cells at indicated time points, using immunoprecipitation (IP) with anti-HER2-ICD (epitope: intracellular domain). C, Marimastat-pretreated T47D cells were stimulated by AMF for 15 min and immunoprecipitation was performed with anti-HER2-ECD (epitope: ectodomain, 9G6 clone). D, HER2 status in EBNA 293 cells transiently transfected with CMV- HER2 (wild type) or vector (-) for 24 hr and then starved and stimulated with AMF for 15 min. E, HER2 transfected and starved EBNA 293 cells were pretreated with Marimastat and stimulated with AMF for 15 min. NS indicates non-specific. F, T47D cells were pretreated with siRNA-HER2 (20 nM) for 48hr and then stimulated by AMF for 5 min after cells starvation. (-) indicates control siRNA. Control indicates bovine serum albumin.
Parental EBNA293 cells did not show AKT/ERK activation after AMF treatment (Supplementary Fig. S2). To verify that AMF indeed triggers phosphorylation and participates in the cleavage of HER2, the p185HER2 expression vector was transfected into EBNA293 cells. The results showed that p-HER2 induction in response to AMF appeared together with cleaved p95HER2 (Fig. 2D) and AMF-induced HER2 cleavage was mediated by MMPs (Fig. 2E), consistent with the above results (Fig. 2B–C). We confirmed the existence of AMF/HER2/AKT axis by HER2 knockdown in T47D cells, in which AMF-induced AKT activation was partially suppressed (Fig. 2F).
AMF interacts with the extracellular domain of HER2
Since the data indicate that AMF induces phosphorylation of HER2, we tested the interaction of exogenously added AMF and HER2, employing a membrane impermeable cross-linker (DTSSP), which establishes extracellular association of proteins. Exogenous AMF was co-immunoprecipitated with HER2 (Fig. 3A, top). To further verify this interaction V5-tagged AMF (AMF-V5) was expressed and purified to rule out any possible background experimental noise from the endogenous AMF. The interaction of exogenous AMF-V5 with HER2 was clearly observed by reciprocal co-immunoprecipitation studies (Fig. 3A, middle and bottom). Additionally, immunofluorescence studies revealed the co-localization of HER2 and exogenous AMF-V5 (Fig. 3B).
Figure 3. AMF interacts with HER2.

A, Serum-starved T47D cells were treated simultaneously with AMF and DTSSP to crosslink extracellular proteins for 1hr at 4°C. Immunoprecipitation of lysate was performed as indicated and analyzed by Western blot of SDS-PAGE in reducing conditions, which reverses the crosslinking and allows for identifying interacting proteins. Immunoprecipitation was performed with anti-HER2-ICD following AMF treatment (top). AMF-V5 treated and cross-linked cells were used for reciprocal immunoprecipitation with anti-HER2-ICD (middle) or anti-V5 (bottom). Control indicates AMF or AMF-V5 obtained from supernatants after their treatment into cells. B, Co-localization of HER2 and treated AMF-V5 in serum-starved SkBr3 cells. Merge is shown as HER2, AMF-V5, and DAPI (nuclear stain, blue). Control indicates conditioned medium of EBNA293 cells transiently transfected with vector (pCDNA3.1/V5) alone. Purified AMF-V5 was treated to cells for 15 min. C, AMF interacts with ectodomain of HER2. EBNA 293 cells transfected with CMV-HER2 or vector were extracellularly cross-linked along with the treatment of EGF, AMF, and AMF-V5 respectively as described. The lysates were immunoprecipitated with anti-HER2 antibodies recognizing different epitopes: ECD, ICD, JM (juxtamembrane). Control indicates bovine serum albumin. D, Prediction of interaction of HER2-ECD with human AMF. Ectodomain of HER2 is displayed in the ball form and human AMF in the ribbon representation. I, II, III, and IV indicate different domains of HER2-ECD.
To elucidate whether AMF interacts with the ectodomain of HER2, HER2-transfected cells were extracellularly cross-linked with either exogenous AMF or AMF-V5. We observed that AMF or AMF-V5 cross-linked HER2 molecules were not immunoprecipitated with anti-HER2-ECD antibodies and immunoprecipitated HER2 levels were reduced compared to Trastuzumab (juxtamembrane) or anti-HER2-ICD antibodies (Fig. 3C), suggesting that AMF-HER2 binding motif is either overlapping or close to the epitope of anti-HER2-ECD antibody. Next, taking advantage of the data obtained from the crystal structures of AMF and the extracellular domain of HER2 (4, 27), we employed the ZDOCK software (http://zdock.bu.edu) to predict their physical interactions. AMF binding to HER2 ectodomain was mapped to the molecular region of HER2 between domain I and III (Fig. 3D)
AMF overcomes the Hereceptin/Trastuzumab suppression of PI3K/MAPK signaling pathways
As HER2 overexpressing cells have a high expression level of endogenous p-HER2 via a ligand independent mechanism (1, 2, 3), we examined how AMF is able to affect endogenous p-HER2 and HER2 in HER2 overexpressing cells. Also, we questioned whether AMF-induced downstream events might be blocked by anti-HER2-ECD antibodies, which can compete with AMF for binding to HER2. As shown in Fig. 4A, the treatment with anti-HER2-ECD antibodies alone showed that it induced p-HER2 levels in concert with HER2 cleavage and AKT/ERK activation, mimicking AMF actions. Combination of AMF and anti-HER2-ECD antibodies shows synergistic effect. Interestingly, HER2 shedding was induced by exogenous AMF regardless of the presence of Herceptin/Trastuzumab in the conditioned media. AMF treatment overcame Herceptin/Trastuzumab mediated suppression of HER2 cleavage and p-AKT/p-ERK levels.
Figure 4. AMF overcomes inhibitory effect of Trastuzumab on cell growth and migration.

A, Breast cancer cells were starved and treated with Trastuzumab or anti-HER2-ECD (9G6, 20 μg/ml) for 16 hr and then stimulated with AMF for 15 min. Quantification of p95HER2 represents the average of two experiments and error bars indicate standard deviation. B. p95HER2 was analyzed in lysate of BT474 cells grown in complete medium and pretreated with Trastuzumab and then stimulated with AMF for 1hr. C. BT474 cells were grown for different durations with Trastuzumab (Tras) and a combination of AMF (90 nM) with Trastuzumab. The cell growth was monitored using MTT assay. D. Effect of AMF on Trastuzumab-inhibited cell migration. SkBr3 cells were pretreated with Trastuzumab or control IgG (10 μg/ml) for 24 hr, and migrated cells were then measured after 16 hr in transwell assays. Quantitative data from three different experiments were presented. *P < 0.05. Error bars represent SEM.
Next, as AMF-induced p95HER2 is an indicator of Trastuzumab resistance through epitope escaping (10), we proceeded to validate the effect of AMF on cell growth and motility with Trastuzumab treatment. We observed AMF-induced HER2 cleavage in cells treated with Trastuzumab in complete medium (Fig. 4B). As a result, exogenous AMF partially overcame the inhibitory effect of Trastuzumab on cell growth and motility (Fig. 4C, D).
AMF secretion is controlled by HER2 expression
Next, we examined the relationship between AMF and HER2 expression. AMF secretion was observed to be positively correlated with the expression of both p95HER2 and p185HER2 proteins (Supplementary Fig. S3). Sequentially, we found that HER2 knockdown inhibits AMF secretion, suggesting that AMF secretion is regulated, at least in part by HER2 signaling in breast carcinoma cells (Fig. 5A). AMF knockdown did not affect HER2 expression, while reduced p-HER2 levels along with suppression of AKT/ERK activation in T47D cells (Fig. 5B). Additionally, the blocking of AMF function partially inhibited the phosphorylation of HER2, resulting in the reduction of p-AKT/p-ERK levels (Fig. 5C) and endogenously secreted AMF does not significantly affect the cleavage of HER2 in HER2 overexpressing cells (Fig. 5C).
Figure 5. Relationship of AMF and HER2 expression in breast cancer tissues.

A, Cells were treated with siRNA-control (-) or siRNA-HER2 for 24 hr and then shifted to serum-free medium for 24 hr (top). Cells were treated with siRNAs for 48 hr and then starved for the indicated time points (bottom). Secreted AMF levels were analyzed in culture medium. B, Western blot analysis of cells treated with control siRNA or siRNA-AMF (20 nM) for 48hr and then shifted into serum-free medium. C, Control IgG or monoclonal anti-AMF antibodies (10μg/ml) were added to serum-free culture medium for 24hr. (-) indicates no treatment. p-HER2 and downstream signaling pathways of HER2 were analyzed. Ten μg of whole cell lysate were loaded for detecting p-HER2 levels and 25 μg of whole cell lysate were used for detecting the p95HER2 fragment. D. Dual-immunohistochemistry of human HER2 positive breast carcinoma and MCF10A DCIS.com xenograft. Merge shows AMF (red), HER2 (green) and DAPI (blue, staining nuclei).
Finally, to further convince that AMF-HER2 relationship is not only an in vitro phenomena, we performed two independent immunohistochemistry studies utilizing human breast cancer tumors and MCF10A DCIS.com xenograft, which is a mouse model of human comedo ductal carcinoma in situ (28). Compared with the surrounding stroma, both HER2 and AMF were found to be strongly expressed in the human breast carcinoma epithelium, and especially co-localized on the cell membranes of MCF10A DCIS.com xenograft (Fig. 5D).
Discussion
The studies presented above show that exogenous AMF contributes to Herceptin/Trastuzumab resistance of human breast carcinoma cells via generation of p95HER2 fragment. Since AMF interacts with HER2 and AMF-interacting HER2 is sensitive to HER2 shedding by MMPs, we hypothesize that the interaction between AMF and HER2 could structurally expose the MMPs enzymatic clip site at HER2 juxtamembrane region. The expression levels and activation of MMPs might be considered a critical limiting step of HER2 shedding due to partial recovery by AMF in Trastuzumab-inhibited cell growth and motility (Fig. 4C–D). Despite the inhibition of MMPs, presence of AMF was sufficient for phosphorylation of HER2 and activation of PI3K/MAPK (Fig. 2C–D), suggesting that AMF can act as a HER2 binding-partner enhancing its activation. Weather AMF-interacting HER2 underdoes heterodimerization with another member of HER2 family for PI3K/MAPK activation is yet to be determined.
Because both AKT and ERK activation occurs mostly in the cellular signaling of receptor tyrosine kinase (RTK), the mechanism in which gp78/AMFR knockdown showed the suppression of AMF-activated ERK but not AMF-induced AKT is poorly understood (Supplementary Fig. S1). Of note, gp78 signaling-suppress ERK activation and also acts as an E3 ligase in the cytoplasm (29).
Transgenic mouse models harboring rat full-length p185HER2 (Neu) recapitulated the initial events of HER2-induced mammary tumorigenesis. A long latency period for tumor initiation is required for the acquisition of activating mutations such as in-frame deletion or insertion at the juxtamembrane region of Neu transgene (30, 31, 32). As these mutations have not been found in human breast cancer, several studies have suggested epigenetic mechanisms for the expression of truncated p95HER2, e.g. alternative translation initiation or an alternative spliced form of HER2 resembling Neu-derived somatic mutations (33, 34, 35). In this respect, p95HER has been found in subgroups of human breast carcinoma patients, resulting in constitutively hyperactive dimers with intermolecular disulfide bonds (26, 36). Transgenic recapitulation studies showed that p95HER2 transgene-induced tumors lead to more aggressive and metastatic phenotype with relatively short latency period compared with full length HER2-induced tumors (37). p95HER2 fragments are suggested to arise through mechanisms of proteolytic shedding by MMPs or translation of HER2 internal initiation codons (26, 38). In the present study, AMF-enhanced HER2 shedding suggests that AMF participates in HER2 oncogenic and aggressive progression through the epigenetic event of AMF secretion. As HER2 overexpression positively controls AMF secretion (Fig. 5A), the positive feedback loop of HER2 overexpression, AMF secretion and p95HER2 generation could be speculated in HER2-induced cancer progression. AMF secretion may also be controlled by other mechanisms, as the secretion level of AMF is higher in high motility cells (MDA-MB-231) compared with low motility cells (T47D), regardless of the similar expression levels of HER2 by both the cell lines (Supplementary Fig. S4).
Although the mechanisms of resistance to Herceptin/Trastuzumab by breast tumors have been delineated at a preclinical level, it is still to be established in patients and remain a challenge for the clinicians. In vitro studies suggested Herceptin/Trastuzumab could inhibit the shedding of p185HER2 (36, 39) and retrospective studies in breast cancer patients revealed a high association between the predisposition of p95HER2 and clinical resistance to Herceptin/Trastuzumab (8, 9). Previously, AMF was identified as a serum tumor marker in breast cancer patients (18). It was also reported that Herceptin could effectively block both ligand-induced and constitutive expression of AMF associated with high HER2 overexpression, implying a role of the AMF pathway in the action of Herceptin (40). Here, we have documented that AMF treatment of breast carcinoma cells suppresses the effect of Herceptin/Trastuzumab by overcoming both p-AKT/p-ERK levels and protecting cell growth through p95HER2 generation (Fig. 4). We suggest that high AMF-secreting HER2 positive cancer cells could initially and intrinsically have the advantage of resistance to Herceptin/Trastuzumab. Therefore, we propose that AMF might be a novel therapeutic target in HER2 overexpressing breast cancer patients and this target should be considered in combination with Heceptin/Trastuzumab therapy. In summary, AMF might play a role in HER2-driven breast cancer progression and initial resistance of Trastuzumab as depicted in Fig. 6.
Figure 6. Schematic representation of positive feedback loop of HER2 overexpression, AMF secretion, and p95HER2 generation in HER2-driven progression and Trastuzumab resistance.
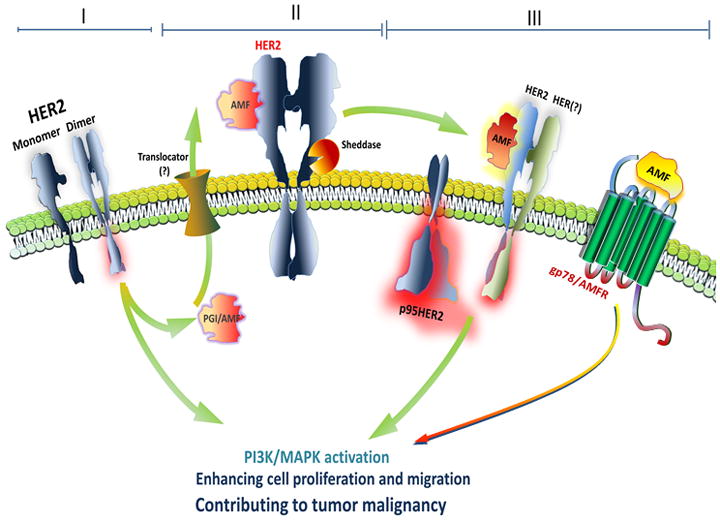
HER2 overexpression (I) enhances AMF secretion through non-classical mechanism due to lack of its signal sequence and thereby AMF interacts with HER2 (II), resulting in HER2 phosphorylation and HER2 cleavage with sheddases (III). Cleaved and constitutively active p95HER2 induces aggressive progression and p95HER2 expressing cells are resistant to Trastuzumab therapy owing to p95HER2 which lacks the epitope of HER2 recognized by Trastuzumab. Interaction of AMF and gp78 might contribute to Trastuzumab resistance.
Supplementary Material
Acknowledgments
We are grateful to Dr. Wei-Zen Wei and Dr. Arun Rishi at Wayne State University for the gift of Trastuzumab, SkBr3 and BT474 cells respectively. We thank Dr. P.V.M. Shekhar for critical evaluation of the manuscript.
Grant support: This work was supported by NIH/National Cancer Institute R01 CA51714 (A. Raz).
References
- 1.Moasser MM. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene. 2007;26:6577–92. doi: 10.1038/sj.onc.1210478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–4. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 3.Moasser M. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–87. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–60. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 5.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–52. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 6.Kita Y, Tseng J, Horan T, Wen J, Philo J, Chang D, et al. ErbB receptor activation, cell morphology changes, and apoptosis induced by anti-Her2 monoclonal antibodies. Biochem Biophys Res Commun. 1996;226:59–69. doi: 10.1006/bbrc.1996.1311. [DOI] [PubMed] [Google Scholar]
- 7.Stancovski I, Hurwitz E, Leitner O, Ullrich A, Yarden Y, Sela M. Mechanistic aspects of the opposing effects of monoclonal antibodies to the ERBB2 receptor on tumor growth. Proc Natl Acad Sci USA. 1991;88:8691–5. doi: 10.1073/pnas.88.19.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pohlmann PR, Mayer IA, Mernaugh A. Resistance to Trastuzumab in Breast Cancer. Clin Cancer Res. 2009;15:7479–91. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 10.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;29:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 11.Nagy P, Friedländer E, Tanner M, Kapanen AI, Carraway KL, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65:473–82. [PubMed] [Google Scholar]
- 12.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Scaltriti M, Rojo F, Ocaña A, Anido J, Guzman M, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99:628–38. doi: 10.1093/jnci/djk134. [DOI] [PubMed] [Google Scholar]
- 14.Sáez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, et al. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin Cancer Res. 2006;12:424–31. doi: 10.1158/1078-0432.CCR-05-1807. [DOI] [PubMed] [Google Scholar]
- 15.Fairbank M, St-Pierre P, Nabi IR. The complex biology of autocrine motility factor/phosphoglucose isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin ligase. Mol Biosyst. 2009;5:793–801. doi: 10.1039/b820820b. [DOI] [PubMed] [Google Scholar]
- 16.Jeffery CJ. Moonlighting proteins--an update. Mol Biosyst. 2009;5:345–50. doi: 10.1039/b900658n. [DOI] [PubMed] [Google Scholar]
- 17.Chaput M, Claes V, Portetelle D, Cludts I, Cravador A, et al. The neurotrophic factorneuroleukin is 90% homologous with phosphohexose isomerase. Nature. 1988;332:454–5. doi: 10.1038/332454a0. [DOI] [PubMed] [Google Scholar]
- 18.Liotta LA, Mandler R, Murano G, Katz DA, Gordon RK, et al. Tumor cell autocrine motility factor. Proc Natl Acad Sci USA. 1986;83:3302–6. doi: 10.1073/pnas.83.10.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumann M, Kappl A, Lang T, Brand K, Siegfried W, Paterok E. The diagnostic validity of the serum tumor marker phosphohexose isomerase (PHI) in patients with gastrointestinal, kidney, and breast cancer. Cancer Invest. 1990;8:351–6. doi: 10.3109/07357909009012053. [DOI] [PubMed] [Google Scholar]
- 20.Tsutsumi S, Hogan V, Nabi NR, Raz A. Overexpression of the autocrine motility factor/phosphoglucose isomerase induces transformation and survival of NIH-3T3 fibroblasts. Cancer Res. 2003;63:242–9. [PubMed] [Google Scholar]
- 21.Araki K, Shimura T, Yajima T, Tsutsumi S, Suzuki H, et al. Phosphoglucose isomerase/autocrine motility factor promotes melanoma cell migration through ERK activation dependent on autocrine production of interleukin-8. J Biol Chem. 2009;284:32305–11. doi: 10.1074/jbc.M109.008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimizu K, Tani M, Watanabe H, Nagamachi Y, Niinaka Y, et al. The autocrine motility factor receptor gene encodes a novel type of seven transmembrane protein. FEBS Lett. 1999;456:295–300. doi: 10.1016/s0014-5793(99)00966-7. [DOI] [PubMed] [Google Scholar]
- 23.Haga A, Komazaki S, Funasaka T, Hashimoto K, Yokoyama Y, et al. AMF/G6PI induces differentiation of leukemic cells via an unknown receptor that differs from gp78. Leuk Lymphoma. 2006;47:2234–43. doi: 10.1080/10428190600773263. [DOI] [PubMed] [Google Scholar]
- 24.Evans CP, Walsh DS, Kohn EC. An autocrine motility factor secreted by the Dunning R-3327 rat prostatic adenocarcinoma cell subtype AT2. 1. Int J Cancer. 1991;49:109–13. doi: 10.1002/ijc.2910490120. [DOI] [PubMed] [Google Scholar]
- 25.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 26.Arribas J, Baselga J, Pedersen K, Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011;71:1515–19. doi: 10.1158/0008-5472.CAN-10-3795. [DOI] [PubMed] [Google Scholar]
- 27.Read J, Pearce J, Li X, Muirhead H, Chirgwin J, Davies C. The crystal structure of human phosphoglucose isomerase at 1. 6 A resolution: implications for catalytic mechanism, cytokine activity and haemolytic anaemia. J Mol Biol. 2001;309:447–63. doi: 10.1006/jmbi.2001.4680. [DOI] [PubMed] [Google Scholar]
- 28.Shekhar MP, Nangia-Makker P, Tait L, Miller F, Raz A. Alterations in galectin-3 expression and distribution correlate with breast cancer progression: functional analysis of galectin-3 in breast epithelial-endothelial interactions. Am J Pathol. 2004;165:1931–41. doi: 10.1016/S0002-9440(10)63245-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. Proc Natl Acad Sci USA. 2001;98:14422–7. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–97. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 31.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol. 1994;14:7068–77. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 34.Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23:62–8. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 35.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–64. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arribas J, Parra-Palau JL, Pedersen K. HER2 fragmentation and breast cancer stratification. Clin Cancer Res. 2010;16:4071–3. doi: 10.1158/1078-0432.CCR-10-1501. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen K, Angelini PD, Laos S, Bach-Faig A, Cunningham MP, Ferrer-Ramón C, Luque-García A, et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol Cell Biol. 2009;29:3319–31. doi: 10.1128/MCB.01803-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsé C, Gauchez AS, Jacot W, Lamy PJ. HER2 shedding and serum HER2 extracellular domain: Biology and clinical utility in breast cancer. Cancer Treat Rev. 2011;38:133–42. doi: 10.1016/j.ctrv.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61:4744–9. [PubMed] [Google Scholar]
- 40.Talukder AH, Bagheri-Yarmand R, Williams RR, Ragoussis J, Kumar R, Raz A. Antihuman epidermal growth factor receptor 2 antibody herceptin inhibits autocrine motility factor (AMF) expression and potentiates antitumor effects of AMF inhibitors. Clin Cancer Res. 2002;8:3285–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.