

Abstract
Metal-catalyzed stereoselective reactions are a central theme in organic chemistry research. In these reactions, the stereoselection is achieved predominantly by introducing chiral ligands at the metal catalyst’s center. For decades, researchers have sought better chiral ligands for asymmetric catalysis and have made great progress. Nevertheless, to achieve optimal stereoselectivity and to catalyze new reactions, new chiral ligands are needed.
Due to their high metal affinity, hydroxamic acids play major roles across a broad spectrum of fields from biochemistry to metal extraction. Dr. K. Barry Sharpless first revealed their potential as chiral ligands for asymmetric synthesis in 1977: He published the chiral vanadium-hydroxamic-acid-catalyzed, enantioselective epoxidation of allylic alcohols before his discovery of Sharpless Asymmetric Epoxidation, which uses titanium-tartrate complex as the chiral reagent. However, researchers have reported few highly enantioselective reactions using metal-hydroxamic acid as catalysts since then.
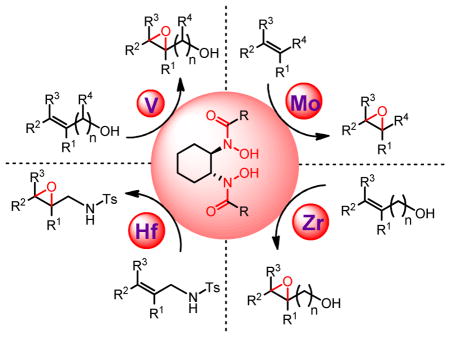
This Account summarizes our research on metal-catalyzed asymmetric epoxidation using hydroxamic acids as chiral ligands. We designed and synthesized a series of new hydroxamic acids, most notably the C2-symmetric bis-hydroxamic acid (BHA) family. V-BHA-catalyzed epoxidation of allylic and homoallylic alcohols achieved higher activity and stereoselectivity than Sharpless Asymmetric Epoxidation in many cases. Changing the metal species led to a series of unprecedented asymmetric epoxidation reactions, such as (i) single olefins and sulfides with Mo-BHA, (ii) homoallylic and bishomoallylic alcohols with Zr- and Hf-BHA, and (iii) N-alkenyl sulfonamides and N-sulfonyl imines with Hf-BHA. These reactions produce uniquely functionalized chiral epoxides with good yields and enantioselectivities.
1. Introduction
Modern asymmetric catalysis has been prosperous for decades thanks to its three major cornerstones: metal catalysis, biocatalysis and organocatalysis. Among the “trio”, asymmetric metal catalysis is the major player in both lab and industry settings.1 In these reactions, the role of asymmetry induction has usually been played by chiral ligands.2 Those ligands interact with the metal center to form catalytically active metal complexes, which catalyze desired stereoselective reactions. By introducing the concept of “privileged chiral catalysts”,3 the metal-ligand-reaction relationship has evolved from single dots, where a specific metal species with a specific category of ligands catalyzes a specific reaction, to a cross-linked network. Now people may choose any combinations of metal species and ligands, and any combination may be able to efficiently catalyze an unexpected reaction.
In light of the central role of chiral ligands in asymmetric metal catalysis, to discover new chiral ligands has been an essential activity of contemporary researchers of asymmetric synthesis. In this account, we present the brief history and development of chiral hydroxamic acid ligands for metal-catalyzed oxidation reactions from our lab and others. Another mission of this article is to promote future attention to this unique class of molecules that may bring interesting new chemistry to life since there have not been many reports using them for asymmetric organic reactions.
2. Structure and Synthesis
Hydroxamic acids resemble amides in structure, with one of the amide N-H substituted by an N-OH group (Figure 1). When deprotonated, the hydroxamate form (O−) is highly coordinative to a variety of metal ions via two oxygen groups.4 In addition, carbon groups R1 and R2 on C=O and N provide chirality for asymmetric induction. Apparently, hydroxamic acids have met the two necessary and sufficient conditions for a chiral ligand: interaction with metal and chirality.
Figure 1.
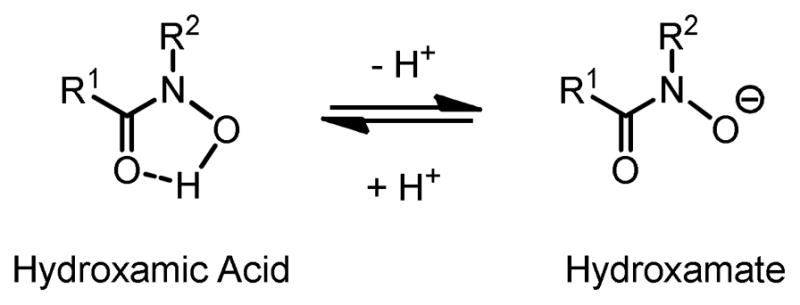
Structure of Hydroxamic Acid
For the synthesis of hydroxamic acids, there are two major strategies: coupling and oxidation (Scheme 1). Amide coupling techniques can be similarly applied to the synthesis of hydroxamic acids. There have been numerous reports about coupling of carboxylic acids and derivatives (halides, anhydrides and esters) with hydroxylamines and protected hydroxylamines mediated by either peptide coupling reagents or bases.4 These methods are in general reliable but sometimes suffer from low yield and low regioselectivity of N-coupling, especially when an N-alkyl group is present due to higher steric repulsion. In addition, synthesis of N-alkyl hydroxylamine also adds a few more steps to the whole synthetic scheme.
Scheme 1.
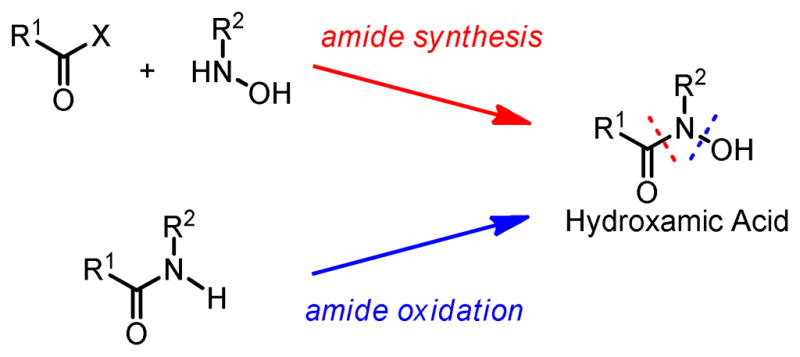
Synthesis of Hydroxamic Acid
The other synthetic strategy for hydroxamic acid is the direct oxidation of amide N-H to N-OH. The advantage of this strategy is that usually the parent amide can be readily obtained. There have been reports on the stoichiometric Molybdenum-mediated oxidation of N-trimethylsilyl amides to their corresponding hydroxamic acids.5 Moderate yields and selectivities have been achieved, but the efficiency of this method also greatly depends on the steric and electronic nature of the substitution groups of the amide.
In addition, N-aryl hydroxamic acids can also be synthesized from the N-Heterocyclic Carbene (NHC) catalyzed coupling of aldehydes and N-arylnitroso compounds.6
In summary, syntheses of hydroxamic acids highly depend on the structure of the target molecules. There has not been an efficient general synthesis of hydroxamic acids developed yet.
3.1 Vanadium-Catalyzed Asymmetric Epoxidation of Alkenyl Alcohols
Chiral hydroxamic acids were first introduced into asymmetric catalysis in 1977 by Sharpless and co-workers as a chiral ligand in V-catalyzed asymmetric epoxidation of allylic alcohols (Scheme 2).7 Later, the proline-derived hydroxamic acid 2 was discovered to form a 1:1 complex with VO(acac)2, an extremely active catalyst for epoxidation of alkenyl alcohols. This new catalyst provided epoxide 4 in 80% ee and 90% yield with tert-butylhydroperoxide (TBHP) as the terminal oxidant.8
Scheme 2.
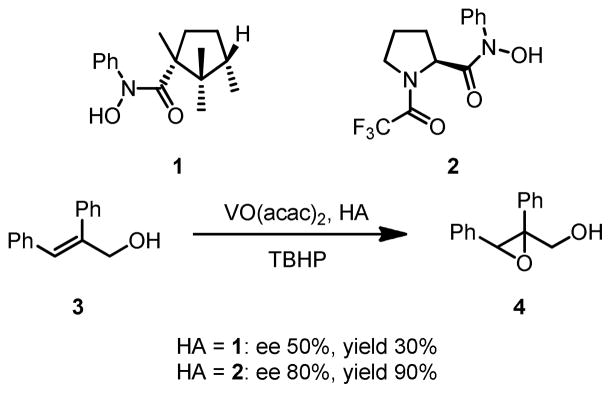
Early Examples of Vanadium-Hydroxamic Acid Catalyzed Epoxidation
Although the initial results looked promising, the vanadium-catalyzed epoxidation soon became less attractive due to the ligand deceleration effect of vanadium catalysts,9 a practical dilemma that adding an excess of chiral ligand is necessary to suppress background reaction and obtain high enantioselectivity, but such excess would favor the formation of catalytically inactive species hence decrease the reactivity. On the contrary, titanium-catalyzed asymmetric epoxidation, the Sharpless Asymmetric Epoxidation (SAE), is ligand-accelerated.9 Adding tartrate ligand to titanium(IV) tetra-alkoxide facilitates the generation of the most active and enantioselective catalytic species. The research of vanadium-catalyzed asymmetric epoxidation had then been halted for almost two decades, while the SAE became quite established during this period.10 Nevertheless, the advantages of vanadium catalysts over titanium catalysts were gradually re-evaluated: less catalyst loading, better moisture tolerance, easier work-up procedures, and higher selectivity for a wide range of substrates, often complementary to the SAE. These features guarantee the indispensable role of the vanadium-hydroxamic acid-catalyzed asymmetric epoxidation in asymmetric synthesis.
The major obstacles facing vanadium catalysts are still the ligand deceleration effect and dynamic ligand exchange. During the catalyst generation phase of the reaction, an equilibrium among metal-ligand complexes is established according to the ratio of metal species and ligand (Scheme 3). 1:1 Complex of metal and ligand (B) was believed to be the active catalyst. To eliminate the racemic background reaction, an excess of ligand has to be applied in order to minimize the content of the achiral vanadium species A; while such excess could also shift the equilibrium away from B to generate unreactive MLn (n>1) species (C and D), therefore hampers the reactivity.
Scheme 3.

Ligand-Deceleration Effect of Vanadium-Hydroxamic Acid Catalysis
The first generation solution to this problem was developed based on the hypothesis that ligand with higher steric hindrance would not favor the formation of metal complexes of two or more ligands. Thus, hydroxamic acids bearing bulky groups on both carboxyl and hydroxylamine components are synthesized and evaluated as catalysts for asymmetric epoxidation of allylic alcohols.
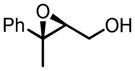
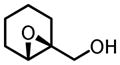
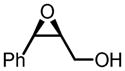
The binaphthol scaffold is one of the most widely used chiral catalyst skeletons in asymmetric catalysis. Axially chiral hydroxamic acids derived from binaphthol scaffold were evaluated in vanadium-catalyzed epoxidation of allylic alcohols (Table 1).11 At low temperature (−20°C) and 5 mol% catalyst loading, up to 94% ee was obtained when using trityl hydroperoxide (THP) as stoichiometric oxidant.
Table 1.
VO(OiPr)3-5-Catalyzed Asymmetric Epoxidation of Allylic Alcohols
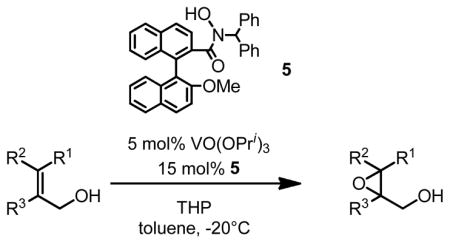
| ||
|---|---|---|
| allylic alcohol | yield(%) | ee(%) |

|
80 | 86 |
|
|
80 | 66 |
|
|
87 | 41 |
|
70 | 78 |
|
|
96 | 91 |
|
61 | 87 |

|
59 | 94 |
|
|
14 | 71 |
|
19 | 38 |
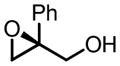
|
53 | 39 |
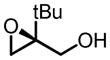
|
16 | 40 |
|
88 | 48 |
|
|
69 | 75 |
|
|
88 | 81 |

|
61 | 50 |
Notably, investigation of the relationship between ligand/metal ratio and reactivity/selectivity showed that increasing the ratio resulted in decrease of the reaction yield and saturation of enantioselectivity for a ratio greater than 1.2 (Figure 2).11b This suggests the active catalyst is a 1:1 complex and that adding excess ligand would reduce its content.
Figure 2.

Reactivity and Selectivity of VO(OiPr)3-5 Catalyzed Epoxidation of 3
The structure of ligand is also shown to be very influential on the reactivity and selectivity. The binaphthyl-derived ligands are relatively successful but the chiral binaphthyl group is rather difficult to modify. In a search for more efficient chiral ligands that can be easily prepared, a combinatorial iterative positional optimization approach was applied to chiral ligand synthesis and screening.12 In this approach, the ligands were designed to consist of three variable parts: L-α-amino acids as the chiral source in place of binaphthyl, dicarboxylimide as Nα-protecting group, and hydroxylamine (Scheme 4). Changing these three components would provide an array of ligands with different steric and electronic profiles, among which 6 has been shown to be the best combination. Allylic alcohols with various substituent patterns were epoxidized with V-6 catalyst in high efficiency and enantioselectivity (Table 2). Furthermore, the catalyst was shown to be effective in even lower catalyst loading (0.1 mol%), with a slight loss of enantioselectivity.
Scheme 4.

Design and Optimization of α-Amino Acid-Based Hydroxamic Acid
Table 2.
VO(OiPr)3-6-Catalyzed Asymmetric Epoxidation of Allylic Alcohols

| |||
|---|---|---|---|
| product | time (h) | yield (%) | ee(%) |

|
6 | 96 | 95 |
|
|
6 | 97 | 95 |
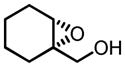
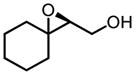
|
5 | 82 | 93 |
|
|
6 | 95 | 81 |
|
|
3 | 97 | 78 |
|
|
70 | 94 | 83 |
|
|
80 | 58 | 87 |
|
|
1 week | 71 | 76 |
|
|
24 | 80 | 82 |
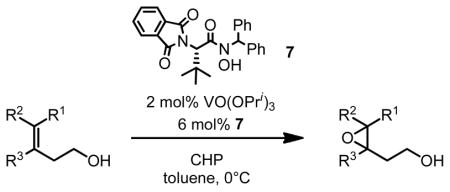
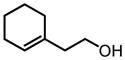
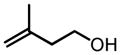
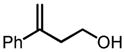
This screening strategy was also applied to the asymmetric epoxidation of vanadium-catalyzed homoallylic alcohols. Results showed that 3-substituted homoallylic alcohols gave high enantioselectivities with 7 as the chiral ligand and cumene hydroperoxide (CHP) as the terminal oxidant, while other substitution patterns gave only moderate enantioselectivities (Table 3).13
Table 3.
VO(OiPr)3-7-Catalyzed Asymmetric Epoxidation of Homoallylic Alcohols
| ||
|---|---|---|
| homoallylic alcohol | yield (%) | ee(%) |
|
|
41 | 52 |
|
|
25 | 40 |
|
|
24 | 46 |
|
|
67 | 36 |
|
61 | 74 |
|
58 | 84 |
|
|
77 | 90 |
|
|
89 | 90 |
|
70 | 89 |
|
|
42 | 91 |
To demonstrate the utility of asymmetric epoxidation of homoallylic alcohols, asymmetric total syntheses of (−)-α-bisabolol and (−)-8-epi-α-bisabolol were accomplished both using this new reaction as the key step (Scheme 5).13
Scheme 5.

Concise and Stereoselective Total Synthesis of (−)-(4S,8S)-α- and (−)-(4S,8R)-epi-α-bisabolol
Reaction conditions: (a) Me2AlCl, (CH2O)n; (b) 2 mol% VO(OiPr)3, 6 mol% D-7, CHP, toluene, 0°C; (c) LiAlH4, THF; (d) TsCl, pyridine, CHCl3; (e) 5 equiv Me2C=CHMgBr, 0.5 equiv CuBr·Me2S, THF.
Although ligands derived from amino acids exhibited high activity and selectivity, the ligand-deceleration effect was still observed. To exclude this deceleration effect, a second generation of ligand, C2-symmetric bishydroxamic acid (BHA) was designed (Scheme 6).14 We hypothesized that this ligand would coordinate with metal as a bidentate bivalent ligand and hence achieve stronger coordination. The steric profile of this ligand may also prevent formation of MLn (n > 1) complexes, leaving a high content of catalytically active 1:1 metal-ligand complex. Ligands with different steric profiles (8a–c) were also prepared according to this general route.
Scheme 6.

General Synthesis of BHA
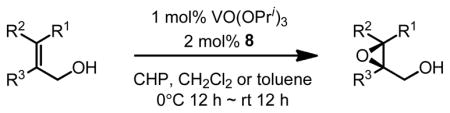
Gratifyingly, vanadium complexes of BHA exhibited extremely high activities and selectivities toward almost all substitution patterns of allylic alcohol substrates (Table 4). In addition, with cumene hydroperoxide as the terminal oxidant, small allylic alcohols could be epoxidized in high yield and enantioselectivity (Table 5). Besides excellent results for a wide scope of substrates, these catalysts also feature several significant advancements superior to other comparable methods. First, the catalyst loading could be as low as 0.2 mol%. Second, the reaction was carried out in open air conditions and aqueous TBHP was used as the oxidant. Third, ligand-deceleration was not observed even when the amount of ligand was the equivalent of three times of the metal.
Table 4.
VO(OiPr)3-BHA Catalyzed Asymmetric Epoxidation of Allylic Alcohols
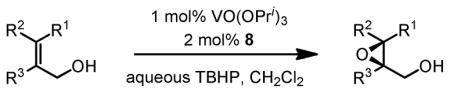
| |||||
|---|---|---|---|---|---|
| ligand | product | T (°C) | time (h) | yield (%) | ee (%) |
| 8a |
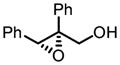
|
−20 | 48 | 91 | 97 |
| 8a |

|
−20 | 48 | 84 | 97 |
| 8a |
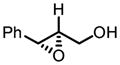
|
−20 | 60 | 53 | 97 |
| 8a |
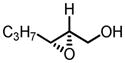
|
−20 | 72 | 56 | 95 |
| 8a |

|
−20 | 72 | 51 | 95 |
| 8a |
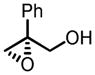
|
0 | 48 | 73 | 95 |
| 8a |

|
−20 | 48 | 79 | 95 |
| 8c |
|
−20 | 48 | 68 | 95 |
| 8b |

|
0 | 72 | 60 | 95 |
| 8b |

|
−20 | 120 | 24 | 97 |
| 8b |

|
−20 | 48 | 64 | 96 |
| 8b |
|
−20 | 48 | 62 | 95 |
Table 5.
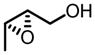
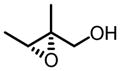
VO(OiPr)3-BHA Catalyzed Asymmetric Epoxidation of Small Allylic Alcohols
| |||
|---|---|---|---|
| ligand | product | yield (%) | ee(%) |
| 8c |

|
78 | 97 |
| 8a |
|
50 | 93 |
| 8b |
|
71 | 92 |
| 8a |
|
68 | 95 |
| 8c |
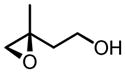
|
73 | 94 |
Furthermore, these catalysts were capable of catalyzing highly selective kinetic resolution of secondary allylic alcohols (Scheme 7). Both the product and starting material were isolated in high yield and enantioselectivity after the reaction.
Scheme 7.
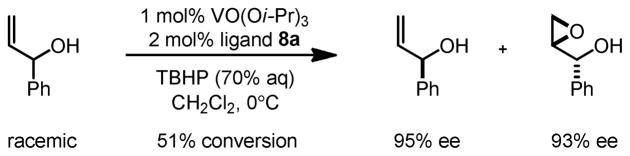
Kinetic Resolution of Allylic Alcohol
We then applied the new vanadium-BHA catalyst system to the epoxidation of homoallylic alcohols, which are more challenging substrates than allylic alcohols.15 BHA ligands with more bulky acyl groups (8d and 8e) had to be prepared in order to improve enantioselectivity (Scheme 8). Reaction conditions were also modified to achieve an optimum synergy. 4-Z- and 4-E-substituted homoallylic alcohols were epoxidized in high enantioselectivity and high yield with the extremely bulky ligand 8e (Table 6). In addition, kinetic resolution of 2,4-disubstituted primary homoallylic alcohols was also performed. As in the cases of allylic alcohols, both the product and starting material were recovered in high yield and enantioselectivity (Table 7).
Scheme 8.
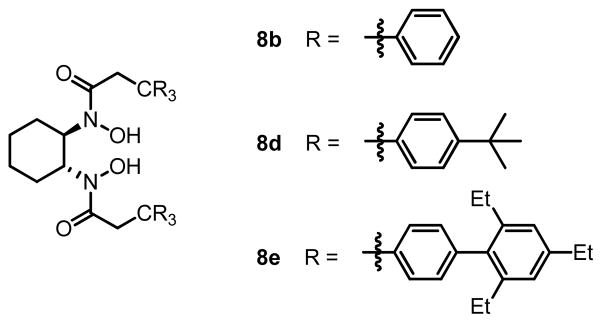
BHA Ligands for Vanadium-Catalyzed Asymmetric Epoxidation of Homoallylic Alcohols
Table 6.
VO(OiPr)3-BHA Catalyzed Asymmetric Epoxidation of Homoallylic Alcohols
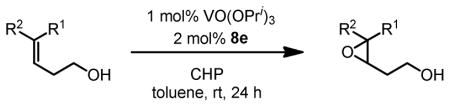
| |||
|---|---|---|---|
| homoallylic alcohol | product | yield (%) | ee (%) |
|
|
|
90 | 96 |
|
|
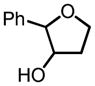
|
85 | 99 |
|
|
|
85 | 93 |
|
|
|
89 | 96 |
|
|
|
92 | 98 |
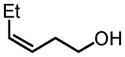
|

|
92 | 95 |
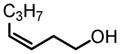
|
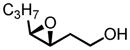
|
90 | 97 |
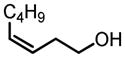
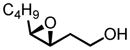
|
|
91 | 99 |
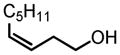
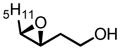
|
|
90 | 99 |
Table 7.
Desymmetrization of meso-Secondary Allylic and Homoallylic Alcohols

| |||||||
|---|---|---|---|---|---|---|---|
| ligand | oxidant | product | T (°C) | time (h) | d.r. | yield (%) | ee(%) |
| 8a | TBHP |
|
0 | 10 | 94:6 | 52 | 95 |
| 8a | TBHP |

|
0 | 12 | 95:5 | 60 | 95 |
| 8a | TBHP |
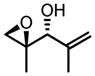
|
−10 | 96 | 99:1 | 62 | 95 |
| 8b | TBHP |
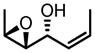
|
−10 | 96 | 87:13 | 73 | 97 |
| 8e | TBHP |
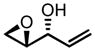
|
0 | 24 | 98:2 | 52 | 95 |
| 8e | CHP |
|
0 | 120 | 92:8 | 51 | 97 |
| 8e | CHP |
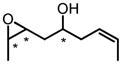
|
0 | 96 | 97:3 | 69 | 97 |
| 8e | CHP |

|
0 | 96 | 60:40 | 40 | 17, 23 |
Its exceptional kinetic resolution activity is an indication that this catalyst system is excellent at differentiating diastereotopic alkene faces. Since desymmetrization of meso-secondary alkenyl alcohols follows the same rule of stereoselection,16 we designed and synthesized several meso-secondary allylic and homoallylic alcohols to evaluate the epoxidation desymmetrization of these substrates (Table 7).17 High diastereoselectivity and enantioselectivity were obtained for a variety of allylic alcohols18 as well as homoallylic alcohols. Previous reports of enantioselective syntheses of syn-1,2-epoxy-6-hepten-4-ol (Table 7, entry 6), which is a heavily investigated drug intermediate of atorvastatin (Liptor®),19 take at least six steps including either poorly diastereoselective Grignard reaction20 or hydrolytic kinetic resolution of its enantiomers.21 With our catalyst, this compound can be obtained with 51% yield, 92:8 desired diastereomer ratio and 97% enantiomer excess in one single step.
The absolute configurations of epoxy alcohol products were determined and models of asymmetric induction of the catalyst were then proposed based on these stereochemistry results (Figure 3).22 The steric repulsion between the bulky carboxylate group and cyclohexyl group forces the hydroxamic acid motif to adopt a trans configuration, in which the carbonyl oxygen is directed away from the metal center. In the transition state, the substrate and oxidant are aligned in a trans manner, with the olefin and peroxide spirally overlapping each other, to minimize steric hindrance. Structure of the ligand plays a crucial role in the transition state. Ligands with different carboxylate groups may be required to match different substrates.
Figure 3.

Postulated Models of the Epoxidation of Allylic and Homoallylic Alcohols by V-BHA Complexes
A number of other research groups have made significant contributions to the development of V-HA catalyst systems and elucidation of reaction mechanism.23
3.2 Molybdenum-Catalyzed Asymmetric Epoxidation of Single Olefins
During the investigation of vanadium-BHA catalyzed epoxidation of allylic alcohols, we interestingly observed that with molybdenum-8c as the catalyst and TBHP as the oxidant, geraniol was epoxidized at both double bonds to provide a 1:1 mixture of 2,3-epoxy and 6,7-epoxygeraniol (Scheme 10).24 This suggests that Mo-BHA is capable of oxidizing isolated single olefins, possibly in an enantioselective fashion.
Scheme 10.

Bias of V and Mo in the Epoxidation of Geraniol
We then examined the enantioselectivity of this reaction. With THP as the oxidant and 8e as the ligand, the reaction can achieve very high enantioselectivity and high yield (Table 8). It is noteworthy that when multiple double bonds were present in the molecule, the one most electron-rich would react most quickly. Mono-epoxidation of squalene, which is essentially a desymmetrization process, also gave good enantioselectivity and yield (Scheme 11).25 Our catalyst system has been the most enantioselective among all Mo-catalyzed epoxidations of olefins thus far.26
Table 8.
Mo-BHA Catalyzed Asymmetric Epoxidation of Olefins

| |||||
|---|---|---|---|---|---|
| ligand | epoxide | oxidant | time (h) | yield (%) | ee (%) |
| 8g |
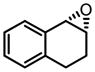
|
THP | 18 | 98 | 95 |
| 8f |
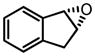
|
THP | 20 | 94 | 92 |
| 8g |
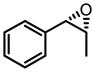
|
THP | 20 | 60 | 90 |
| 8d |
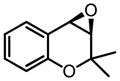
|
TBHP | 20 | 92 | 96 |
| 8d |
|
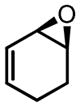
CHP | 22 | 77 | 64 |
| 8d |
|
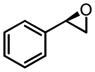
CHP | 40 | 95 | 85 |
| 8d |
|
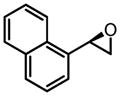
CHP | 36 | 84 | 82 |
| 8d |

|
CHP | 40 | 95 | 85 |
| 8d |
|
CHP | 40 | 92 | 50 |
| 8d |

|
TBHP | 36 | 44 | 62 |
| 8d |

|
CHP | 36 | 92 | 85 |
| 8d |
|
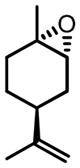
CHP | 46 | 94 | 66 (de) |
| 8h |
|
CHP | 42 | 82 | 76 |
| 8d |

|
TBHP | 17 | 13 | 43 |
Scheme 11.
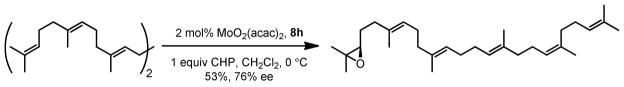
Regio- and Enantioselective Oxidation of Squalene
The mechanism of this Mo-BHA catalyzed epoxidation is believed to follow concerted metal peroxide oxygen transfer mechanism.27 A possible transition state of the oxygen transfer event is shown (Figure 4). Sterics played an important role as in the V-BHA catalyzed epoxidation.
Figure 4.
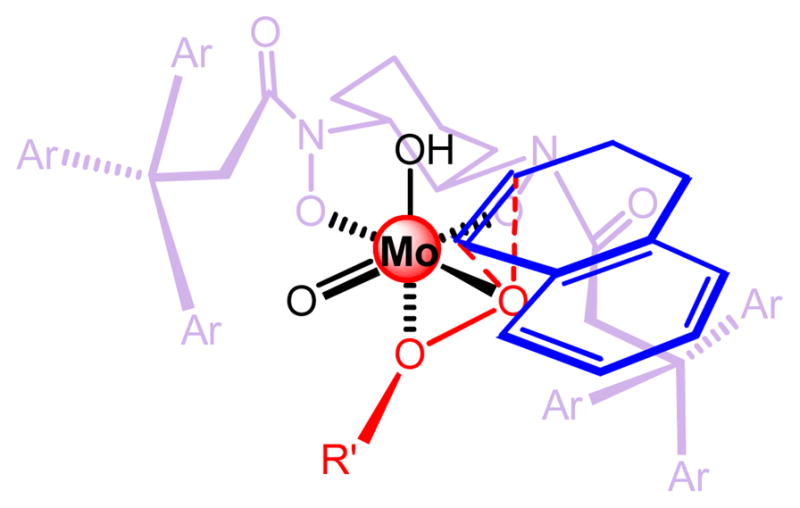
Postulated Models of the Epoxidation of Olefins by Mo-BHA Complexes
3.3 Molybdenum-Catalyzed Asymmetric Oxidation of Sulfides and Disulfides
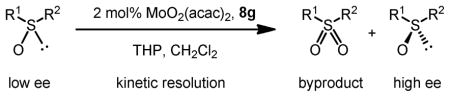
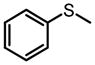
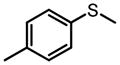
The same Molybdenum-BHA catalysts were also shown to catalytically oxidize sulfides and disulfides to provide enantiomer-enriched sulfoxides.28 With 8g as ligand and THP as oxidant, alkylarylsulfides were converted to sulfoxide with good yield and enantioselectivity (Table 9). By adding an excess of oxidant, the enantioselectivity of a product can be improved by its kinetic resolution to sulfone at the expense of yield (Table 10). In addition, disulfides can also be oxidized to the corresponding chiral sulfoxide (Scheme 12).
Table 9.
Mo-BHA Catalyzed Asymmetric Oxidation of Sulfides

| |||
|---|---|---|---|
| sulfide | time (h) | yield (%) | ee(%) (config.) |
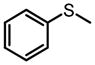
|
16 | 81 | 79 (S) |
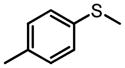
|
20 | 75 | 81 (S) |
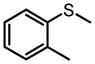
|
19 | 76 | 75 (S) |

|
18 | 81 | 82 (S) |
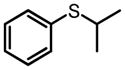
|
24 | 66 | 62 (R) |
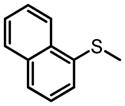
|
17 | 82 | 86 (S) |
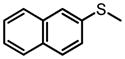
|
19 | 83 | 72 (S) |
Table 10.
Kinetic Resolution Helps Improve Enantioselectivity
| |||
|---|---|---|---|
| sulfide | sulfoxide:sulfone | yield (%) | ee(%) (config.) |
|
81:19 | 68 | 92 (S) |
|
74:26 | 55 | 94 (S) |

|
72:28 | 51 | 96 (S) |

|
76:24 | 50 | 93 (S) |
Scheme 12.
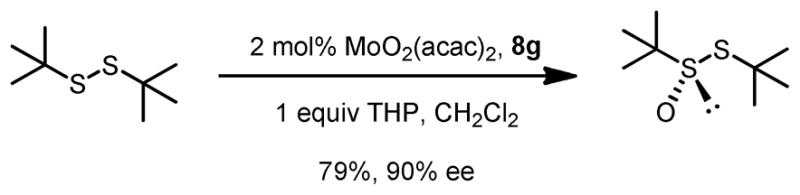
Mo-BHA Catalyzed Asymmetric Oxidation of Disulfide
3.4 Zirconium- and Hafnium-Catalyzed Asymmetric Epoxidation of Alkenyl Alcohols
After the development of vanadium-BHA catalyzed epoxidation of homoallylic alcohols, we were interested in whether the substrate scope could be expanded and catalyst loading decreased. Since further modification of the ligand for vanadium would be difficult, changing the metal center may lead to new reactivities. The choice of metal candidates is not difficult to make29 since titanium has been proven a very efficient epoxidation catalyst30 and zirconium has also been shown to perform the asymmetric epoxidation of homoallylic alcohols.31 Indeed, in the initial screening of metal ion species, Zr(OtBu)4-8a stood out in the M-BHA catalyzed epoxidation of homoallylic alcohol among all candidates providing the highest enantioselectivity and reactivity given otherwise identical reaction conditions. It should be noted that ligand 8a is much smaller than 8e, the best ligand for vanadium-catalyzed epoxidation of homoallylic alcohols. Later on, we found that Hf(OtBu)4 is a slightly better metal source than Zr(OtBu)4.32
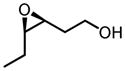
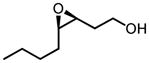
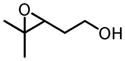
Screening of optimum reaction conditions demonstrated the significant difference between V-BHA and Zr/Hf-BHA epoxidation. The new Zr/Hf-BHA reaction performs best at 1:1 ratio of metal to ligand.33 The reaction could be improved by polar aprotic additives, probably because coordination of polar molecules to the metal center could stabilize and regenerate catalytically active monomeric Zr/Hf-BHA complex from polymeric metal complexes.34 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) was shown to be the most effective additive. Moreover, the reaction should be conducted in an inert atmosphere with the addition of molecular sieves, as has been done in the Sharpless Asymmetric Epoxidation.35 With all these special conditions, the reaction provides high yield and enantioselectivity for a variety of homoallylic alcohol substrates, especially those with 4-Z-substituents or 3-substituents (Table 11).32
Table 11.
Zr/Hf(OtBu)4-BHA Catalyzed Epoxidation of Homoallylic Alcohols

| |||
|---|---|---|---|
| epoxy alcohol | metal | yield (%) | ee(%) |
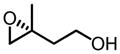
|
Zr | 61 | 91 |
| Hf | 81 | 97 | |
|
| |||
|
|
Hf | 37 | 63 |
|
| |||
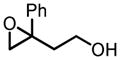
|
Zr | 67 | 92 |
| Hf | 69 | 98 | |
|
| |||
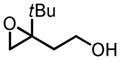
|
Zr | 67 | 63 |
| Hf | 70 | 71 | |
|
| |||
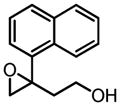
|
Hf | 31 | 91 |
|
| |||
|
|
Hf | 47 | 73 |
|
| |||
|
Zr | 45 | 93 |
| Hf | 82 | 94 | |
|
| |||
|
Zr | 80 | 92 |
| Hf | 83 | 96 | |
|
| |||
|
Hf | 41 | 71 |
|
| |||

|
Zr | 72 | 76 |
| Hf | 81 | 89 | |
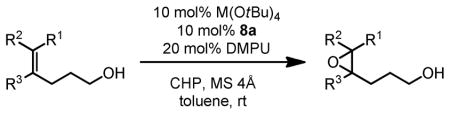
Similarly, bishomoallylic alcohols with 4-aryl substituents can be epoxidized to corresponding epoxides without intramolecular cyclization in most cases (Table 12), although 10 mol% of catalyst has to be applied. Unfortunately, other types of bishomoallylic alcohols usually gave poor results.32
Table 12.
Hf(OtBu)4-BHA Catalyzed Epoxidation of Bishomoallylic Alcohols
| |||
|---|---|---|---|
| bishomoallylic alcohol | product | yield (%) | ee (%) |
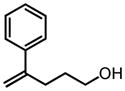
|
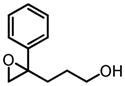
|
75 | 99 |
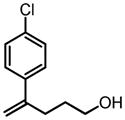
|
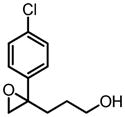
|
57 | 98 |

|

|
73 | 97 |

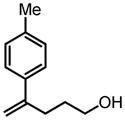
|
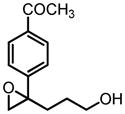
|
79 | 97 |
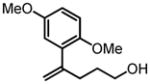
|
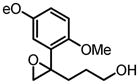
|
25 | 97 |
|
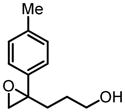
|
53 | 99 |

|

|
47 | 59 |

|
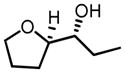
|
43 | 95 |
Non-linearity experiments indicate the catalyst should be a mononuclear complex. For the high enantioselectivity obtained in the epoxidation of longer alkenyl alcohols, we hypothesize that more space around the metal center would facilitate enantioselectivity because longer carbon chain would then fold more freely and be controlled by the ligand more easily (Figure 5). There are a few reasons for this: (1) the ionic radii for Zr4+ (0.72 Å) and Hf4+ (0.71 Å) are significantly larger than Ti4+ (0.61 Å) and V5+ (0.54 Å);36 (2) they do not come with a terminal oxo as in V=O; (3) Zr and Hf are penta-coordinate 37 in the transition state while V is hexa-coordinate;15 (4) ligand 8a is much smaller than 8e. All these factors contribute to a larger chiral space around the metal center, which is the basis of our hypothesis. Absolute configuration of products can be well explained with these models.38 Unlike V-BHA which gave opposite facial selectivity for allylic and homoallylic alcohols (Figure 3), Zr/Hf-BHA gave same facial preference for both homoallylic alcohols and bishomoallylic alcohols with similar substitution pattern.
Figure 5.
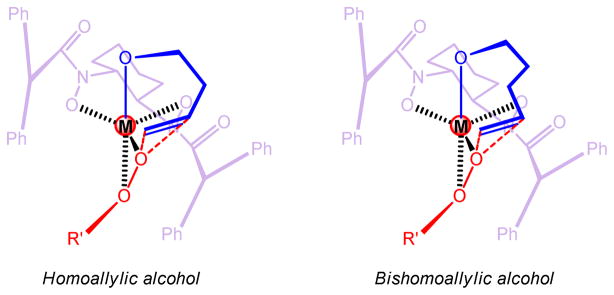
Postulated Models of the Epoxidation of Homoallylic and Bishomoallylic Alcohols by Zr/Hf- BHA Complexes (M = Zr or Hf)
3.5 Hf-Catalyzed Asymmetric Epoxidation of N-Alkenyl Sulfonamides and N-Tosyl Imines
Since the interaction between hydroxyl and metal has been utilized so well,39 we postulated that nitrogen may act as a directing group in the metal-catalyzed epoxidation of allylic amine derivatives, which has not been successfully developed yet. Screening of the N-protection group revealed that the sulfonamide protection group allowed the epoxidation of N-allyl sulfonamide in high enantioselectivity. Further optimization of the reaction conditions showed that the electron-rich protection group is more effective than the electron-deficient protection group. Meanwhile, additives needed to be changed. Molecular sieves and DMPU were detrimental to the reaction. On the other hand, a metal oxide such as MgO powder showed superior catalytic activity enhancement when added.40
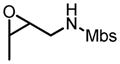
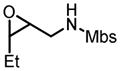
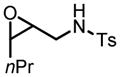
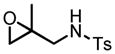
This reaction condition is suitable for substrates with a variety of substitution patterns on both the alkene and the sulfonamide (Table 13).40 In general, 3-Z-substituted N-allylic sulfonamides gave better selectivity. To further demonstrate the synthetic utility of this reaction, an easily cleaved protection group was also tested and excellent result was obtained (Table 13, entry 22).
Table 13.
Hf(OtBu)4-BHA Catalyzed Epoxidation of N-Alkenylsulfonamides
| |||
|---|---|---|---|
| entry | product | yield(%) | ee(%) |
| 1 |

|
46 | 90 |
| 2 |

|
32 | 84 |
| 3 |
|
49 | 93 |
| 4 |

|
74 | 64 |
| 5 |
|
92 | 91 |
| 6 |
|
86 | 77 |
| 7 |

|
98 | 87 |
| 8 |
|
90 | 89 |
| 9 |

|
91 | 70 |
| 10 |
|
56 | 46 |
| 11 |
|
40 | 34 |
| 12 |
|
48 | 20 |
| 13 |

|
97 | 92 |
| 14 |

|
69 | 90 |
| 15 |
|
84 | 62 |
| 16 |

|
92 | 42 |
| 17 |
|
26 | 31 |
| 18 |

|
88 | 9 |
| 19 |

|
9 | 67 |
| 20 |

|
21 | 45 |
| 21 |
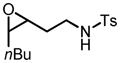
|
36 | 31 |
| 22 |
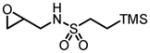
|
88 | 87 |
Ts = p-toluenesulfonyl; Mbs = 4-methoxybenzenesulfonyl; TMS = trimethylsilyl.
To probe the activation mode of this reaction, we conducted a series of control experiments (Scheme 13). The NH group was replaced by O and CH2 groups to see if the reaction still proceeded. Gratifyingly, catalytic reaction of this substrate series showed that the functional group at this position did not affect the enantioselectivity so much. Reactions for all three substrates proceeded in a comparable magnitude of enantioselectivity. We hypothesize that the catalyst activates the substrate via one of the sulfonyl oxygens.41 This new activation mode has not been extensively investigated and it might open new avenues to Lewis acid catalysis.
Scheme 13.
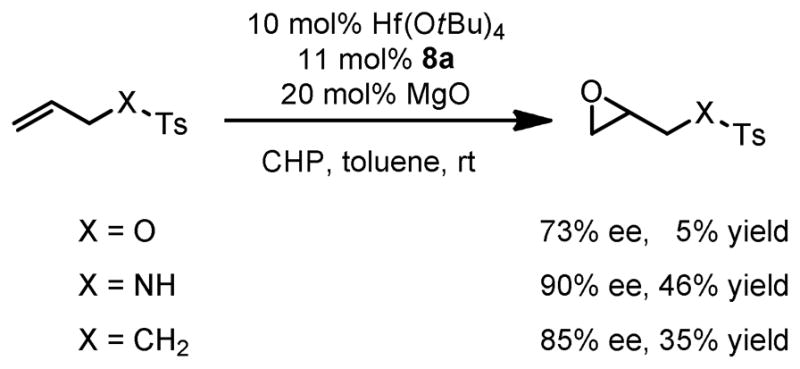
Epoxidation of Allylic Sulfonate, Sulfonamide, and Sulfone
Since the sulfonyl group was demonstrated to be a directing group in epoxidation, other functional groups susceptible to oxidation can be incorporated in a molecule with this group to generate a substrate for Hf-BHA catalyzed oxidation. N-Tosyl imines are the latest addition of these substrates.42 With similar conditions of epoxidation of N-allyl sulfonamides, N-tosyl imines were epoxidized to their corresponding oxaziridines in very high enantioselectivity and high yield (Table 14).40 With these results, we are more convinced that the actual interaction between Hf-BHA and imine substrate is via the sulfonyl oxygen.
Table 14.
Hf-BHA Catalyzed Oxaziridination of N-Tosyl Imines
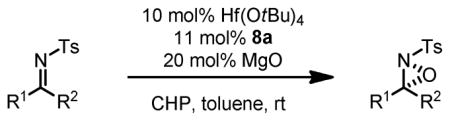
| ||
|---|---|---|
| product | yield (%) | ee(%) |
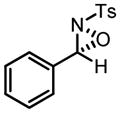
|
78 | 98 |
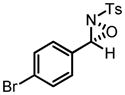
|
84 | 95 |

|
82 | 91 |
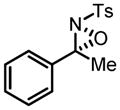
|
38 | 98 |
Ts = p-toluenesulfonyl
3.6 Miscellaneous Reactions Involving Chiral Hydroxamic Acids as Catalyst
Although hydroxamic acid is a particularly suitable ligand for oxidation reactions thanks to its resistance to oxidation in the reaction condition,7 there have been reports for its usage in other types of reactions, especially recently. Adolfsson and co-workers reported chiral amino acid derivatives including hydroxamic acids were good ligands for rhodium- and iridium-catalyzed asymmetric transfer hydrogenation of ketones.43 Good enantioselectivities were obtained, even in aqueous media. More recently, Bode and co-workers reported a kinetic resolution of cyclic secondary amines using chiral hydroxamic acid as one of the co-catalysts, acting as an acyl transferring reagent.44
4. Summary
In the journey searching for chiral hydroxamic acid ligands, several new metal-catalyzed reactions have been developed in our group: V-catalyzed epoxidation of allylic and homoallylic alcohols revived by mono-hydroxamic acid and followed by bishydroxamic acid; Mo-catalyzed epoxidation of isolated olefins, sulfides and disulfides; Zr- and Hf-catalyzed epoxidation of homoallylic alcohols and bishomoallylic alcohols; and Hf-catalyzed epoxidation of compounds containing the sulfonyl directing group. Since the application hydroxamic acid ligand should not be limited only to asymmetric oxidation, we believe the potential of this special type of ligand – hydroxamic acid – remains far too unexplored.
Scheme 9.
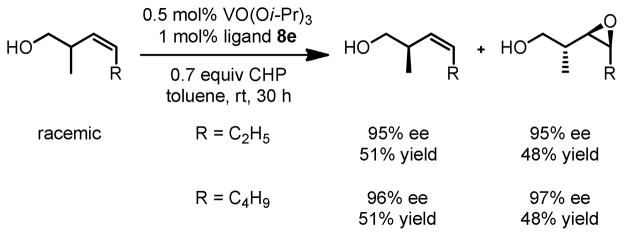
Kinetic Resolution of Homoallylic Alcohols
Acknowledgments
Japan Science and Technology Agency (JST), The University of Chicago, the National Institutes of Health (R01-GM068433), GAANN, and Merck Research Laboratories are greatly appreciated for providing financial support for the research. H. Y. would also like to thank all students and scholars who worked on these projects.
Biographies
Dr. Zhi Li was born in Jilin, China in 1983. He obtained B.S. degree from University of Science and Technology of China in 2006 under the supervision of Professor Tianpa You, and received PhD from the University of Chicago under the supervision of Professor Hisashi Yamamoto in 2011. His PhD research was the development of V, Zr, Hf-chiral bis-hydroxamic acid complexes and their applications in asymmetric epoxidation. Currently he is a Postdoctoral Fellow at Northwestern University with Professor Tobin Marks.
Hisashi Yamamoto was born in Kobe, Japan in 1943. He received his B.S. degree from Kyoto University and PhD from Harvard University under Professor Hitoshi Nozaki and Professor E. J. Corey, respectively. In 1972 he was appointed as Assistant Professor at Kyoto University, and in 1977 Associate Professor of Chemistry at the University of Hawaii. In 1980, he returned to Japan to Nagoya University where he became Professor in 1983. In 2002, he moved to the United States as the Arthur Holly Compton Distinguished Professor at The University of Chicago. In 2012 he retired from the University of Chicago and was appointed with Chubu University, Japan, as the Chairman of the Center of Homogeneous Catalysis. His honors include: Prelog Medal in 1993, Le Grand Prix de la Fondation Maison de la Chimie in 2002, the National Prize of the Purple Medal (Japan) in 2002, the Yamada Prize in 2004, the Tetrahedron Prize in 2006, The Japan Academy Prize in 2007, the ACS Award for Creative Work in Synthetic Organic Chemistry in 2009, and the Ryoji Noyori Prize in 2011.
References
- 1.Rouhi AM. Chiral Chemistry. Chem Eng News. 2004:47–62. [Google Scholar]
- 2.Bauer EB. Chiral-at-metal complexes and their catalytic applications in organic synthesis. Chem Soc Rev. 2012;41(8):3153–3167. doi: 10.1039/c2cs15234g. [DOI] [PubMed] [Google Scholar]
- 3.Yoon TP, Jacobsen EN. Privileged Chiral Catalysts. Science. 2003;299(5613):1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 4.Codd R. Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord Chem Rev. 2008;252(12–14):1387–1408. [Google Scholar]
- 5.Matlin SA, Sammes PG, Upton RM. The oxidation of trimethylsilylated amides to hydroxamic acids. J Chem Soc, Perkin Trans. 1979;1:2481–2487. [Google Scholar]
- 6.Wong FT, Patra PK, Seayad J, Zhang Y, Ying JY. N-Heterocyclic Carbene (NHC)-Catalyzed Direct Amidation of Aldehydes with Nitroso Compounds. Org Lett. 2008;10(12):2333–2336. doi: 10.1021/ol8004276. [DOI] [PubMed] [Google Scholar]
- 7.Michaelson RC, Palermo RE, Sharpless KB. Chiral hydroxamic acids as ligands in the vanadium catalyzed asymmetric epoxidation of allylic alcohols by tert-butyl hydroperoxide. J Am Chem Soc. 1977;99(6):1990–1992. [Google Scholar]
- 8.Sharpless KB, Verhoeven TR. Metal-Catalyzed, Highly Selective Oxygenations of Olefins and Acetylenes with tert-Butyl Hydroperoxide. Practical Considerations and Mechanisms. Aldrichim Acta. 1979;12:63–73. [Google Scholar]
- 9.Berrisford DJ, Bolm C, Sharpless KB. Ligand-Accelerated Catalysis. Angew Chem, Int Ed Engl. 1995;34(10):1059–1070. [Google Scholar]
- 10.(a) Katsuki T, Sharpless KB. The first practical method for asymmetric epoxidation. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]; (b) Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]; (c) Katsuki T, Martin VS. Asymmetric Epoxidation of Allylic Alcohols: the Katsuki–Sharpless Epoxidation Reaction. Org React. 1996;48:1–299. [Google Scholar]
- 11.(a) Murase N, Hoshino Y, Oishi M, Yamamoto H. Chiral Vanadium-Based Catalysts for Asymmetric Epoxidation of Allylic Alcohols. J Org Chem. 1999;64:338–339. [Google Scholar]; (b) Hoshino Y, Murase N, Oishi M, Yamamoto H. Design of Optically Active Hydroxamic Acids as Ligands in Vanadium-Catalyzed Asymmetric Epoxidation. Bull Chem Soc Jpn. 2000;73(7):1653–1658. [Google Scholar]
- 12.Hoshino Y, Yamamoto H. Novel α-Amino Acid-Based Hydroxamic Acid Ligands for Vanadium-Catalyzed Asymmetric Epoxidation of Allylic Alcohols. J Am Chem Soc. 2000;122:10452–10453. [Google Scholar]
- 13.Makita N, Hoshino Y, Yamamoto H. Asymmetric Epoxidation of Homoallylic Alcohols and Application in a Concise Total Synthesis of (−)-α-Bisabolol and (−)-8-epi-α-Bisabolol. Angew Chem, Int Ed. 2003;42:941–943. doi: 10.1002/anie.200390250. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Basak A, Kosugi Y, Hoshino Y, Yamamoto H. Enantioselective Epoxidation of Allylic Alcohols by a Chiral Complex of Vanadium: An Effective Controller System and a Rational Mechanistic Model. Angew Chem, Int Ed. 2005;44:4389–4391. doi: 10.1002/anie.200500938. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, Yamamoto H. Vanadium-Catalyzed Asymmetric Epoxidation of Homoallylic Alcohols. J Am Chem Soc. 2007;129:286–287. doi: 10.1021/ja067495y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber SL, Schreiber TS, Smith DB. Reactions that proceed with a combination of enantiotopic group and diastereotopic face selectivity can deliver products with very high enantiomeric excess: experimental support of a mathematical model. J Am Chem Soc. 1987;109(5):1525–1529. [Google Scholar]
- 17.Li Z, Zhang W, Yamamoto H. Vanadium-Catalyzed Enantioselective Desymmetrization of meso-Secondary Allylic Alcohols and Homoallylic Alcohols. Angew Chem, Int Ed. 2008;47(39):7520–7522. doi: 10.1002/anie.200802523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Smith DB, Wang Z, Schreiber SL. The asymmetric epoxidation of divinyl carbinols: theory and applications. Tetrahedron. 1990;46(13–14):4793–4808. [Google Scholar]; (b) Brückner R. Asymmetric Epoxidation of Pentadienols. In: Christmann M, Bräse S, editors. Asymmetric Synthesis – The Essentials. 2. WILEY-VCH Verlag GmbH & Co; Weinheim: 2008. pp. 10–16. completely revised ed. [Google Scholar]
- 19.Butler DE, Deering CF, Millar A, Nanninga TN, Roth BD. Process for trans-6-[2-(substituted-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis. 5,245,047. US Patent. 1993
- 20.Kocieński PJ, Yeates C, Street SDA, Campbell SF. The 3,4-dihydro-2H-pyran approach to (+)-milbemycin β3. Part 1. An alternative synthesis of (2S,4S,6R,8R,9S)-2-formylmethyl-4-(dimethyl-t-butylsilyloxy)-8,9-dimethyl-1,7-dioxaspiro[5.5]undecane. J Chem Soc, Perkin Trans. 1987;1:2183–2187. [Google Scholar]
- 21.Kim YJ, Tae J. Resolution of 2-Silyloxy-1-oxiranyl-4-pentenes by HKR: Total Synthesis of (5S,7R)-Kurzilactone. Synlett. 2006;(1):61–64. [Google Scholar]
- 22.Barlan AU, Zhang W, Yamamoto H. Development and application of versatile bis-hydroxamic acids for catalytic asymmetric oxidation. Tetrahedron. 2007;63:6075–6087. doi: 10.1016/j.tet.2007.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For recent reviews, see: Bolm C. Vanadium-catalyzed asymmetric oxidations. Coord Chem Rev. 2003;237:245–256.Xia QH, Ge HQ, Ye CP, Liu ZM, Su KX. Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation. Chem Rev. 2005;105(5):1603–1662. doi: 10.1021/cr0406458.Licini G, Conte V, Coletti A, Mba M, Zonta C. Recent advances in vanadium catalyzed oxygen transfer reactions. Coord Chem Rev. 2011;255(19–20):2345–2357.da Silva JAL, da Silva JJRF, Pombeiro AJL. Oxovanadium complexes in catalytic oxidations. Coord Chem Rev. 2011;255(19–20):2232–2248.
- 24.Barlan AU, Basak A, Yamamoto H. Enantioselective Oxidation of Olefins Catalyzed by a Chiral Bishydroxamic Acid Complex of Molybdenum. Angew Chem, Int Ed. 2006;45(35):5849–5852. doi: 10.1002/anie.200601742. [DOI] [PubMed] [Google Scholar]
- 25.(a) Corey EJ, Noe MC, Wen-Chung S. A short and convergent enantioselective synthesis of (3S)-2,3-oxidosqualene. Tetrahedron Lett. 1993;34(38):5995–5998. [Google Scholar]; (b) Raina S, Singh VK. Reaction of epoxides with activated DMSO reagent. General method for synthesis of α-chlorocarbonyl compounds: Application in asymmetric synthesis of (3S)-2,3-oxidosqualene. Tetrahedron. 1995;51(8):2467–2476. [Google Scholar]
- 26.Brito JA, Royo B, Gomez M. An overview of chiral molybdenum complexes applied in enantioselective catalysis. Catal Sci Technol. 2011;1(7):1109–1118. [Google Scholar]
- 27.Deubel DV, Frenking G, Gisdakis P, Herrmann WA, Rösch N, Sundermeyer J. Olefin Epoxidation with Inorganic Peroxides. Solutions to Four Long-Standing Controversies on the Mechanism of Oxygen Transfer. Acc Chem Res. 2004;37(9):645–652. doi: 10.1021/ar0400140. [DOI] [PubMed] [Google Scholar]
- 28.Basak A, Barlan AU, Yamamoto H. Catalytic enantioselective oxidation of sulfides and disulfides by a chiral complex of bis-hydroxamic acid and molybdenum. Tetrahedron: Asymmetry. 2006;17(4):508–511. [Google Scholar]
- 29.Jørgensen KA. Transition-metal-catalyzed epoxidations. Chem Rev. 1989;89(3):431–458. [Google Scholar]
- 30.Rossiter BE, Sharpless KB. Asymmetric epoxidation of homoallylic alcohols. Synthesis of (−)-.gamma.-amino-.beta.-(R)-hydroxybutyric acid (GABOB) J Org Chem. 1984;49:3707–3711. [Google Scholar]
- 31.(a) Ikegami S, Katsuki T, Yamaguchi M. Asymmetric Epoxidation of Homoallylic Alcohols Using Zirconium Tetrapropoxide, Dicyclohexyltartramide, and t-Butyl Hydroperoxide System. Chem Lett. 1987:83–84. [Google Scholar]; (b) Okachi T, Murai N, Onaka M. Catalytic Enantioselective Epoxidation of Homoallylic Alcohols by Chiral Zirconium Complexes. Org Lett. 2003;5(1):85–87. doi: 10.1021/ol027261t. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Yamamoto H. Zirconium(IV)- and Hafnium(IV)-Catalyzed Highly Enantioselective Epoxidation of Homoallylic and Bishomoallylic Alcohols. J Am Chem Soc. 2010;132:7878–7880. doi: 10.1021/ja100951u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fouché KF, le Roux HJ, Phillips F. Complex formation of Zr(IV) and Hf(IV) with hydroxamic acids in acidic solutions. J Inorg Nucl Chem. 1970;32:1949–1962. [Google Scholar]
- 34.Vogl EM, Gröger H, Shibasaki M. Towards Perfect Asymmetric Catalysis: Additives and Cocatalysts. Angew Chem, Int Ed. 1999;38(11):1570–1577. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1570::AID-ANIE1570>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 35.Hanson RM, Sharpless KB. Procedure for the catalytic asymmetric epoxidation of allylic alcohols in the presence of molecular sieves. J Org Chem. 1986;51(10):1922–1925. [Google Scholar]
- 36.Shannon R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr, Sect A. 1976;32(5):751–767. [Google Scholar]
- 37.Lubben TV, Wolczanski PT. Dioxygen activation by Group 4 tritox alkyls [tritox = (tert-Bu)3CO−]: insertion and oxygen atom transfer. J Am Chem Soc. 1987;109:424–435. [Google Scholar]
- 38.LI Z. PhD Thesis. The University of Chicago; Chicago, USA: 2011. New Asymmetric Epoxidation Reactions Enabled by Chiral Metal-Bishydroxamic Acid Catalysts. [Google Scholar]
- 39.Hoveyda AH, Evans DA, Fu GC. Substrate-directable chemical reactions. Chem Rev. 1993;93(4):1307–1370. [Google Scholar]
- 40.Olivares-Romero JL, Li Z, Yamamoto H. Hf(IV)-Catalyzed Enantioselective Epoxidation of N-Alkenyl Sulfonamides and N-Tosyl Imines. J Am Chem Soc. 2012;134(12):5440–5443. doi: 10.1021/ja211880s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Tsui GC, Lautens M. Linear-Selective Rhodium(I)-Catalyzed Addition of Arylboronic Acids to Allyl Sulfones. Angew Chem, Int Ed. 2010;49(47):8938–8941. doi: 10.1002/anie.201004345. [DOI] [PubMed] [Google Scholar]; (b) Csatayová K, Davies SG, Lee JA, Ling KB, Roberts PM, Russell AJ, Thomson JE. Chemo- and diastereoselective cyclopropanation of allylic amines and carbamates. Tetrahedron. 2010;66(43):8420–8440. [Google Scholar]
- 42.Lykke L, Rodríguez-Escrich C, Jørgensen KA. Catalytic Enantioselective Oxaziridination. J Am Chem Soc. 2011;133(38):14932–14935. doi: 10.1021/ja2064457. [DOI] [PubMed] [Google Scholar]
- 43.(a) Ahlford K, Ekström J, Zaitsev AB, Ryberg P, Eriksson L, Adolfsson H. Asymmetric Transfer Hydrogenation of Ketones Catalyzed by Amino Acid Derived Rhodium Complexes: On the Origin of Enantioselectivity and Enantioswitchability. Chem Eur J. 2009;15(42):11197–11209. doi: 10.1002/chem.200900548. [DOI] [PubMed] [Google Scholar]; (b) Ahlford K, Adolfsson H. Amino acid derived amides and hydroxamic acids as ligands for asymmetric transfer hydrogenation in aqueous media. Catal Commun. 2011;12(12):1118–1121. [Google Scholar]
- 44.Binanzer M, Hsieh SY, Bode JW. Catalytic Kinetic Resolution of Cyclic Secondary Amines. J Am Chem Soc. 2011;133(49):19698–19701. doi: 10.1021/ja209472h. [DOI] [PubMed] [Google Scholar]