

Abstract
Exocytosis of neutrophil granules contributes to acute lung injury (ALI) induced by infection or inflammation, suggesting that inhibition of neutrophil exocytosis in vivo could be a viable therapeutic strategy. This study was conducted to determine the effect of a cell-permeable fusion protein that inhibits neutrophil exocytosis (TAT-SNAP-23) on ALI using an immune complex deposition model in rats. The effect of inhibition of neutrophil exocytosis by intravenous administration of TAT-SNAP-23 on ALI was assessed by albumin leakage, neutrophil infiltration, lung histology, and proteomic analysis of bronchoalveolar lavage fluid (BALf). Administration of TAT-SNAP-23, but not TAT-Control, significantly reduced albumin leakage, total protein levels in the BALf, and intra-alveolar edema and hemorrhage. Evidence that TAT-SNAP-23 inhibits neutrophil exocytosis included a reduction in plasma membrane CD18 expression by BALf neutrophils and a decrease in neutrophil granule proteins in BALf. Similar degree of neutrophil accumulation in the lungs and/or BALf suggests that TAT-SNAP-23 did not alter vascular endothelial cell function. Proteomic analysis of BALf revealed that components of the complement and coagulation pathways were significantly reduced in BALf from TAT-SNAP-23-treated animals. Our results indicate that administration of a TAT-fusion protein that inhibits neutrophil exocytosis reduces in vivo ALI. Targeting neutrophil exocytosis is a potential therapeutic strategy to ameliorate ALI.
Keywords: Complement, proteomics, immune complex
INTRODUCTION
Acute lung injury (ALI) is a life threatening complication associated with sepsis, pneumonia, aspiration, multiple blood transfusions, trauma, and acute pancreatitis. Despite improvements in management of critically ill patients, mortality remains about 40%, and survivors often do not return to a normal life (1). Neutrophils are the first immune cells to accumulate in the lung during ALI, recruited by a complex network of cytokines and chemokines produced by tissue macrophages (2). Evidence supporting a causative role for neutrophils in lung injury includes a correlation between the degree of neutrophil accumulation in bronchoalveolar lavage fluid (BALf) and the severity of ALI in humans, and a reduction in the severity of ALI in animal models following neutrophil depletion or inhibition of neutrophil accumulation in the lungs (3–7). The major systems by which neutrophils induce lung injury include the release of granule components, formation of neutrophil extracellular traps (NETs), and generation of reactive oxygen species (ROS) (2, 8–10). A number of neutrophil granule components, including serine proteases, matrix metalloproteinases, lactoferrin, LL-37, azurocidin, and defensins, have been shown to contribute to ALI (2) and direct evidence for a role of neutrophil exocytosis was provided by studies in which injection of granule products produced ALI (8).
Although a role for neutrophils in ALI is well established, strategies to control neutrophil activation to prevent tissue and organ injury have not followed. We recently developed a TAT-fusion protein containing a SNARE domain from SNAP-23 (TAT-SNAP-23) that inhibited secretory vesicle, gelatinase granule, and specific granule exocytosis in human neutrophils (11). Using that TAT-fusion protein we showed that inhibition of neutrophil exocytosis reduced phagocytosis-stimulated hydrogen peroxide production by 60% and inhibited tumor necrosis factor (TNF) α and platelet activating factor-induced priming of the respiratory burst by up to 80% (11). To determine if administration of TAT-SNAP-23 was able to inhibit inflammatory injury in vivo, the present study used a model of ALI induced by immune complex (IC) deposition in rats. That model is dependent on complement activation, cytokine production, and chemokine-induced neutrophil infiltration (12, 13). Our results show that intravenous administration of TAT-SNAP-23 inhibited the development of increased vascular permeability without affecting neutrophil accumulation in the lungs, and that effect was associated with reduced accumulation of complement components.
MATERIALS AND METHODS
Rodent Model of ALI
ALI was induced in pathogen-free male Long-Evans rats (275 to 300 g) as previously described (3–7). Briefly, rabbit polyclonal IgG anti-bovine serum albumin (BSA; 2.5 mg, MP-Biomedics, Solon, OH) was injected intratracheally in 0.3 ml of PBS, pH 7.4. Immediately thereafter, 10 mg of BSA (Sigma-Aldrich, St. Louis, MO) with 250 μg of FITC-BSA (Invitrogen, Carlsbad, CA) in 0.5 ml of PBS was injected intravenously. Two h later, 0.05 mg/kg of TAT-SNAP-23 or a control TAT-fusion protein of similar molecular size (TAT-Control) (11) was injected intravenously. Animals were euthanized four h after initiation of injury. In separate studies intravenous administration of TAT-SNAP-23 or TAT-Control showed no side effects after 24 h. Animal care was performed following the National Institute of Health guidelines. Animal studies were approved by the University of Louisville Institutional Animal Care and Use Committee.
In Vitro Assays
BALf was collected by instilling and withdrawing 5 ml of sterile PBS three times via an intratracheal cannula. Lung injury was quantified by measuring leakage of FITC-BSA into lung parenchyma by fluorometry and by measuring the BALf albumin concentration. Cells were recovered from BALf by centrifugation and Wright-stained following a cytospin. A cell differential count was performed and the total number of neutrophils calculated. BALf was stored at −80 C for mass spectrometry analysis and measurement of cytokine/chemokine levels (Multi-Analyte ELISA array, SABiosciences, Frederick, MD). For immunoblot analysis, BALf cells were lysed in 40 μl of ice-cold lysis buffer as previously described (11, 14). Lysate or BALf proteins were separated by 4–12% gradient SDS-PAGE, followed by immunoblot analysis for His-tag (1:1000, Abcam, Cambridge, MA), C-reactive protein (CRP, Santa Cruz, Santa Cruz, CA, 1:1000), or complement C3 (1:1000, Pierce, Rockford, IL). Lung homogenates were separated by 4–12% gradient SDS-PAGE, followed by immunoblot analysis for caspase-3 (1:500, Cell Signaling Technology, Donvers, MA). Protein signal was visualized by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). Plasma membrane expression of CD18 was measured in BAL cells by FITC-conjugated monoclonal anti-CD18 (Abcam, Cambridge, MA); secretory vesicle and specific granule exocytosis by isolated human neutrophils was determined by measuring the increase in plasma membrane expression of FITC-conjugated monoclonal anti-CD35 (secretory vesicles, clone E11; Pharmingen, San Diego, CA), and FITC-conjugated monoclonal anti-CD66b (specific granule, clone CLB-B 13.9; Accurate Chemical, Westbury, NY) in human neutrophils. All the plasma membrane markers were analyzed by flow cytometry as previously described (11, 14).
Human neutrophils were isolated from healthy donors using plasma-Percoll gradients as previously described (11, 14, 16). Wright staining performed on cytospins of isolated cells confirmed that >95% of the cells were neutrophils. Trypan blue exclusion staining confirmed >97% of cells were viable. The Institutional Review Boards of the University of Louisville approved the use of human donors for the study.
Lung histology
In a separate group of rats, lungs were fixed by intratracheal instillation of 10% buffered formaldehyde. Sections of embedded lung were stained by hematoxilin and eosin. The extent and location of neutrophils in the lungs was determined by immunohistochemistry for myeloperoxidase (MPO) using a polyclonal anti-rat MPO antibody (1:500; Abcam, Cambridge, MA).
Mass spectrometry analysis of BALf
To achieve greater sensitivity for identification of low-abundance proteins, BALf supernatants were immunodepleted of seven high-abundance serum proteins (albumin, IgG, transferrin, fibrinogen, IgA, alpha-2 macroglobulin, IgM) using a ProteomeLab™ IgY R7 LC-2 column according to manufacturer guidelines. Proteins in the flow-through fractions were reduced, alkylated, and digested with trypsin, as previously described (14, 15). Tryptic peptides were separated by 2DLC, and MS/MS data generated by nanospray ionization into a LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific Waltham, MA). The acquired mass spectrometry data were searched against a rat refseq protein database modified to include bovine serum albumin using the SEQUEST (version 27 revision 11) algorithm, as described previously (14, 15). The database analysis was performed with SequestSorcerer (Sage-N Research, San Jose, CA) and high-probability peptide and protein identifications were assigned from the SEQUEST results using the ProteinProphet (tools.proteomecenter.org/software.php) and SageN Sorcerer statistical platforms. Scaffold 3 proteomic analysis software (ProteomeSoftware, Inc, Portland, OR) was used for quantitative comparison using a label-free spectral counting method (14, 15). Qualitative comparison of protein expression patterns was performed by Ingenuity Pathways Analysis software (http://ingenuity.com) (16).
Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis was performed using ANOVA with the Tukey-Kramer multiple-comparison test. Differences were considered statistically significant when P < 0.05
RESULTS
Administration of TAT-SNAP-23 ameliorates ALI
To determine an effective dose of TAT-SNAP-23, a preliminary study was performed in which 0.01 mg/kg, 0.05 mg/kg, or 0.1 mg/kg was injected intravenously 2 h after IC deposition in the lung. BALf was collected at 4 h and vascular permeability was measured by the amount of FITC-BSA fluorescence. Induction of ALI resulted in a 5-fold increase in fluorescence in BALf that was reduced by 54% following administration of 0.01 mg/kg of TAT-SNAP-23, by 57% with 0.05 mg/kg, and 35% with 0.1 mg/kg (data not shown). Based on those results, 0.05 mg/kg TAT-SNAP-23 was used in all subsequent experiments.
To determine the effect of inhibiting exocytosis with TAT-SNAP-23 on ALI, four groups of rats were used. A control group received intratracheal and intravenous saline, while the other 3 groups received anti-BSA intratracheally and BSA and FITC-labeled BSA intravenously. One group also received an intravenous injection of 0.05 mg/kg TAT-SNAP-23 and a second received 0.05 mg/kg TAT-Control 2 h after IC deposition. At 4 h rats were euthanized, and bronchoalveolar lavage was performed or lung tissue obtained for histology. Figure 1A shows the BALf fluorescence resulting from leakage of FITC-labeled BSA from blood into the lungs. As seen in the preliminary study, ALI was associated with a significant increase in BALf fluorescence that was significantly reduced following administration of TAT-SNAP-23, but not TAT-control. Figure 1B shows total protein in BALf from rats with ALI with and without treatment with TAT-Control or TAT-SNAP-23. Those results confirm that treatment with TAT-SNAP-23, but not TAT-Control, significantly reduced the increased vascular permeability associated with ALI. A differential cell count was performed in the BALf cells recovered from the four groups. The average number of neutrophils was very low in the control animals (0.07 × 106 ± 0.02 × 106, n=8 animals). About 95% of the cells recovered from rats with ALI were neutrophils, as previously described (12, 13). ALI was associated with a significant increase in total neutrophil count (5.2 × 106 ± 2 × 106, n=8 animals) that was not affected by treatment with TAT-SNAP-23 (8 × 106 ± 2.8 × 106, n=8 animals) or TAT-Control (9 × 106 ± 2.2 × 106, n=8 animals). No statistical difference was found between the number of cells recovered from BALf of ALI, or TAT-SNAP-23, or TAT-Control treated animals. Thus, TAT-SNAP-23 treatment significantly reduced the increased vascular permeability and total protein levels following IC deposition, while neutrophil infiltration was not affected.
Figure 1. Lung injured animals treated with TAT-SNAP-23 showed amelioration of lung injury without altering neutrophil infiltration into the lungs.
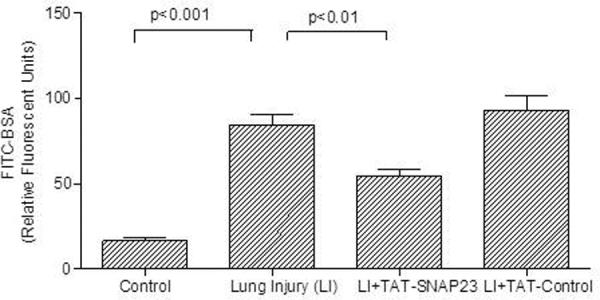
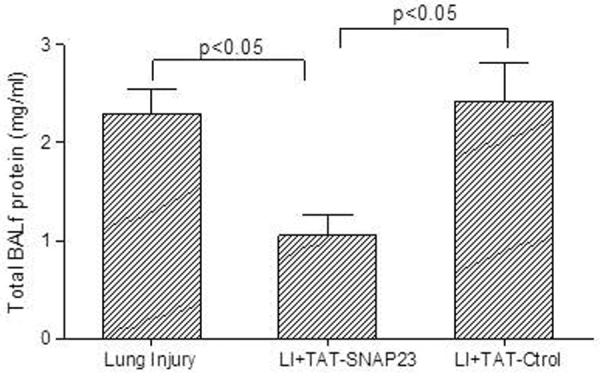

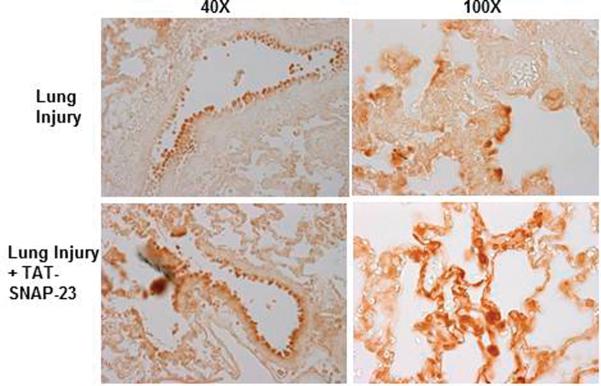
(A) Fluorescence of FITC-BSA was measured in BALf samples from 4 animal groups: control, lung injury (LI), lung injury followed by TAT-SNAP-23 (LI + TAT-SNAP-23), or lung injury followed by TAT-Control (LI + TAT-Control). Data were expressed as mean relative fluorescent units ± SEM, n= 6 animals per group. (B) Total protein was measured in BALf samples from 3 animal groups: lung injury (LI), lung injury followed by TAT-SNAP-23 (LI + TAT-SNAP-23), or lung injury followed by TAT-Control (LI + TAT-Control). Data were expressed as mean ± SEM, n= 4 animals per group. (C) Hematoxilin & Eosin (H&E) staining was performed on lung sections from 4 animal groups: control, lung injury, lung injury followed by TAT-SNAP-23, or lung injury followed by TAT-Control; original magnification 10 × and 100 ×. (D) Immunohistochemistry for myeloperoxidase (MPO) was performed on lung sections from lung injured animals, and lung injured treated with TAT-SNAP-23; original magnification 40 × and 100 ×.
Figure 1C shows representative lung histology from four groups. Rats administered saline showed a normal lung structure, while rats with untreated ALI demonstrated intraalveolar edema and hemorrhage and neutrophil infiltration. Treatment with TAT-SNAP-23 resulted in a reduction in lung edema and hemorrhage, although infiltrating neutrophils were still observed. Lung histology in rats treated with TAT-Control was similar to that seen in rats with untreated ALI. To confirm the observation that neutrophil infiltration was not impaired in TAT-SNAP-23 treated animals, immunohistochemistry for MPO was performed. Figure 1D shows that the number and location of neutrophils was similar with or without TAT-SNAP-23 treatment. Thus, histology supported the BALf findings that TAT-SNAP-23, but not TAT-control, inhibited the increased vascular permeability produced by IC deposition, while neutrophil migration into lungs was not affected.
To determine if neutrophils infiltrating the lungs contained TAT-fusion proteins, immunoblot analysis for the His-tag present in the TAT-fusion proteins was performed in cells contained in BALf. Figure 2 shows that cell lysates from rats treated with TAT-SNAP-23 and TAT-Control contained proteins at the appropriate molecular size that stained for the His-tag (11). As the majority of cells contained in BALf were neutrophils, we interpret those results to indicate that TAT-fusion proteins entered circulating neutrophils that subsequently migrated into the lungs.
Figure 2. BAL cells have TAT-SNAP-23 or TAT-Control.

BAL cells collected from 3 animal groups: lung injury (LI), lung injury treated with TAT-SNAP-23 (LI + TAT-SNAP-23), or lung injury followed by TAT-Control (LI + TAT-Control), were lysed and proteins were separated by SDS-PAGE followed by immunoblot analysis for His-tag. A representative immunoblot from three different and independent experiments is shown.
Effect of TAT-SNAP-23 on neutrophil exocytosis and generation of inflammatory stimuli
To determine if intravenous administration of TAT-SNAP-23 inhibited neutrophil exocytosis, plasma membrane expression of a granule marker CD18 (β2 integrin) by BALf neutrophils was analyzed by flow cytometry. Table 1 shows that neutrophils from rats with ALI showed a significant increase in the plasma membrane expression of CD18, indicating that exocytosis had occurred in infiltrating neutrophils. BALf neutrophils obtained from animals with ALI that were also treated with TAT-SNAP-23, but not TAT-Control, showed a small but significant reduction in the plasma membrane expression of CD18 (Table 1).
Table 1.
Administration of TAT-SNAP-23 reduced CD18 plasma membrane expression in BAL cells.
|
|
||
|---|---|---|
| CD18MCF (mean ± SEM) | N=number of rats | |
| Control | 305 ± 37** | 5 |
| IC-ALI | 611 ± 20 | 7 |
| TAT-SNAP-23-IC-ALI | 562 ± 16* | 9 |
| TAT-Control-IC-ALI | 619 ± 12 | 9 |
p<0.05 compared to TAT-Control;
p<0.001 compared to IC-ALI
Although the inhibitory effect of TAT-SNAP-23 on CD18 expression suggests that intravenous administration of TAT-SNAP-23 partially inhibited neutrophil exocytosis, other possibilities are that TAT-SNAP-23 treatment reduced expression of neutrophil stimuli in BALf. To determine if BALf from TAT-SNAP-23 treated animals contained stimuli of neutrophil exocytosis, in vitro degranulation assays were performed with isolated human neutrophils incubated with BALf from the four different animal groups. Secretory vesicle and specific granule exocytosis was determined by an increased plasma membrane expression of CD35 and CD66b, respectively. Figures 3A and 3B show that BALf obtained from rats with ALI with or without treatment with TAT-SNAP-23 or TAT-Control stimulated exocytosis equivalent to that stimulated by fMLF, while human neutrophils incubated with BALf from control animals showed no increase in CD35 and CD66b expression compared to basal levels. These data indicate that there are sufficient stimuli in the BALf of TAT-SNAP-23 treated animals to activate neutrophils; however those animals showed less severe tissue injury and reduced upregulation of CD18. In vitro degranulation assays using human neutrophils pre-treated with TAT-SNAP-23 followed by incubation with the different BALf showed inhibition of secretory vesicle and specific granule exocytosis (data not shown). No difference in TNFα, and IL-6 levels were observed in BALf from rats with ALI with or without treatment with TAT-SNAP-23 or TAT-Control (data not shown). No change in caspase-3 levels was observed in lung homogenates from rats with ALI with or without treatment with TAT-SNAP-23 or TAT-Control (data not shown). Taken together, the results suggest that administration of TAT-SNAP-23 resulted in amelioration of ALI by reducing neutrophil exocytosis, while generation of pro-inflammatory stimuli, apoptosis, and infiltration of neutrophils into the lungs was not affected.
Figure 3. Effect of BALf from TAT-SNAP-23 treated animals' on human neutrophil exocytosis.


Human neutrophils (4 × 106 cell/ml) were incubated with buffer (Basal), stimulated with fMLF (300 nM, 5 min), or with BALf (1:100 dilution, 30 min) from 4 animal groups: control, lung injury (LI), lung injury followed by TAT-SNAP-23 (LI + TAT-SNAP-23), or lung injury followed by TAT-Control (LI + TAT-Control). (A), exocytosis of secretory vesicles was measured as the increase in plasma membrane expression of CD35. (B), exocytosis of specific granule was measured as the increase in plasma membrane expression of CD66b. Data are expressed as mean ± SEM of the mean channel fluorescence (MCF) for 7 independent experiments.
Proteomic analysis of BALf
To examine potential mechanisms by which inhibition of neutrophil exocytosis reduced ALI, identification and quantification of proteins contained in BALf from rats with untreated ALI or ALI treated with TAT-SNAP-23 or TAT-Control was performed by mass spectrometry. A total of 117 proteins were identified from the three groups that had spectral counts greater than 1 (see Supplemental Table 1 which contains the accession number and spectral counts for all the proteins identified in the BALf). Those proteins were subjected to Ingenuity Pathways Knowledge Base (IPKB) analysis to assign functional and canonical pathways. Table 2 shows the seven functional and canonical pathways contained the largest number of identified proteins. This analysis indicates that a number of proteins present in BALf from rats with ALI are associated with pathways related to an immune or inflammatory response. Functional pathways included antigen presentation (23 proteins), humoral immune response (9 proteins), inflammatory response (30 proteins), and immune cell trafficking (23 proteins). Canonical pathways included the acute phase response (24 proteins), the complement system (12 proteins), and the coagulation system (6 proteins).
Table 2.
Distribution of proteins by functional and canonical pathwaysa
| Function | # of proteins | Canonical | # of proteins |
|---|---|---|---|
| Antigen presentation | 23 | Acute phase response signaling | 24 |
| Humoral immune response | 9 | Complement system | 9 |
| Inflammatory response | 30 | Coagulation system | 4 |
| Cellular movement | 24 | Neuroprotective role of THOP1 in Alzheimer's disease | 4 |
| Hematological system development and function | 26 | Hepatic fibrosis/hepatic stellate cell activation | 4 |
| Immune cell trafficking | 23 | LXR/RXR activation | 3 |
| Cell-to-cell signaling and interaction | 23 | Intrinsic prothrombin activation pathway | 2 |
Proteins were categorized by functional and canonical pathways based on Ingenuity analysis.
To determine if TAT-SNAP-23 treatment altered protein expression in BALf, the spectral counts of all 117 proteins were normalized to the total protein concentration. Of the 117 proteins, 78 (67%) proteins were reduced by at least 50% in the TAT-SNAP-23 treated rats, 32 proteins (28%) had similar spectral counts among the three groups, and 6 proteins (5%) showed at least a 50% increase in the spectral counts in TAT-SNAP-23 treated animals (see Supplemental Table 1). Of the 78 proteins reduced in BALf from TAT-SNAP-23 treated rats, 12 proteins were components of the complement cascade, 3 proteins components of the coagulation pathway, 4 were neutrophil granule proteins, and one was an acute phase protein (Table 3). Validation of the proteomic data was performed by immunoblot analysis for two identified proteins, the acute phase protein C-reactive protein and the complement protein C3. As shown in Figure 4, a reduction of C-reactive protein (Fig 4A) and C3 (Fig 4B) was observed in BALf from TAT-SNAP-23 treated animals compared to TAT-Control or untreated ALI.
Table 3.
Administration of TAT-SNAP-23 resulted in a reduction in BALf proteins identified by proteomic analysis.
| Spectral counts normalized to total protein | ||||
|---|---|---|---|---|
| Identified Proteins | Accession Number | LI | LI + TAT-SNAP-23 | LI + TAT-Control |
| Classical Complement Pathway Proteins | ||||
| Complement C3 | gi|158138561 | 434.63 | 160.9 | 387.7 |
| Complement factor H | gi|77861917 | 56.23 | 22.03 | 43.76 |
| Complement factor B | gi|218156285 | 51.5 | 19.3 | 44.83 |
| Complement C9 | gi|16924006 | 29.6 | 12.03 | 27.76 |
| Complement C4 precursor | gi|158186621 | 33.63 | 12.5 | 25.2 |
| Predicted: Complement C5 | gi|109468076 | 15.23 | 6.2 | 11.26 |
| Complement factor I precursor | gi|13162353 | 13.03 | 4.6 | 13.4 |
| Complement C8 alpha chain | gi|157821013 | 8 | 3.63 | 8.13 |
| Complement C8 gamma chain | gi|157817131 | 5.06 | 2.7 | 5.13 |
| Complement C6 precursor | gi|28570180 | 4 | 2.56 | 7.86 |
| Complement factor D precursor | gi|117647198 | 3.36 | 1.93 | 4.06 |
| Complement C4, gene 2 | gi|50657362 | 22.16 | 7.1 | 15.03 |
| Coagulation Factors | ||||
| Plasminogen | gi|16758216 | 129 | 56.06 | 132.1 |
| Prothrombin | gi|161333847 | 15.56 | 8.33 | 14.83 |
| Coagulation factor XII | gi|62078741 | 4.4 | 2.73 | 5 |
| Acute Phase Proteins | ||||
| C-reactive protein precursor | gi|8393197 | 60.43 | 32.7 | 65.76 |
| Neutrophil Granule Proteins | ||||
| Myeloperoxidase | gi|157820285 | 43.6 | 22.36 | 31.33 |
| Lysozyme C-1 precursor | gi|40254796 | 52.26 | 30.46 | 53.63 |
| Neutrophil collagenase precursor | gi|11560008 | 4.16 | 2 | 7.2 |
| Vitamin D-binding protein | gi|6978879 | 109.2 | 47.7 | 111.5 |
Note: LI= Lung Injury
Figure 4. Less expression of CRP and C3 protein in the BALf of TAT-SNAP-23 treated animals.
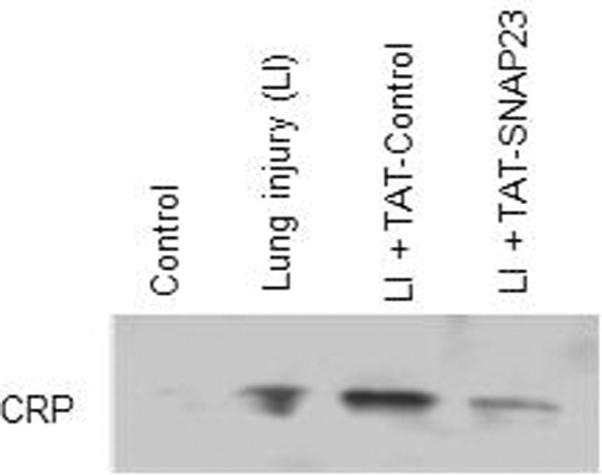
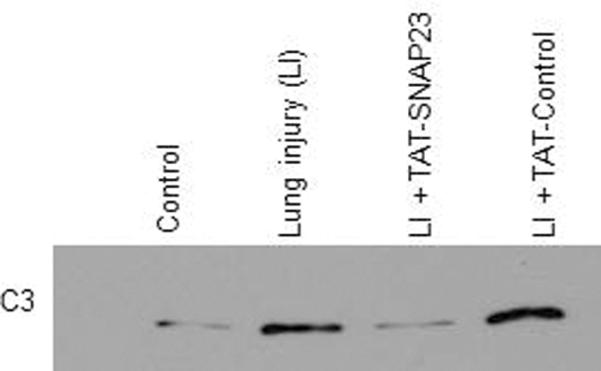
Immunoblot for C-reactive protein (CRP), and complement component 3 (C3) was performed on the BALf from 4 animal groups: control, lung injury (LI), lung injury followed by TAT-SNAP-23 (LI + TAT-SNAP-23), or lung injury followed by TAT-Control (LI + TAT-Control). Proteins present in the BALf were separated by SDS-PAGE and the immunoblot analysis performed with anti-CRP (A), or anti-C3 (B).
DISCUSSION
The major finding of the present study is that in vivo administration of a novel fusion protein containing a SNARE domain from SNAP-23 resulted in amelioration of injury in a neutrophil-dependent ALI rat model. Using a well characterized rat model of neutrophil-dependent ALI induced by alveolar deposition of IC (3–7, 12, 13, 17–19), intravenous administration of that fusion protein inhibited the development of lung injury, as measured by a reduction in albumin leakage and by reduced alveolar hemorrhage and edema on histologic examination. Additionally, proteomic analysis of BALf suggested that one mechanism by which inhibition of neutrophil exocytosis reduced ALI is via reduced activation of the complement and coagulation cascades. The data suggest that targeting SNARE protein-mediated exocytosis may be an effective therapeutic strategy for neutrophil-mediated diseases.
SNARE proteins mediate exocytosis in a variety of cell types, including neuronal cells, leukocytes, endothelial and epithelial cells (20, 21). We recently showed that incubation of isolated human neutrophils with a novel TAT-fusion protein containing the SNARE domain of SNAP-23 significantly inhibited stimulated exocytosis of secretory vesicles, gelatinase granules, and specific granules, but not azurophilic granules (11). That inhibition of exocytosis was associated with a reduction in phagocytosis-stimulated respiratory burst activity and with inhibition of TNFα-and platelet activating factor-induced priming of fMLF-stimulated superoxide release. Based on the ability of TAT-SNAP-23 to bind SNARE protein complexes in vitro, the mechanism of inhibition of exocytosis was proposed to be interference with SNARE protein coiled-coil formation (11). The current study provides evidence that TAT-SNAP-23 inhibited neutrophil exocytosis in vivo. Neutrophils obtained from BALf of TAT-SNAP-23 treated rats contained TAT-SNAP-23 and showed reduced plasma membrane expression of CD18. The increase in plasma membrane expression of CD18 in stimulated neutrophils results from exocytosis of gelatinase and specific granules (22, 23). Additionally, quantitative proteomic analysis of BALf found reduced levels of several neutrophil granule proteins, including myeloperoxidase, collagenase, and lysozyme.
As SNARE proteins are involved in protein trafficking in all cells, the possibility that TAT-SNAP-23 inhibited ALI through an action on cells other than neutrophils was considered. N-ethylmaleimide-sensitive factor (NSF), like SNAP-23, is another molecule involved in vesicular trafficking (21). Morrel et al. showed that intravenous administration of a TAT-fusion protein containing an inhibitory peptide derived from NSF inhibited vascular endothelial cell exocytosis and blocked neutrophil infiltration into the peritoneum in a mouse model of inflammatory peritonitis (24). If TAT-SNAP-23 acted similarly, reduced neutrophil recruitment into the lungs would be predicted. The presence of similar numbers of neutrophils in the lungs and BALf of TAT-SNAP-23 treated and untreated animals in our study argues against an effect of TAT-SNAP-23 on vascular endothelial cell exocytosis. Additionally, the presence of similar levels of cytokines, including TNFα and IL-6, in BALf of all three groups of rats with alveolar IC deposition indicates that administration of TAT-SNAP-23 had no effect on cytokine production and release by resident macrophages, lung parenchymal cells, and infiltrating neutrophils. In vitro studies performed with isolated human neutrophils exposed to BALf from all three groups of rats showed that the reduced plasma membrane expression of CD18 in rat neutrophils derived from BALf was not due to the absence of adequate pro-inflammatory stimuli. Taken together, our data suggest that TAT-SNAP-23 inhibits development of ALI by blocking exocytosis of neutrophils that have infiltrated into the lung in response to the inflammatory response initiated by immune complex deposition.
The proteomic analysis of BALf identified complement and coagulation proteins, as well as C-reactive protein, and the neutrophil-derived proteins myeloperoxidase, collagenase, gelatinase, and vitamin-D binding protein in rats with ALI. Quantitative analysis found that a number of proteins in the complement and coagulation systems were reduced in BALf from TAT-SNAP-23-treated animals. Complement activation is necessary for neutrophil recruitment and IC-induced ALI in the rodent model used in this study (3–7, 17–19, 25). Complement activation and generation of C5a results in activation of alveolar macrophages stimulating the release of pro-inflammatory cytokines such as TNF-α, IL-1 and chemokines such as MIP-2 and KC (12, 13, 19). Additionally, C5a and the membrane attack complex (C5b-9) are involved in up-regulation of endothelial cell adhesion molecules which participate in neutrophil migration into the lung parenchyma (17). This pro-inflammatory environment results in enhanced neutrophil recruitment and activation leading to tissue damage. It has been shown that intratracheal administration of a neutralizing antibody to C5a or inducing the IC-ALI in a C5 knockout mice resulted in attenuation of neutrophil infiltration and amelioration of lung injury (26–28). Our data show that blocking neutrophil exocytosis by administration of TAT-SNAP-23 results in reduction of complement components and amelioration of lung injury. Previous reports showed that intra-tracheal injection of neutrophil granule components produced ALI (8) and that specific granule contents, including serine proteases, matrix metalloproteinases, LL-37, lactoferrin, defensins, and azurocidin, may contribute to ALI (2). An interaction of those granule components with the complement cascade has not been examined. Our findings suggest the novel hypothesis that neutrophil exocytosis participates in ALI through release of granule components that, among other actions, regulate complement activation. Neutrophils and alveolar macrophages cleave C5 to generate active C5a, and serine proteases participate in C5a generation (29, 30). Our findings suggest that inhibition of neutrophil exocytosis not only limits the release of granule components capable of directly inducing tissue injury, but also prevents activation of an amplification loop which results from activation of the complement cascade.
The ability of BALf from both treated and untreated groups to stimulate equal neutrophil exocytosis in vitro indicates IC deposition results in the generation of equivalent amounts of neutrophil stimuli. We have reported previously that TNFα is a potent stimulus of neutrophil exocytosis (11). The presence of substantial amounts of TNFα in TAT-SNAP-23–treated animals supports the conclusion that TAT-SNAP-23 inhibited ALI by blocking neutrophil exocytosis. Although the mechanism by which exocytosis contributed to complement activation was not examined in our study, Camous et al. recently described a novel mechanism by which the alternative complement pathway is activated on the surface of neutrophils (31). Neutrophils primed by TNFα and stimulated by fMLF demonstrated increased plasma membrane expression of properdin released from granules, which activated the alternative complement pathway resulting in plasma membrane deposition of C3. We reported previously that properdin is a component of neutrophil specific granules (22). Ritis et al. reported that activation of neutrophil C5a receptors triggers activation of the extrinsic coagulation cascade via release of tissue factor (32). It has been shown that high levels of C5a result in upregulation of factor H, a key complement inhibitor, suggesting that C5a controls its own generation to maintain homeostasis (33). Flierl et al. reported that neutralization of C5a via systemic infusion of anti-C5a resulted in reduction of factor H, and amelioration of trauma-induced ALI (34). Similarly, in the present study inhibition of neutrophil exocytosis resulted in a reduction in both C5 and factor H and amelioration of IC-induced ALI. Thus, neutrophil degranulation might contribute to complement activation leading to tissue damage through a mechanism that remains to be determined.
A proteomic study performed on BALf of patients with acute respiratory distress syndrome (ARDS) found higher levels of complement components C3, C4 and C9 compared to normal subjects (35). In the same study, BALf from ARDS patients showed increasing expression of C3 as the disease progressed (35). Additionally, this study showed increased S100 proteins (S100 A8 and S100 A9) at Day 1 in BALf of ARDS patients compared to control subjects. We reported previously that S100 A8 and S100 A9 proteins are components of neutrophil specific and gelatinase granules (22). Those reports and our data support a pathophysiologically relevant link between neutrophil exocytosis and activation of the complement and coagulation cascade following IC deposition. Understanding the mechanisms involved in regulation of neutrophil exocytosis may lead to new therapeutic strategies to improve the outcome of ALI.
Supplementary Material
Acknowledgments
The authors thank Karen Brinkley, Junyi Le, Shweta Tandon, Timmothy M. Creed, Terri Manning, and Daniel Wilkey for their expert technical help.
Sources of support: This work was supported by grants from the Department of Veterans Affair Merit Review Board (to K.R.M.), the American Heart Association (BGIA 0765387B to S.M.U.), University of Louisville School of Medicine (to S.M.U.), and the National Institutes of Health (K99/R00 HL087924 to S.M.U., R01 AI075212 to M.J.R.).
Footnotes
The authors have no financial conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage-or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1137–L1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 2.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17(3–4):293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lentsch AB, Ward PA. Regulation of experimental lung inflammation. Respir Physiol. 2001;128(1):17–22. doi: 10.1016/s0034-5687(01)00260-2. [DOI] [PubMed] [Google Scholar]
- 4.Ward PA, Lentsch AB. Endogenous regulation of the acute inflammatory response. Mol Cell Biochem. 2002;234/235(1–2):225–228. [PubMed] [Google Scholar]
- 5.Guo RF, Lentsch AB, Sarma JV, Sun L, Riedemann NC, McClintock SD, McGuire SR, Rooijen NV, Ward PA. Activator protein-1 activation in acute lung injury. Am J Pathol. 2002;161(1):275–282. doi: 10.1016/S0002-9440(10)64179-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lentsch AB, Czermak BJ, Jordan JA, Ward PA. Regulation of acute lung inflammatory injury by endogenous IL-13. J Immunol. 1999;162(2):1071–1076. [PubMed] [Google Scholar]
- 7.Mulligan MS, Varani J, Warren JS, Till GO, Smith CW, Anderson DC, Todd RF, Ward PA. Role of β2 integrins of rat neutrophils in complement- and oxygen radical-mediated acute inflammatory injury. J Immunol. 1992;148(6):1847–1857. [PubMed] [Google Scholar]
- 8.Soehnlein O, Oehmck S, Ma X, Rothfuchs AG, Frithiof R, van Rooijen N, Morgelin M, Herwald H, Lindbom L. Neutrophil degranulation mediates severe lung damage triggered by streptococcal M1 protein. Eur Respir J. 2008;32(2):405–412. doi: 10.1183/09031936.00173207. [DOI] [PubMed] [Google Scholar]
- 9.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infection. 2003;5(14):1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew A-A, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uriarte SM, Rane MJ, Luerman GC, Ward RA, Nauseef WM, McLeish KR. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol. 2011;187(1):391–400. doi: 10.4049/jimmunol.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao H, Neff T, Ward PA. Regulation of lung inflammation in the model of IgG immune-complex injury. Annu Rev Pathol. 2006;1:215–242. doi: 10.1146/annurev.pathol.1.110304.100155. [DOI] [PubMed] [Google Scholar]
- 13.Guo RF, Ward PA. Mediators and regulation of neutrophil accumulation in inflammatory responses in lung: insights from the IgG immune complex model. Free Radic Biol Med. 2002;33(3):303–310. doi: 10.1016/s0891-5849(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 14.Luerman GC, Powell DW, Uriarte SM, Cummins TD, Merchant ML, Ward RA, McLeish KR. Identification of Phosphoproteins Associated with Human Neutrophil Granules Following Chemotactic Peptide Stimulation. Mol Cell Proteomics. 2011;10(3):M110.001552. doi: 10.1074/mcp.M110.001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merchant M, Powell D, Wilkey D, Cummins T, Deegens J, Rood I, McAfee J, Fleischer C, Klein E, Klein J. Microfiltration isolation of human urinary exosomes for characterization by mass spectrometry. “Microfiltration isolation of human urinary exosomes for characterization by MS.”. Proteomics Clin Appl. 2010;4:84–96. doi: 10.1002/prca.200800093. [DOI] [PubMed] [Google Scholar]
- 16.Uriarte SM, Powell DW, Luerman GC, Merchant ML, Cummins TD, Jog NR, Ward RA, McLeish KR. Comparison of Proteins Expressed on Secretory Vesicle Membranes and Plasma Membranes of Human Neutrophils. J Immunol. 2008;180(8):5575–5581. doi: 10.4049/jimmunol.180.8.5575. [DOI] [PubMed] [Google Scholar]
- 17.Czermak BJ, Lentxch AB, Bless NM, Schmal H, Friedl HP, Ward PA. Role of complement in in vitro and in vivo lung inflammatory reactions. J Leukoc Biol. 1998;64:40–48. doi: 10.1002/jlb.64.1.40. [DOI] [PubMed] [Google Scholar]
- 18.Sarma VJ, Huber-Lang M, Ward PA. Complement in lung disease. Autoimmunity. 2006;39(5):387–394. doi: 10.1080/08916930600739456. [DOI] [PubMed] [Google Scholar]
- 19.Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–159. doi: 10.1007/978-1-4614-0106-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323(5913):474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol. 2001;2(2):98–106. doi: 10.1038/35052017. [DOI] [PubMed] [Google Scholar]
- 22.Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. 2005;4(10):1503–1521. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee G, Rasmusson B, Linner JG, Quinn MT, Parkos CA, Magnusson K-E, Jesaitis AJ. Organization and mobility of CD11b/CD18 and targeting of superoxide on the surface of degranulated human neutrophils. Arch Biochem Biophys. 1998;357(1):164–172. doi: 10.1006/abbi.1998.0807. [DOI] [PubMed] [Google Scholar]
- 24.Morrell CN, Matsushita K, Lowenstein CJ. A novel inhibitor of N-ethylmaleimide-sensitive factor decreases leukocyte trafficking and peritonitis. J Pharmacol Exp Ther. 2005;314(1):155–161. doi: 10.1124/jpet.104.082529. [DOI] [PubMed] [Google Scholar]
- 25.Åsberg AE, Mollnes TE, Videm V. Complement activation by neutrophil granulocytes. Scand J Immunol. 2008;67(4):354–361. doi: 10.1111/j.1365-3083.2008.02077.x. [DOI] [PubMed] [Google Scholar]
- 26.Larsen GL, Mitchell BC, Henson PM. The pulmonary response of C5 sufficient and deficient mice to immune complexes. Am Rev Resp Medicine. 1981;123:434–439. doi: 10.1164/arrd.1981.123.4.434. [DOI] [PubMed] [Google Scholar]
- 27.Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, Hugli TE, Miyasaka M, Warner RL, Johnson KJ, Ward PA. Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest. 1996;98:503–512. doi: 10.1172/JCI118818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nature Medicine. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 29.Huber-Lang M, Younkin EM, Sarma JV, Riedemann N, McGuire SR, Lu KT, Kunkel R, Younger JG, Zetoune FS, Ward PA. Generation of C5a by phagocytic cells. Am J Pathol. 2002;161:1849–1859. doi: 10.1016/S0002-9440(10)64461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ward PA, Hill JH. C5 chemotactic fragments produced by an enzyme in lysosomal granules of neutrophils. J Immunol. 1970;104:535–543. [PubMed] [Google Scholar]
- 31.Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117(4):1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 32.Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794–4802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 33.Schlaf G, Nitzi F, Heine I, Hardeland R, Schieferdecker HL, Götze O. C5a anaphylatoxin as a product of complement activation up-regulates the complement inhibitory factor H in rat Kupffer cells. Eur J Immunol. 2004;34:3257–3266. doi: 10.1002/eji.200324806. [DOI] [PubMed] [Google Scholar]
- 34.Flierl MA, Perl M, Rittirsch D, Barti C, Schreiber H, Fleig V, Schlaf G, Liener U, Brueckner UB, Gebhard F, Huber-Lang MS. The role of C5a in the innate immune response after experimental blunt chest trauma. Shock. 2008;29:25–31. doi: 10.1097/shk.0b013e3180556a0b. [DOI] [PubMed] [Google Scholar]
- 35.Chang DW, Hayashi S, Gharib SA, Vaisar T, Trevor King S, Tsuchiya M, Ruzinski JT, Park DR, Matute-Bello G, Wurfel MM, Bumgarner R, Heinecke JW, Martin TR. Proteomic and computational analysis of bronchoalveolar proteins during the course of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2008;178:701–709. doi: 10.1164/rccm.200712-1895OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.