

Abstract
Many of the best-studied actin regulatory proteins use non-covalent means to modulate the properties of actin. Yet, actin is also susceptible to covalent modifications of its amino acids. Recent work is increasingly revealing that actin processing and its covalent modifications regulate important cellular processes. In addition, numerous pathogens express enzymes that specifically use actin as a substrate to regulate their hosts cells. Actin post-translational alterations have been linked to different normal and disease processes and the effects associated with metabolic and environmental stressors. Herein, we highlight specific co- and post-translational modifications of actin and discuss the role that these modifications play in regulating actin dynamics.
Introduction
The actin cytoskeleton enables diverse cellular behaviors and so a critical goal is to understand the factors that specify its organization and dynamic properties. Small molecules such as Mg2+ and ATP have long been known to modulate actin and an increasing number of specific proteins are well-known for their actin regulatory abilities [1]. These effectors, along with environmental (e.g., reactive metals such as copper) and well-known biologically-produced (e.g., cytochalasins) toxins, physically associate and use non-covalent mechanisms to control actin. However, actin is also regulated through the covalent alteration of its amino acids. Although less-well appreciated, these co- and post-translational modifications (PTMs) of actin are widely employed, occur through enzymatic and non-enzymatic mechanisms, regulate the monomer-polymer equilibrium and organization of actin, and direct both physiological and pathological processes. Characterizing these modifications constitutes a rapidly expanding area within cell biology and actin studies. Below, we provide an overview of these actin PTMs and discuss recent progress on characterizing actin regulatory enzymes.
Acetylation
When actin was initially sequenced by amino acid hydrolysis, it was found to be N-terminally acetylated [2]. Subsequent studies unraveled actin’s N-terminal acetylation machinery and revealed processing by specialized enzymes, including aminopeptidases that remove the N-terminal Met and on occasion the second amino acid, and acetyltransferases that sequentially modify the first, second, or third actin residue (Figures 1–3, Tables 1, S1–3). While such acetylation appears non-essential in lower eukaryotes [3], maturation and maintenance of actin’s structural and functional properties in multiple species often requires N-terminal acetylation (Table S2). Acetylation also facilitates actomyosin interactions in muscles and may determine actin ubiquitylation and metabolic fate (Table S2).
Figure 1. Structural model of the actin subunits and their fit within the filament structure and intersubunit interactions.
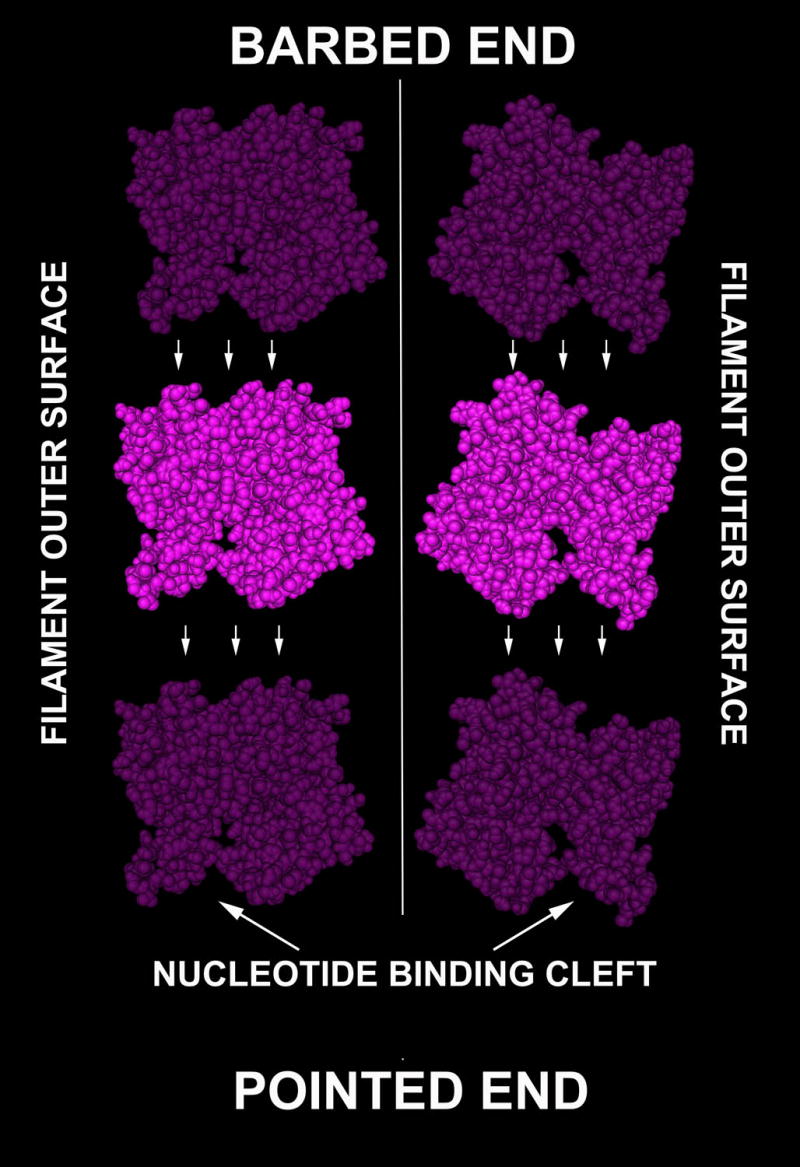
Front and back view of the same actin subunit is shown in each left and right panel, respectively. The key functional areas are indicated. Structure shown is based on alpha cardiac actin, PDB identifier 2A42.
Figure 3. Structural model of the actin molecule (front and back view) with individually mapped sites for several major post-translational modifications.
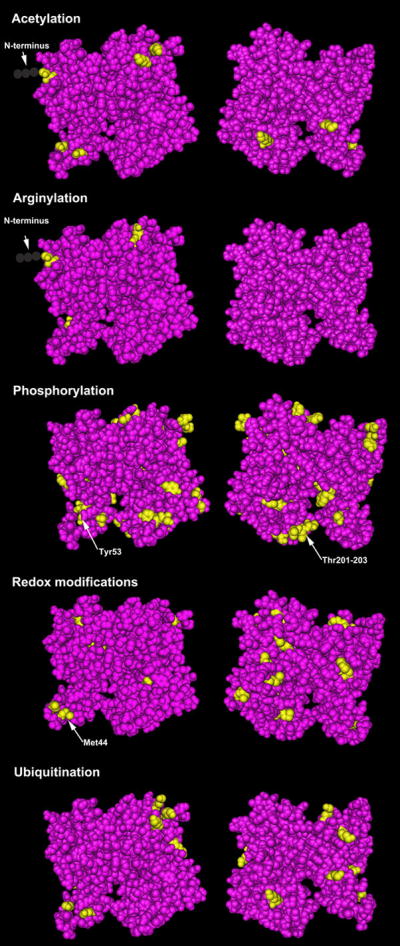
Residues found to undergo modifications are highlighted in yellow in each set of panels. Selected modification sites with defined effects are indicated by arrows in the corresponding panels. Structure shown is based on alpha cardiac actin, PDB identifier 2A42.
Table 1.
Specific Post-translational modifications of actin
| Modification | Known Sites on Actin (*=known functionally important modification) |
|---|---|
| Acetylation | Met-1*, Asp/Glu-2*, Asp-3*, Lys-50, Lys-61, Lys-68, Lys-191, Lys-326, Lys-328 |
| ADP-Ribosylation | Arg-28, Arg-95*, Thr-148*, Arg-177*, Arg-206*, Arg-372* |
| Arginylation | Asp-3*, Ser-52, Ile-87, Phe-90, Gly-152, Leu-295, Asn-299 |
| Carbonylation | His-40, His-87, His-173, Cys-374* |
| Crosslinking | Lys-50 (with Glu-270)* |
| Glutathionylation | Cys-217, Cys-374* |
| Isoaspartylation | Asp-25, Asp-179 |
| Malonylation | Lys-61 |
| Methylation | His-73*, Ile-87, Asn-299, Lys-326 |
| Nitration | Tyr-53, Tyr-69*, Tyr-91, Tyr-198, Tyr-218, Tyr-240, Tyr-294, Tyr-362 |
| Nitrosylation | Cys-217, Cys-257, Cys-285, Cys-374* |
| O-GlcNAcylation | Ser-52, Ser-155, Ser-199, Ser-232, Ser-323, Ser-368 |
| Oxidation | Met-44*, Met-47, Trp-79, Met-82, Trp-86, Met-178, Met-190, Cys-217, Met-227, Cys-257, Met-269, Cys-272*, Cys-285*, Met-325, Trp-340, Met- 355, Trp-356, Cys-374* |
| Phosphorylation | Ser-14, Ser-33, Ser-52, Tyr-53*, Ser-60, Thr-66, Tyr-69, Thr-89, Tyr-91, Thr-148, Thr-160, Thr-162, Tyr-166, Tyr-169, Thr-186, Tyr-188, Tyr-198, Ser-199, Thr/Ser-201*, Thr-202*, Thr-203*, Tyr-218*, Thr-229, Ser- 233, Ser-239, Tyr-240, Thr-262, Tyr-294, Thr-297, Tyr-306, Thr-318, Ser- 323, Ser-324, Tyr-362, Ser-365 |
| SUMOylation | Lys-68*, Lys-284* |
| Transglutamination | Gln-41* |
| Ubiquitylation | Lys-50, Lys-61, Lys-68, Lys-84, Lys-113, Lys-118, Lys-191, Lys-291, Lys- 315, Lys-326, Lys-328 |
Recent advances have also exposed other actin acetylation sites (Figure 3, Tables 1, S1–3) and diverse processes mediated by multiple acetyltransferases and deacetylases [4–6]. For example, the histone deacetylase HDAC6 specifically associates with actin and participates in actin rearrangements in vivo [7–9] and nuclear actin directly interacts with histone acetyltransferase during transcriptional regulation [10]. Thus, accumulating evidence suggests that acetylation may play important roles in modulating actin’s role in cell movement, intracellular transport, and transcriptional regulation.
ADP-Ribosylation
Both eukaryotes and bacterial pathogens express different ADP-ribosyltransferases that transfer ADP-ribose moieties from nicotinamide adenine dinucleotide (NAD) to specific actin residues (Figure 2, Tables 1, S2–3; reviewed in [11,12]). Perhaps the best-characterized ADP-ribosyltransferase is the Clostridium botulinum C2 toxin which ADP-ribosylates non-muscle G-actin on Arg-177, inducing cell rounding and actin network disruption (Table S2). This ADP-ribosylation inhibits actin polymerization. Moreover, ADP-ribosylated G-actin appears to cap the barbed end of actin, thereby inhibiting polymerization of non-modified actin. ADP-ribosylation of Arg-177 also reduces actin ATP hydrolysis and the nucleation activity of the Gelsolin-actin complex (Table S2). In contrast, another bacterial ADP-ribosyltransferase Photorhabdus luminescens TccC3, modifes a different residue on actin, Thr-148, and increases F-actin levels (Table S2). Eukaryotic ADP-ribosyltransferases affect actin in different ways – from the effects of Transferase A, which ribosylates Arg-95 and Arg-372 and delays filament formation, to arginine-specific ADP-ribosyltransferase, which modifies Arg-206 in G-actin to alter polymerization (Table S2). Enzymes that reverse actin ADP-ribosylation (ADP-ribosylactin Hydrolases) have also been described although they remain poorly understood [13].
Figure 2.

Chemistries of the major actin modifications.
Arginylation
Life at actin’s N-terminus has proven to be even more complex after a recent discovery that actin is arginylated (Figures 1–3, Tables 1, S2–3; [14,15]). Arginylation is an enigmatic modification mediated by arginyltransferase Ate1 that has emerged as a global biological regulator. In-depth analysis reveals that actin and actin-binding proteins constitute a large subset of intracellular arginylation targets [16] and preventing arginylation leads to cell migration and myofibril contractility defects [14,17–19]. Ate1 knockout mice die with heart, vasculature [20] and neural crest morphogenesis [18] defects, reminiscent of those resulting from cell migration abnormalities in various mouse models (reviewed in [21]). In cells, lack of arginylation collapses the leading edge lamella and reduces F-actin without significantly affecting total actin levels [22].
While decreased F-actin levels and other cellular defects may be mediated by multiple arginylation-dependent events, the leading edge collapse is due to a lack of N-terminally arginylated β-actin [14]. Re-introduction of N-terminally arginylated β-actin into Ate1 knockout cells rescues their leading edge defect unlike a non-arginylated version. Remarkably, this effect appears to be highly specific to β and not γ-actin. A specialized mechanism selectively removes arginylated γ-actin by targeting differences in actin coding sequences and translation rates [23], thus ensuring that no arginylated γ-actin is found in vivo. The reasons for this bias relate to the mechanisms maintaining the functional distinction between different actins, and are not fully understood.
Since N-terminal arginylation neighbors a residue typically modified by acetylation (Asp-2), these two N-terminal modifications are likely to be mutually exclusive. Given acetylation’s prevalence and the strict regulation of actin arginylation, a specialized mechanism (undoubtedly regulated by particular intracellular events) likely co-translationally targets actin for either acetylation or arginylation.
Actin is also modified by internal arginylation (Figure 3, Tables 1, S2), which is predicted to affect polymerization and actin-binding protein interactions. It is unclear whether this arginylation occurs within intact proteins (which is chemically possible for some residues) or following proteolysis, which may generate functional or degradatory actin fragments. Further identifying actin arginylation sites and the mechanisms and physiological consequences of these modifications constitute exciting directions for future studies.
Crosslinking
Another means that bacteria use to modify actin is through crosslinking. The diarrheal pathogen Vibrio cholerae induces cell rounding and F-actin loss by crosslinking actin into oligomers with the MARTX and VgrG-1 enzymes (Tables 1, S2–3; Reviewed in [12]). Other types of crosslinking proteins including transglutaminases (TGases) also affect actin, altering Gln-41 and inducing crosslinking with specific amino acid residues (Tables 1, S1–3). TGases also crosslink actin to other proteins and small molecules, making them useful tools for studying actin (Table S1). In contrast to the actin crosslinking catalyzed by MARTX and VgrG-1, TGase-mediated crosslinking often stabilizes actin, making it more resistant to depolymerization, fragmentation, and proteolytic degradation, and has been linked to insulin secretion [24], pollen tube growth [25], growth factor signaling [26,27], apoptosis [28], and neuronal and endothelial cell form and function [29,30]. For example, the pollen TGase is thought to control the transition between short/unstable and stable bundles of F-actin at the apical and base domain boundary of growing pollen tubes [25]. Specific actin regulatory proteins including myosins are also indirectly affected by TGases and exhibit decreased binding and ATPase activity towards transglutaminated actin [25].
Glucose-mediated Modifications
Actin is covalently modified by sugars. The best known of these modifications, N- and O-linked glycosylation, are mainly confined to proteins within secretory/membrane-associated pathways/organelle lumens and thus would not typically modify actin. However, rats exposed to alcohol in utero exhibit glycosylated actin [31], suggesting that under some condition such glycosylation can occur.
Actin is readily susceptible to O-GlcNAcylation (O-GlcNAc) (Figure 2, Tables 1, S2–3), which occurs within the cytoplasm and nucleus and is controlled by two conserved enzymes that add or remove O-GlcNAc in response to stimuli. Although poorly understood, actin O-GlcNAcylation has been implicated in modulating cardiac [32] and skeletal [33] muscle contraction, in cardioprotection [34], and in tissue changes in diabetics [35], as has another glucose-mediated actin PTM, glycation (the non-enzymatic attachment of glucose to lysine residues). Notably, purified actin is susceptible to glycation, as is actin in diabetic patients and animal models [36–39], where glucose decreases polymerization and lowers F-actin levels [39–41]. Since F-actin normally deforms cells and facilitates their passage through vessels, it is interesting in light of the effects of actin glycation that diabetic retinopathy is characterized by decreased cell deformability and retinal microcapillary blockage [39].
Methylation
Actin from many species is normally methylated on His-73 (Figure 2, Tables 1, S2–3). Recent structural analyses indicate that His-73 methylation regulates actin’s interdomain flexibility and stability (Table S2). These effects are thought to be due in part to methylated His-73 slowing the inorganic phosphate (Pi) release after ATP hydrolysis, leading to F-actin stabilization. Actin methylating enzymes have been identified [42,43], although it is unclear if they methylate His-73. Methylation also occurs in some species on a specific actin lysine residue (Lys-326; Tables 1, S2–3) and is post-translationally added to arginylated actin residues (Tables 1, S2–3; [44]). Interestingly, decreased actin methylation occurs following cellular transformation with the Src oncogene [45], further indicating that understanding the mechanisms that regulate actin methylation and its effects constitute important research directions.
Oxidation, Nitrosylation, and other Redox-related Modifications
Shortly after identifying actin, Straub and his colleagues noted that the “addition of an oxidizing agent prevents that polymerization of globular actin, [and] it even destroys polymerized actin” [46]. Since that time numerous reactive oxygen, nitrogen, and lipid reagents have revealed that actin is susceptible to Redox-mediated PTMs including oxidation, nitrosylation, nitration, carbonylation, and glutathionylation (Figures 1–3, Tables 1, S1–3). Yet, this susceptibility to Redox intermediates varies considerably depending on actin’s state, including its nucleotide and salt binding condition and/or whether actin is present in filamentous versus globular form (Table S1). Thus, particular conditions might make actin more susceptible to Redox regulation in vivo and may even contribute to disease processes. For instance, it has long been noted that oxidative stress affects cellular actin organization and generates Redox-related actin PTMs (Table S1). Likewise, numerous pathologies/diseases are associated with Redox-related actin PTMs (Table S1). However, similar PTMs occur under normal conditions and following activation of specific signaling pathways (Table S1). Actin may even serve to protect against oxidative stressors including those that result from normal oxidative metabolism (Table S1). Thus, Redox-mediated actin PTMs are still poorly understood and do not simply induce a common effect – and so critical goals are to characterize the sites on actin that are modified by Redox intermediates and the specific effects they elicit.
Actin has five cysteines that are highly susceptible to Redox intermediates – including reactive oxygen, nitrogen, and lipid species (Tables 1, S1–2). Cys-374 is the most susceptible, undergoing oxidation, glutathionylation, carbonylation, and nitrosylation. For instance, oxidation of Cys-374 occurs upon air-exposure, aging, and freeze-thawing of purified actin, inducing disulfide bond formation between actin monomers (Table S2). Reducing agents such as DTT are typically added to purified actin to reverse these effects. Redox modification of Cys-374 has also been linked to intramolecular disulfide bond formation, decreased polymerization rates, increased critical concentrations, filament weakening, and abnormal actin function within irreversible sickled cells (Table S2). However, Cys-374 is used to label actin (e.g., pyrene-actin) and its modification in some cases allows for relatively normal actin properties (Table S1). Thus, this Redox-modified Cys-374 needs further characterization, as do other Redox-susceptible cysteines including Cys-272 and Cys-285, whose modification has been linked to decreased polymerization and altered interactions with actin regulatory proteins (Tables 1, S2–3).
Some of the 16 methionines within actin are also susceptible to oxidation (Tables 1, S2–3) and this oxidation has long been postulated to functionally impair actin [47,48]. Indeed, the specific oxidation of the Met-44 residue of actin has recently been found to disassemble F-actin and inhibit its polymerization [49]. Met-44 is within the D-loop of actin that is critical for actin subunit contacts. Oxidizing Met-44 introduces a negative charge into the monomer-monomer contact region and severs F-actin [49]. As with other Redox sensitive residues within actin that exhibit different Redox susceptibility depending on actin conformation and ionic conditions (as discussed above, Table S1), Met-44 is buried within F-actin and poorly accessible to diffusible oxidants [48–52]. This raises the possibility that particular in vivo conditions might make Met-44 more susceptible to Redox-regulation and it is interesting that Met-44 is oxidized in vivo in response to oxidative stress (Table S2). Specific tyrosine, histidine, and tryptophan residues of actin are also susceptible to Redox-induced modifications, although the functional significance of these modifications are unknown (Tables 1, S2–3).
While a range of non-specific chemicals and Redox intermediates are known to affect actin, it has been unclear whether Redox processes that specifically target actin also exist. For example, enzymes that generate reactive oxygen, nitrogen, and lipid species have been linked to actin modification (Table S1), but because of the non-specific nature of these enzymes that diffusibly release reactive species, actin constitutes only one of a number of proteins that are modified. Likewise, because of its abundance, actin is more likely to be non-specifically modified and may even “soak-up” reactive species and/or serve to protect the cell from Redox intermediates. Yet, some of these enzymes including nitric oxide synthase (NOS), NADPH oxidase, and 5-lipooxygeanse associate/co-localize [53–65] and have their activity regulated by actin [66–69]. Still, the specific roles of these diffusible Redox generators in regulating actin remain poorly understood.
Recent work has identified a Redox enzyme, the multi-domain flavoprotein monooxygenase Mical, that specifically and directly modifies actin [49,70]. Mical, which is a cytosolic enzyme that mediates semaphorin-dependent cellullar guidance, uses its C-terminus to associate with the semaphorin receptor plexin and its N-terminus monooxygenase (Redox) enzymatic domain to bind to F-actin [71,72]. Mical is activated by F-actin binding and specifically oxidizes Met-44 and Met-47 to disassemble F-actin and decrease polymerization [49]. Interestingly, Mical’s active site likely “fits” between monomers to access and modify these poorly-accessible residues [48–52]. Mutating Met-44 disrupts Mical-mediated actin regulation in vitro and in vivo [49] and leads to actin accumulation defects in both model organisms and human patients that mimic the effects of mutating Mical [49,72,73].
Phosphorylation
Phosphorylation is a major post-translational regulator, so it is perhaps no surprise that actin is heavily phosphorylated. Yet, despite the identification of 35 different phosphorylation sites (Figures 1–3, Tables 1, S1–3), actin phosphorylation in higher eukaryotes is still poorly understood.
Actin phosphorylation has been best characterized in Dictyostelium and in the slide mold Physarum (Table S2), which may have their own machinery for phosphorylation-dependent actin regulation. In Dictyostelium, phosphorylation occurs on Tyr-53, a conserved residue adjacent to the DNase I binding loop (D-loop) (residues 40–50) that has been implicated in actin subunit-subunit contacts (Table S2; [74]). Such phosphorylation interferes with polymerization, likely due to stabilization of actin’s D-loop by hydrogen bonding with the phosphate group. This actin phosphorylation appears to be critical for Dictyostelium to respond to specific signals and transition into a dormancy state.
In Physarum, a Ca2+-dependent actin kinase (actin-fragmin kinase, AFK) modifies a cluster of actin threonine residues (Thr 201–203), facilitating filament elongation by weakening interactions with fragmin, a gelsolin-related protein that controls filament length (Table S2). Protein phosphatases PP1 and PP2A specifically reverse this phosphorylation and its effects [75]. These threonine residues are phylogenetically conserved on the surface facing the pointed end of filaments (Figures 1, 3), suggesting that their phosphorylation may facilitate subunit addition to the pointed end. As in Dictyostelium, this phosphorylation is regulated during stimuli-induced transitions to dormancy and likely represents an adaptation mechanism to modulate sporulation and germination through rapid actin rearrangements (Table S1). An analogous form of regulation may mediate actin rearrangements in mammals through casein kinase I, which phosphorylates actin and exhibits AFK-like characteristics, including Ser/Thr specificity and Ca2+ dependence (Table S1).
In mammals, a vast collection of actin phosphorylation sites is emerging from proteomics of numerous cell types, disease conditions, and following various stimuli (Tables S1–2). For example, insulin stimulates actin phosphorylation on Tyr and Ser residues by unknown kinases, leading to impaired DNase I binding. Phosphorylation by PAK1, a p21-activated kinase, is linked to stress fiber dissolution and F-actin redistribution. Phosphorylation triggered by membrane-associated and Src kinases also exert negative effects on actin polymerization. Yet, few reports examine direct kinase-mediated actin phosphorylation. The most extensive work in this regard involves phosphorylation by cAMP-dependent protein kinase (PKA) and Ca2+/phosphoinositide-dependent protein kinase (PKC), which produce opposing effects on actin: phosphorylation by PKA interferes with polymerization, while PKC-mediated phosphorylation induces it [76,77]. These results suggest a dynamic interplay between signaling cascades mediated by these two kinases in actin-dependent morphological changes.
Ubiquitylation and SUMOylation
Protein ubiquitylation is characterized by the covalent attachment of ubiquitin/ubiquitin-like proteins to specific target proteins and is best known for tagging proteins for degradation (polyubiquitylation). Actin, too, is ubiquitylated (Figures 1–3, Tables 1, S2–3) by specific ubiquitin ligases including MuRF-1, UbcH5, and Trim32 [78–81], which decrease actin levels [81]. Such polyubiquitin-mediated actin degradation is linked to muscle remodeling and atrophy [79–81], although myofibrillar proteins like myosin appear to protect actin from this degradation [78,80]. Ubiqiuitin-dependent actin degradation also occurs co-translationally on slowly synthesized arginylated actin, representing a means to degrade incorrectly arginylated actin [23] and perhaps other PTM actins. In contrast to polyubiquitylation, monoubiquitylation is thought to confer stability and differential subcellular localization to actin (Tables 1, S2–3; [82,83]) and occurs in many organisms including Drosophila, where muscle actin is monoubiquitylated on every seventh filament subunit, which may regulate muscle contraction [84]. Actin is also modified by the small ubiquitin-like modifier (SUMO) (Tables 1, S2–3), which retains actin in the nucleus [85–87]. Other small ubiquitin-like actin modifications including ISGylation [88] have also been identified but their effects are unknown.
Other Actin Post-translational Modifications
Actin, like other proteins, is susceptible to isomerization of aspartyl residues (isoaspartylation) and deamidation of asparaginyl residues, modifications linked to protein aging and inactivation that occur through non-enzymatic processes (Tables 1, S2–3;[89]). Actin is also a substrate for enzymes such as protein L-isoaspartyl methyltransferase (PIMT) that convert isoaspartyl residues back to aspartyl residues, and PIMT-KO mice exhibit a higher prevalence of isoapartylated actin [89,90]. Fatty acylation/alkanoylation, which includes such modifications as myristoylation and palmitoylation, also occur on actin, although it is unknown if these are enzyme-driven modifications [91,92]. Likewise, actin undergoes non-enzymatic cysteine acylation [93] and interacts directly with membrane lipids (Table S1). Actin also undergoes proteolytic cleavage (Table S1) and in some cases proteolysis and fatty acylation may occur together. For example, actin is a caspase substrate and such digestion triggers actin N-myristoylation, mitochondrial targeting, and participation in apoptosis [94]. Sulfation of actin also occurs [95] and while it is unclear how sulfation affects actin, sulfotransferases bind and may use F-actin to mobilize and allow the sulfation of hydroxysteroids, such as cholesterol, at specific intracellular locations [96]. Recent work has also revealed that actin is susceptible to S-sulfhydration of its cysteines, a modification that enhances polymerization and results from cystathionine gamma-lyase (CSE)-mediated hydrogen sulfide (H2S) generation [97]. Finally, numerous other covalent protein modifications [98], including those chemically-induced (e.g., succinylation; Table S1), and newly-discovered naturally occurring ones (e.g, malonylation; Tables 1, S2–3), may also play in vivo roles in regulating actin through enzymatic and non-enzymatic means.
Conclusions
As more and more studies identify new post-translational regulatory mechanisms and protein modifications, actin – one of the most essential and abundant intracellular proteins – has emerged as a major target of such PTMs. These observations further increase actin’s functional and regulatory complexity, although much remains to be understood of these modifications and their physiological/pathological roles. Additional complexity may result from PTMs of the same or neighboring sites on actin. For example, modifications such as phosphorylation, nitration, and O-GlcNAcylation can alternatively affect the same residues, while others work sequentially, such as methylation of arginylated actin residues (Tables 1, S2–3; [44]). Different PTMs may also facilitate or oppose one another by regulating access to modifying enzymes and/or chemical groups. Notably, numerous neighboring residues within actin’s subdomain 2 are post-translationally modified, including Met-44 (oxidation) and Tyr-53 (phosphorylation) (Tables 1, S2–3), indicating that this important region is extensively regulated, perhaps by inter-related or mutually exclusive means. Understanding the individual effects and relationships of different PTMs constitutes an exciting future direction. Likewise, developing new methods for detecting and characterizing specific PTMs will also further our understanding of the roles and hierarchical relationship between the multiple PTMs of actin.
Supplementary Material
Acknowledgments
We thank Klaus Aktories, Emil Reisler, and Peter Rubenstein for helpful comments on the review. Because of space and reference limitations, we apologize for not being able to fully discuss and cite much of the literature in the main text and have therefore included and referred to Supplemental Tables 1 and 2. Supported by grants from the NIH (NS073968) and the Welch Foundation (I-1749) to J.R.T. and by R01HL084419 to A.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1**.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. In-depth review of actin based mechanical support and motility including discussions of different non-covalent actin modifying proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vandekerckhove J, Weber K. Mammalian cytoplasmic actins are the products of at least two genes and differ in primary structure in at least 25 identified positions from skeletal muscle actins. Proc Natl Acad Sci U S A. 1978;75:1106–1110. doi: 10.1073/pnas.75.3.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook RK, Sheff DR, Rubenstein PA. Unusual metabolism of the yeast actin amino terminus. J Biol Chem. 1991;266:16825–16833. [PubMed] [Google Scholar]
- 4.Mottet D, Castronovo V. Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis. 2008;25:183–189. doi: 10.1007/s10585-007-9131-5. [DOI] [PubMed] [Google Scholar]
- 5.Anderson KA, Hirschey MD. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012;52:23–35. doi: 10.1042/bse0520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. doi: 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Kovacs JJ, Hubbert C, Yao TP. The HDAC complex and cytoskeleton. Novartis Found Symp. 2004;259:170–177. discussion 178–181, 223–175. [PubMed] [Google Scholar]
- 8.Maas A. Viewpoint: Angela Maas, MD, PhD. Interview by Jennifer Taylor. Circulation. 2007;116:f91–93. doi: 10.1161/CIRCULATIONAHA.107.186290. [DOI] [PubMed] [Google Scholar]
- 9.Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 10.Obrdlik A, Kukalev A, Louvet E, Farrants AK, Caputo L, Percipalle P. The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol Cell Biol. 2008;28:6342–6357. doi: 10.1128/MCB.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheterline P, Clayton J, Sparrow J. Actin. Protein Profile. 1995;2:1–103. [PubMed] [Google Scholar]
- 12**.Aktories K, Lang AE, Schwan C, Mannherz HG. Actin as target for modification by bacterial protein toxins. FEBS J. 2011;278:4526–4543. doi: 10.1111/j.1742-4658.2011.08113.x. In-depth review covering the current state of investigations into enzyme-driven ADP-ribosylation and crosslinking of actin. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto H, Fujita H, Matsuyama S, Tsuyama S. Purification, characterization, and localization of an ADP-ribosylactin hydrolase that uses ADP-ribosylated actin from rat brains as a substrate. J Biol Chem. 1997;272:28116–28125. doi: 10.1074/jbc.272.44.28116. [DOI] [PubMed] [Google Scholar]
- 14.Karakozova M, Kozak M, Wong CCL, Bailey AO, Yates JR, Mogilner A, Zebroski H, Kashina A. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 2006;313:192–196. doi: 10.1126/science.1129344. [DOI] [PubMed] [Google Scholar]
- 15.Kashina AS. Differential arginylation of actin isoforms: the mystery of the actin N-terminus. Trends Cell Biol. 2006;16:610–615. doi: 10.1016/j.tcb.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Wong CC, Xu T, Rai R, Bailey AO, Yates JR, 3rd, Wolf YI, Zebroski H, Kashina A. Global analysis of posttranslational protein arginylation. PLoS Biol. 2007;5:e258. doi: 10.1371/journal.pbio.0050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rai R, Wong CC, Xu T, Leu NA, Dong DW, Guo C, McLaughlin KJ, Yates JR, 3rd, Kashina A. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development. 2008;135:3881–3889. doi: 10.1242/dev.022723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurosaka S, Leu NA, Zhang F, Bunte R, Saha S, Wang J, Guo C, He W, Kashina A. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 2010;6:e1000878. doi: 10.1371/journal.pgen.1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurosaka S, Leu NA, Pavlov I, Han X, Ribeiro PA, Xu T, Bunte R, Saha S, Wang J, Cornachione A, et al. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J Mol Cell Cardiol. 2012 doi: 10.1016/j.yjmcc.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, Varshavsky A. An essential role of N-terminal arginylation in cardiovascular development. Science. 2002;297:96–99. doi: 10.1126/science.1069531. [DOI] [PubMed] [Google Scholar]
- 21.Kurosaka S, Kashina A. Cell biology of embryonic migration. Birth Defects Res C Embryo Today. 2008;84:102–122. doi: 10.1002/bdrc.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha S, Mundia MM, Zhang F, Demers RW, Korobova F, Svitkina T, Perieteanu AA, Dawson JF, Kashina A. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol Biol Cell. 2010;21:1350–1361. doi: 10.1091/mbc.E09-09-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23*.Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 2010;329:1534–1537. doi: 10.1126/science.1191701. The authors found that the specificity of N-terminal arginylation to beta actin is regulated through its mRNA coding sequence, coupling differential translation speeds of beta and gamma actin to their aginylation-dependent ubiquitination and selective removal of incorrectly arginylated gamma actin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo L, Marsella C, Nardo G, Massignan T, Alessio M, Piermarini E, La Rosa S, Finzi G, Bonetto V, Bertuzzi F, et al. Transglutaminase 2 transamidation activity during first-phase insulin secretion: natural substrates in INS-1E. Acta Diabetol. 2012 doi: 10.1007/s00592-012-0381-6. [DOI] [PubMed] [Google Scholar]
- 25.Del Duca S, Serafini-Fracassini D, Bonner P, Cresti M, Cai G. Effects of post-translational modifications catalysed by pollen transglutaminase on the functional properties of microtubules and actin filaments. Biochem J. 2009;418:651–664. doi: 10.1042/BJ20081781. [DOI] [PubMed] [Google Scholar]
- 26.Katoh S, Inoue T, Kohno H, Ohkubo Y. Transglutaminase-modified actin decreases epidermal growth factor binding to its receptor in rat liver membrane. Biomedical Res. 1994;15:1–8. [Google Scholar]
- 27.Katoh S, Nakagawa N, Yano Y, Satoh K, Kohno H, Ohkubo Y. Transglutaminase induced by epidermal growth factor negatively regulates the growth signal in primary cultured hepatocytes. Biochem J. 1996;313 (Pt 1):305–309. doi: 10.1042/bj3130305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemes Z, Jr, Adany R, Balazs M, Boross P, Fesus L. Identification of cytoplasmic actin as an abundant glutaminyl substrate for tissue transglutaminase in HL-60 and U937 cells undergoing apoptosis. J Biol Chem. 1997;272:20577–20583. doi: 10.1074/jbc.272.33.20577. [DOI] [PubMed] [Google Scholar]
- 29.Baumgartner W, Weth A. Transglutaminase 1 stabilizes beta-actin in endothelial cells correlating with a stabilization of intercellular junctions. J Vasc Res. 2007;44:234–240. doi: 10.1159/000100422. [DOI] [PubMed] [Google Scholar]
- 30.Dolge L, Aufenvenne K, Traupe H, Baumgartner W. Beta-actin is a target for transglutaminase activity at synaptic endings in chicken telencephalic cell cultures. J Mol Neurosci. 2012;46:410–419. doi: 10.1007/s12031-011-9601-8. [DOI] [PubMed] [Google Scholar]
- 31.Fofana B, Yao XH, Rampitsch C, Cloutier S, Wilkins JA, Nyomba BL. Prenatal alcohol exposure alters phosphorylation and glycosylation of proteins in rat offspring liver. Proteomics. 2010;10:417–434. doi: 10.1002/pmic.200800969. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedou J, Cieniewski-Bernard C, Leroy Y, Michalski JC, Mounier Y, Bastide B. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. J Biol Chem. 2007;282:10360–10369. doi: 10.1074/jbc.M606787200. [DOI] [PubMed] [Google Scholar]
- 34.Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–1182. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- 35.Akimoto Y, Miura Y, Toda T, Wolfert MA, Wells L, Boons GJ, Hart GW, Endo T, Kawakami H. Morphological changes in diabetic kidney are associated with increased O-GlcNAcylation of cytoskeletal proteins including alpha-actinin 4. Clin Proteomics. 2011;8:15. doi: 10.1186/1559-0275-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLean WG, Pekiner C, Cullum NA, Casson IF. Posttranslational modifications of nerve cytoskeletal proteins in experimental diabetes. Mol Neurobiol. 1992;6:225–237. doi: 10.1007/BF02780555. [DOI] [PubMed] [Google Scholar]
- 37.Brown MR, Keith TJ, Knull HR. Nonenzymatic incorporation of glucose and galactose into brain cytoskeletal proteins in vitro. Neurochem Int. 1992;21:177–183. doi: 10.1016/0197-0186(92)90144-g. [DOI] [PubMed] [Google Scholar]
- 38.Pekiner C, Cullum NA, Hughes JN, Hargreaves AJ, Mahon J, Casson IF, McLean WG. Glycation of brain actin in experimental diabetes. J Neurochem. 1993;61:436–442. doi: 10.1111/j.1471-4159.1993.tb02143.x. [DOI] [PubMed] [Google Scholar]
- 39.Sulochana KN, Indra C, Rajesh M, Srinivasan V, Ramakrishnan S. Beneficial role of amino acids in mitigating cytoskeletal actin glycation and improving F-actin content: in vitro. Glycoconj J. 2001;18:277–282. doi: 10.1023/a:1013666829851. [DOI] [PubMed] [Google Scholar]
- 40.Kuleva NV, Kovalenko ZS. Change in the functional properties of actin by its glycation in vitro. Biochemistry (Mosc) 1997;62:1119–1123. [PubMed] [Google Scholar]
- 41.Resmi H, Akhunlar H, Temiz Artmann A, Guner G. In vitro effects of high glucose concentrations on membrane protein oxidation, G-actin and deformability of human erythrocytes. Cell Biochem Funct. 2005;23:163–168. doi: 10.1002/cbf.1129. [DOI] [PubMed] [Google Scholar]
- 42.Vijayasarathy C, Rao BS. Partial purification and characterisation of S-adenosylmethionine:protein-histidine N-methyltransferase from rabbit skeletal muscle. Biochim Biophys Acta. 1987;923:156–165. doi: 10.1016/0304-4165(87)90139-5. [DOI] [PubMed] [Google Scholar]
- 43.Raghavan M, Lindberg U, Schutt C. The use of alternative substrates in the characterization of actin-methylating and carnosine-methylating enzymes. Eur J Biochem. 1992;210:311–318. doi: 10.1111/j.1432-1033.1992.tb17423.x. [DOI] [PubMed] [Google Scholar]
- 44**.Saha S, Wong CC, Xu T, Namgoong S, Zebroski H, Yates JR, 3rd, Kashina A. Arginylation and methylation double up to regulate nuclear proteins and nuclear architecture in vivo. Chem Biol. 2011;18:1369–1378. doi: 10.1016/j.chembiol.2011.08.019. The authors found that posttranslationally added Arg on actin and some other proteins can further undergo Arg-methylation, thus setting the first precedent of “double-PTMs” occurring simultaneously on the same protein sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiou YY, Fu SL, Lin WJ, Lin CH. Proteomics analysis of in vitro protein methylation during Src-induced transformation. Electrophoresis. 2012;33:451–461. doi: 10.1002/elps.201100280. [DOI] [PubMed] [Google Scholar]
- 46.Feuer G, Molnar F, Pettko E, Straub FB. Studies on the composition and polymerization of actin. Hung Acta Physiol. 1948;1:150–163. [PubMed] [Google Scholar]
- 47.Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, DalleDonne I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000;9:1774–1782. doi: 10.1110/ps.9.9.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic Biol Med. 2002;32:927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 49**.Hung RJ, Pak CW, Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science. 2011;334:1710–1713. doi: 10.1126/science.1211956. Using purified proteins and model in vivo systems, the authors describe a new biochemical process that regulates actin. They find that actin is a specific substrate of the Redox enzyme, Mical, which oxidizes the Met-44 residue within the D-loop of the subdomain 2 portion of actin, simultaneously severing filaments and decreasing polymerization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan JQ, Almo SC, Reisler E, Chance MR. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 2003;42:11992–12000. doi: 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
- 51.Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry. 2005;44:3166–3175. doi: 10.1021/bi048021j. [DOI] [PubMed] [Google Scholar]
- 52*.Takamoto K, Kamal JK, Chance MR. Biochemical implications of a three-dimensional model of monomeric actin bound to magnesium-chelated ATP. Structure. 2007;15:39–51. doi: 10.1016/j.str.2006.11.005. Along with Refs 38 and 39, this study reveals the susceptibility of different forms of monomeric and F-actin to hydroxyl radicals. Among other findings, the results reveal that specific actin residues are protected when actin is present in the filamentous state. [DOI] [PubMed] [Google Scholar]
- 53.Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314:1765–1776. doi: 10.1016/j.yexcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 54.Webb JL, Harvey MW, Holden DW, Evans TJ. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect Immun. 2001;69:6391–6400. doi: 10.1128/IAI.69.10.6391-6400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamura M, Kai T, Tsunawaki S, Lambeth JD, Kameda K. Direct interaction of actin with p47(phox) of neutrophil NADPH oxidase. Biochem Biophys Res Commun. 2000;276:1186–1190. doi: 10.1006/bbrc.2000.3598. [DOI] [PubMed] [Google Scholar]
- 56.Tamura M, Kanno M, Endo Y. Deactivation of neutrophil NADPH oxidase by actin-depolymerizing agents in a cell-free system. Biochem J. 2000;349:369–375. doi: 10.1042/0264-6021:3490369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamura M, Itoh K, Akita H, Takano K, Oku S. Identification of an actin-binding site in p47phox an organizer protein of NADPH oxidase. FEBS Lett. 2006;580:261–267. doi: 10.1016/j.febslet.2005.11.080. [DOI] [PubMed] [Google Scholar]
- 58.Shao D, Segal AW, Dekker LV. Subcellular localisation of the p40phox component of NADPH oxidase involves direct interactions between the Phox homology domain and F-actin. Int J Biochem Cell Biol. 2010;42:1736–1743. doi: 10.1016/j.biocel.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rasmussen I, Pedersen LH, Byg L, Suzuki K, Sumimoto H, Vilhardt F. Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC Immunol. 2010;11:44. doi: 10.1186/1471-2172-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 61.Gu Y, Xu YC, Wu RF, Souza RF, Nwariaku FE, Terada LS. TNFalpha activates c-Jun amino terminal kinase through p47(phox) Exp Cell Res. 2002;272:62–74. doi: 10.1006/excr.2001.5404. [DOI] [PubMed] [Google Scholar]
- 62.Wu RF, Gu Y, Xu YC, Nwariaku FE, Terada LS. Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem. 2003;278:36830–36840. doi: 10.1074/jbc.M302251200. [DOI] [PubMed] [Google Scholar]
- 63.Kang LT, Vanderhoek JY. Mono (S) hydroxy fatty acids: novel ligands for cytosolic actin. J Lipid Res. 1998;39:1476–1482. [PubMed] [Google Scholar]
- 64.Kang LT, Phillips TM, Vanderhoek JY. Novel membrane target proteins for lipoxygenase-derived mono(S)hydroxy fatty acids. Biochim Biophys Acta. 1999;1438:388–398. doi: 10.1016/s0167-4838(99)00100-4. [DOI] [PubMed] [Google Scholar]
- 65.Lepley RA, Fitzpatrick FA. 5-Lipoxygenase contains a functional Src homology 3-binding motif that interacts with the Src homology 3 domain of Grb2 and cytoskeletal proteins. J Biol Chem. 1994;269:24163–24168. [PubMed] [Google Scholar]
- 66.Su Y, Kondrikov D, Block ER. Beta-actin: a regulator of NOS-3. Sci STKE. 2007;2007:pe52. doi: 10.1126/stke.4042007pe52. [DOI] [PubMed] [Google Scholar]
- 67.Morimatsu T, Kawagoshi A, Yoshida K, Tamura M. Actin enhances the activation of human neutrophil NADPH oxidase in a cell-free system. Biochem Biophys Res Commun. 1997;230:206–210. doi: 10.1006/bbrc.1996.5881. [DOI] [PubMed] [Google Scholar]
- 68.Miller YI, Chang MK, Funk CD, Feramisco JR, Witztum JL. 12/15-lipoxygenase translocation enhances site-specific actin polymerization in macrophages phagocytosing apoptotic cells. J Biol Chem. 2001;276:19431–19439. doi: 10.1074/jbc.M011276200. [DOI] [PubMed] [Google Scholar]
- 69.Provost P, Doucet J, Hammarberg T, Gerisch G, Samuelsson B, Radmark O. 5-Lipoxygenase interacts with coactosin-like protein. J Biol Chem. 2001;276:16520–16527. doi: 10.1074/jbc.M011205200. [DOI] [PubMed] [Google Scholar]
- 70.Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N, Huang Z, van Berkel WJ, Terman JR. Mical links semaphorins to F-actin disassembly. Nature. 2010;463:823–827. doi: 10.1038/nature08724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002;109:887–900. doi: 10.1016/s0092-8674(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 72.Hung R-J, Terman JR. Extracellular inhibitors, repellents, and Semaphorin/Plexin/MICAL-mediated actin filament disassembly. Cytoskeleton. 2011;68:415–433. doi: 10.1002/cm.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laing NG, Dye DE, Wallgren-Pettersson C, Richard G, Monnier N, Lillis S, Winder TL, Lochmuller H, Graziano C, Mitrani-Rosenbaum S, et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1) Hum Mutat. 2009;30:1267–1277. doi: 10.1002/humu.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bender N, Fasold H, Kenmoku A, Middelhoff G, Volk KE. The selective blocking of the polymerization reaction of striated muscle actin leading to a derivative suitable for crystallization. Modification of Tyr-53 by 5-diazonium-(1H)tetrazole. Eur J Biochem. 1976;64:215–218. doi: 10.1111/j.1432-1033.1976.tb10290.x. [DOI] [PubMed] [Google Scholar]
- 75.Waelkens E, Gettemans J, De Corte V, De Ville Y, Goris J, Vandekerckhove J, Merlevede W. Microfilament dynamics: regulation of actin polymerization by actin-fragmin kinase and phosphatases. Adv Enzyme Regul. 1995;35:199–227. doi: 10.1016/0065-2571(94)00013-s. [DOI] [PubMed] [Google Scholar]
- 76.Prat AG, Bertorello AM, Ausiello DA, Cantiello HF. Activation of epithelial Na+ channels by protein kinase A requires actin filaments. Am J Physiol. 1993;265:C224–233. doi: 10.1152/ajpcell.1993.265.1.C224. [DOI] [PubMed] [Google Scholar]
- 77.Ohta Y, Akiyama T, Nishida E, Sakai H. Protein kinase C and cAMP-dependent protein kinase induce opposite effects on actin polymerizability. FEBS Lett. 1987;222:305–310. doi: 10.1016/0014-5793(87)80391-5. [DOI] [PubMed] [Google Scholar]
- 78.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 79*.Polge C, Heng AE, Jarzaguet M, Ventadour S, Claustre A, Combaret L, Bechet D, Matondo M, Uttenweiler-Joseph S, Monsarrat B, et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011;25:3790–3802. doi: 10.1096/fj.11-180968. The authors identified that actin is polyubiquitinated in vitro and in muscles, leading to degradation. These observations suggest that the polybiquitylation pathway may contribute to muscle wasting conditions. [DOI] [PubMed] [Google Scholar]
- 80*.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. The authors find that the ubiquitin ligase, muscle RING-finger 1 (MuRF1), binds and ubiquitylates a number of muscle proteins including actin. However, in vivo, actin and other thin filament components decreased by a mechanism not requiring MuRF1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol. 2005;354:413–424. doi: 10.1016/j.jmb.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 82.Field SJ, Pinder JC, Clough B, Dluzewski AR, Wilson RJ, Gratzer WB. Actin in the merozoite of the malaria parasite, Plasmodium falciparum. Cell Motil Cytoskeleton. 1993;25:43–48. doi: 10.1002/cm.970250106. [DOI] [PubMed] [Google Scholar]
- 83.Dantan-Gonzalez E, Rosenstein Y, Quinto C, Sanchez F. Actin monoubiquitylation is induced in plants in response to pathogens and symbionts. Mol Plant Microbe Interact. 2001;14:1267–1273. doi: 10.1094/MPMI.2001.14.11.1267. [DOI] [PubMed] [Google Scholar]
- 84.Burgess S, Walker M, Knight PJ, Sparrow J, Schmitz S, Offer G, Bullard B, Leonard K, Holt J, Trinick J. Structural studies of arthrin: monoubiquitinated actin. J Mol Biol. 2004;341:1161–1173. doi: 10.1016/j.jmb.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 85.Vertegaal AC, Ogg SC, Jaffray E, Rodriguez MS, Hay RT, Andersen JS, Mann M, Lamond AI. A proteomic study of SUMO-2 target proteins. J Biol Chem. 2004;279:33791–33798. doi: 10.1074/jbc.M404201200. [DOI] [PubMed] [Google Scholar]
- 86.Rosas-Acosta G, Russell WK, Deyrieux A, Russell DH, Wilson VG. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol Cell Proteomics. 2005;4:56–72. doi: 10.1074/mcp.M400149-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87**.Hofmann WA, Arduini A, Nicol SM, Camacho CJ, Lessard JL, Fuller-Pace FV, de Lanerolle P. SUMOylation of nuclear actin. J Cell Biol. 2009;186:193–200. doi: 10.1083/jcb.200905016. The authors find that specific sites on nuclear actin are modified by SUMOylation, which they find to be critical for nuclear localization of actin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giannakopoulos NV, Luo JK, Papov V, Zou W, Lenschow DJ, Jacobs BS, Borden EC, Li J, Virgin HW, Zhang DE. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem Biophys Res Commun. 2005;336:496–506. doi: 10.1016/j.bbrc.2005.08.132. [DOI] [PubMed] [Google Scholar]
- 89.Cimmino A, Capasso R, Muller F, Sambri I, Masella L, Raimo M, De Bonis ML, D’Angelo S, Zappia V, Galletti P, et al. Protein isoaspartate methyltransferase prevents apoptosis induced by oxidative stress in endothelial cells: role of Bcl-Xl deamidation and methylation. PLoS One. 2008;3:e3258. doi: 10.1371/journal.pone.0003258. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90.Zhu JX, Doyle HA, Mamula MJ, Aswad DW. Protein repair in the brain, proteomic analysis of endogenous substrates for protein L-isoaspartyl methyltransferase in mouse brain. J Biol Chem. 2006;281:33802–33813. doi: 10.1074/jbc.M606958200. [DOI] [PubMed] [Google Scholar]
- 91.Stadler J, Gerisch G, Bauer G, Deppert W. In vivo acylation of Dictyostelium actin with palmitic acid. EMBO J. 1985;4:1153–1156. doi: 10.1002/j.1460-2075.1985.tb03753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rioux V, Galat A, Jan G, Vinci F, D’Andrea S, Legrand P. Exogenous myristic acid acylates proteins in cultured rat hepatocytes. J Nutr Biochem. 2002;13:66–74. doi: 10.1016/s0955-2863(01)00196-6. [DOI] [PubMed] [Google Scholar]
- 93.Bano MC, Jackson CS, Magee AI. Pseudo-enzymatic S-acylation of a myristoylated yes protein tyrosine kinase peptide in vitro may reflect non-enzymatic S-acylation in vivo. Biochem J. 1998;330 (Pt 2):723–731. doi: 10.1042/bj3300723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Utsumi T, Sakurai N, Nakano K, Ishisaka R. C-terminal 15 kDa fragment of cytoskeletal actin is posttranslationally N-myristoylated upon caspase-mediated cleavage and targeted to mitochondria. FEBS Lett. 2003;539:37–44. doi: 10.1016/s0014-5793(03)00180-7. [DOI] [PubMed] [Google Scholar]
- 95.Schick BP, Jacoby JA. Sulfation of guinea pig megakaryocyte and platelet proteins. J Cell Physiol. 1994;159:356–364. doi: 10.1002/jcp.1041590219. [DOI] [PubMed] [Google Scholar]
- 96.Kurogi K, Sakakibara Y, Kamemoto Y, Takahashi S, Yasuda S, Liu MC, Suiko M. Mouse cytosolic sulfotransferase SULT2B1b interacts with cytoskeletal proteins via a proline/serine-rich C-terminus. FEBS J. 2010;277:3804–3811. doi: 10.1111/j.1742-4658.2010.07781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97*.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. The authors find that under physiological conditions actin is sulfhydrated, which enhances actin polymerization in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh CT. Posttranslational modifications of proteins. Expanding nature’s inventory. Englewood, CO: Roberts and Company Publishers; 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.