

Abstract
Background
Incomplete penetrance and variable expression of Hypertrophic Cardiomyopathy (HCM) is well appreciated. Common genetic polymorphisms variants that may affect HCM penetrance and expression have been predicted but are not well established.
Methods and Results
We performed a case-control genome wide association (GWA) study to identify common HCM-associated genetic polymorphisms and then asked whether such common variants were more represented in HCM or could explain the heterogeneity of HCM phenotypes. We identified an intronic FHOD3 variant (rs516514) associated with HCM (OR = 2.45 (95% CI 1.76–3.41), p=1.25 × 10−7) and validated this finding in an independent cohort. Next, we tested FHOD3-V1151I (rs2303510), a non-synonymous variant in partial linkage disequilibrium (LD) with rs516514, and we detected an even stronger association with HCM (p=1.76 × 10−9). While HCM patients were more likely to carry these FHOD3 alleles subjects homozygous for FHOD3-1151I had similar HCM phenotypes as carriers of the V1151 allele. FHOD3 expression is increased in the setting of HCM and both alleles of FHOD3-V1151I were detected in HCM myectomy tissue. Previously FHOD3 was found to be required for formation of the sarcomere and here we demonstrate that its fly homolog fhos is required for normal adult heart systolic contraction.
Conclusions
Here we demonstrate the association of a common non-synonymous FHOD3 genetic variant with HCM. This discovery further strengthens the potential role of gene mutations and polymorphisms that alter the amino acid sequence of sarcomere proteins and HCM.
Keywords: contractility, genome-wide analysis, hypertrophic cardiomyopathy
Introduction
Approximately one hundred years ago, primary, or idiopathic, left ventricular hypertrophy developing in the absence of a defined volume or pressure load called hypertrophic cardiomyopathy (HCM) was first described1. HCM commonly presents during the second decade of life, yet the presentation may be delayed into adulthood2, with a phenotype that includes thickened or enlarged ventricular walls and obstruction of blood flow at the left ventricular (LV) outflow tract. Excessive LV wall thickening and marked obstruction of the LV outflow tract in the setting of HCM are associated with an increased risk of death3–6. HCM is a leading cause of sudden death in young athletes7.
Mutations in genes that change the amino acid sequence of protein components of the thick and thin sarcomere filaments can cause HCM8, 9. Mutations are identified in approximately 65% of cases when related family members also have HCM (familial HCM)10. Prospective screening of eight cardiac contractile protein genes identified mutations in almost half of HCM patients with sporadic HCM9, 11. Clinical testing of the currently defined genes associated with HCM is now part of routine clinical practice10.
Similar to other mono-genetic “Mendelian” disorders HCM has a variable degree of phenotype penetrance such that some people with a normal heart structure can carry a contractile gene mutation that causes HCM in related family members. Factors that protect mutation carriers from manifesting LV hypertrophy and outflow tract obstruction are poorly understood. In addition to incomplete penetrance variable left ventricular wall thickening and outflow tract obstruction between patients carrying the same mutation is well described12. Variability in HCM disease penetrance and expression can only be partially explained by currently known HCM-causing cardiac sarcomere gene mutations9, as consistent and mutation-specific effects have not been identified13.
Several lines of evidence support a role for genetic factors in HCM beyond contractile gene mutations ultimately responsible for triggering HCM itself. First, chromosomal loci associated with differences in LV mass have been found within a large HCM family carrying a single contractile gene mutation12. Second, mice engineered with a human HCM-causing gene mutation exhibit strain-specific HCM phenotypes14. Third, extreme HCM phenotypes, including the development of advanced heart failure and the need for heart transplantation, are associated with the presence of more than one contractile gene mutation11, 15, 16. Finally, in the absence of HCM LV mass is heritable17–19, and a genome-wide association (GWA) study identified several loci represented by single nucleotide polymorphisms (SNPs) that explain a portion of the variance of LV phenotypes20.
Here, we describe a GWA study for HCM. We did not expect a GWA study to identify rare “private” mutations responsible for HCM in a single family because GWA testing is not powered to identify mutations. Rather, we hypothesized that GWA testing may identify common genetic polymorphisms overrepresented in HCM patients relative to controls if those variants somehow modified the risk of a patient having clinical HCM. We tested this hypothesis with a two-stage approach starting with a GWA study of the Tufts HCM Genetics Cohort followed by confirmation of findings in the independent Mayo Clinic HCM Genetics Cohort. Finally, we used biological assays to determine if the identified gene has a biologically plausible role in myocardial phenotypes.
Materials and Methods
Tufts HCM Cohort, Genome-Wide Array Analysis
The Tufts Medical Center/Tufts University Institutional Review Board approved all studies and study procedures following participant informed consent, described to and signed by voluntary participants in accordance with the principles expressed in the Declaration of Helsinki. All subjects were unrelated and were identified through their diagnosis of HCM, which was confirmed by echocardiography. DNA was collected and purified by manufacturer protocols (PaxGene); DNA concentration was measured using the PicoGreen assay (Invitrogen). Genomic DNA was genotyped on the Illumina 370CNV array by deCode (Reykjavik, Iceland). The Tufts HCM replication cohort was genotyped using TaqMan assays for rs516514 and rs2303510 (Applied Biosystems Assays on Demand) using a 7900HT. Genotypes for 823 control individuals (all of CEU ancestry, average age 43, range 30 to 88) were obtained from Illumina Genotype Control Database (iControlDB, www.illumina.com). These individuals were genotyped on the same platform as our experimental study was with the exception of copy number probes present on the 370CNV array variant that were not included in the control population study; all non-overlapping probes were set to missing for the purposes of this study.
We estimated21 that we had 80% power to detect SNPs associated with HCM at an odds ratio of ≥ 2.0 with 153 individuals assuming that the associated SNP is has a minor allele frequency of 0.4. Population structure in the Tufts cohort was analyzed using the software package Structure (version 2.3.2.1), which found no evidence of significant allele frequency divergence at any value of K within the control and experimental groups when considered singly or as a whole. The PCA-population based covariates had no effect on the reported results. GWA study association analyses were carried out using PLINK v1.0721. Total genotyping call rate across all individuals (174 cases, 823 controls) was 0.98, and 311,399 SNPs were available for analysis. Logistic analysis was performed for the binary disease trait (e.g. HCM/no-HCM) with covariate adjustments for gender, presence of known HCM mutations, and age. Raw p-values were adjusted relative to number of tests performed and data-derived genomic inflation values (λ = 1.01 in the initial association analysis). Imputation of variants throughout the associated region was accomplished using the HapMap CEU (release 23a) and plink as described22. Within the 0.1 Mb that defines the area of highest LD with rs516514 (defined as r2≥0.8), concordance rates between imputed and assayed identity reached 100%, averaged 97.1%, and had a median value of 99.1%.
Mayo Clinic HCM Cohort Analysis
The Mayo Clinic Institutional Review Board approved all studies and study procedures following participant informed consent, described to and signed by voluntary participants in accordance with the principles expressed in the Declaration of Helsinki. Subjects were recruited from the Mayo Clinic Hypertrophic Cardiomyopathy Clinic from 1998–2009 and all individuals were unrelated. The diagnosis of HCM was made via physical examination, ECG, and echocardiography. DNA was purified from whole blood collected from each HCM subject using the AutoGen FlexStar automated DNA extraction system (AutoGen, Holliston, MA). Genomic DNA was genotyped with the ABI 7900HT Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Duplicate internal controls and a blank control were included. The call rate was 97.0% for rs516514 and 98.3% for rs2303510. The Mayo control cohort for this study consisted of patients with normal ECG identified that had either peripheral arterial disease (n= 1,648 cases) or no evidence of peripheral artery disease (n= 1,675), had European ancestry and were recruited between October 2006 and May 2009. All participants gave their written consent for participation in the studies and the use of their data for future research. In the Mayo Control Cohort rs516514 was genotyped in Illumina Human660W-quadv1-A genotyping platform and rs2303510 genotypes were imputed by MACH123 based on HapMap II CEU database (release 21)24.
Clinical and echocardiographic data were extracted from the electronic medical record for each patient in the Mayo Clinic HCM Cohort. Each patient had been analyzed previously for mutations in the 9 most common HCM-susceptibility genes (MYH7, MYBPC3, TNNT2, TNNI3, TNNC1, TPM1, ACTC, MYL3, MYL2). T-tests were performed to assess the relationship between the predictor variable genotype (CC+CT versus TT) and continuous outcome variables (age at diagnosis, mean septal thickness, mean resting gradient). Chi-squared tests were performed to assess the relationship between the predictor variable genotype (CC+CT versus TT) and categorical variables (obstruction, family history of HCM and sudden cardiac death, myectomy, ICD). These analyses were conducted using JMP 8 (SAS Institute Inc., Cary, NC, USA).
Tissue Expression Studies
The Gene Expression Omnibus datasets derived from multiple human organs were searched through the NCBI website (http://www.ncbi.nlm.nih.gov/geo/). Quantitative real-time PCR (qRT-PCR) was performed with RNA extracted from 17 control donor left ventricular tissues and 19 HCM myectomy tissues. cDNA was generated from each RNA sample using the iScript cDNA Synthesis Kit (Bio-Rad). TaqMan Gene Expression Assays (Applied Biosystems) for FHOD3 and endogenous control GAPDH were run in triplicate on the ABI Prism 7900HT Real Time System (Applied Biosystems). The standard curve for the FHOD3 primer and probe set had an R2 =0.99921 and a slope of −3.6. For GAPDH the R2=0.99984 and the slope was −4.1. qRT-PCR data was analyzed after calculating 2−ΔCt for the average ΔCt value (FHOD3-GAPDH) for the triplicate replicates of each sample. A t-test was performed to test expression level differences. A fold-change was calculated by the 2−ΔΔCt method25.
Western blot analysis of FHOD3 protein expression was performed for 6 donor heart tissues and 6 HCM myectomy tissues. Briefly, total protein extracted from heart tissue by homogenization with ReadyPrep Mini Grinders (Bio-Rad) in an LDS buffer was separated by 10% SDS-PAGE and transferred from the gel to Amersham Hybond-P polyvinylidene difluoride membranes (GE Healthcare). The membranes were probed with an anti-FHOD3 polyclonal rabbit antibody (HPA024696, Sigma-Aldrich) and HRP-conjugated goat anti-rabbit secondary antibody (ab6721, abcam) before imaging with Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare) using a Molecular Dynamics SI Personal Densitometer (GE Healthcare) and Image Quant TL Software (GE Healthcare). FHOD3 expression was normalized to the actin as detected by a 1:10,000 dilution of monoclonal mouse anti-actin (alpha-sarcomeric) primary antibody (A2172, Sigma-Aldrich) and 50ng/mL of Cy3 donkey anti-mouse cy3 secondary antibody (715-165-150, Jackson ImmunoResearch Laboratories, Inc.). Fold-change was calculated as the ratio of average normalized relative expression in cases divided by controls. A t-test was performed to determine if the difference in expression achieved statistical significance.
Drosophila Studies
The UAS-fhos RNAi line (transformant ID 108347; construct ID 108238) was obtained from the Vienna Drosophila RNAi Center, the tinC-GFP; tinC-Gal4 flies were generated as previously described26. All fly stocks maintained on standard yeast protein media at room temperature. Fhos knockdown is under the well-established Gal4/UAS binary system27. Analysis of fhos knockdown was analyzed using Actin5C-Gal4 which produces ubiquitous Gal4 expression. qRT-PCR was performed with a 7900HT (Applied Biosystems) using Power SYBR Green Reagents (Applied Biosystems, cat # 4367659) and custom oligonucleotide primers specific for Act5C (CAGATCATGTTCGAGACCTTCAA and ATCTTCATCAGGTAGTCGGTCAA)28 and fhos (AACGGTCAAGCTCTTCTGGA and GGCAACTCTGGGTTGGATAA). Cardiac function in adult Drosophila was measured using an OCT microscopy system (Bioptigen, Inc. Durham, NC)29, 30. Multiple 3 second OCT m-modes were recorded and images acquired in the transverse orientation to center of the heart chamber were processed using ImageJ software using a 125 micron standard. After m-mode acquisition, the flies were examined in the transverse B-mode orientation to assure consistent measurements from the heart chamber. End-diastolic (EDD), end-systolic (ESD), and heart rate were determined from 3 consecutive heart beats. Fractional shortening (FS) was calculated as [EDD-ESD]/EDDx100. Comparisons of EDD chamber dimensions were determined by a Student’s t-test using GraphPad Prism statistical software (GraphPad Software Inc.).
Results
HCM genome-wide association study identifies rs516514
The Tufts HCM Cohort is comprised of consecutive patients treated for HCM at Tufts Medical Center. Echocardiography of the cohort demonstrated an expected3, 31 degree of inter-individual heterogeneity of maximal left ventricular wall thickness as well as resting and inducible outflow tract obstruction (Table 1). Analysis of eight contractile genes by denaturing high-performance chromatography identified a contractile gene mutation with a high probability of being disease-causing in 27.4% of cases, which is similar to other cohorts32. The Tufts HCM Cohort was genotyped on the Illumina Hap370CNV array. We obtained a genetic dataset including all available Caucasian control individuals (n = 823) from the Illumina iControlDB database, genotyped on the Illumina Hap300 array, which is an identical genotyping platform as the 370CNV but lacking copy number variation probes. The control cohort had a similar age distribution as the Tufts HCM Cohort. After applying standard quality control techniques, 311,399 SNPs were available for analysis across the combined case and control datasets. Structure analysis of ancestry informative markers present on the arrays proved the cases and controls to be well matched for the European ancestry; no significant subpopulations were found between (or within) case or control groups. After applying an additive logistic regression test to the case and control groups followed by a correction for multiple testing, two separate chromosomal loci were found to be associated with HCM defined by having a Bonferroni-corrected p-value less than 0.05 (Table 2). We were unable to confirm the association of rs12341266 with HCM in the Mayo Clinic cohort (data not shown). Here, we report the association of rs516514, which is located within an intron of the FHOD3 gene, with HCM (OR = 2.45 (1.76–3.41), p=1.25 × 10−7, Figure 1A). The minor allele frequency (MAF) for rs516514 reported by HapMap was ~0.48 in all tested racial and ethnic groups, which is considerably higher than the prevalence of HCM itself33; the observed MAF in HCM affected individuals was 0.61 (control MAF 0.44). We then genotyped 136 additional cases whose DNA had been collected after the GWA samples. Analysis of this second set of cases against the same control cohort again demonstrated an association of rs516514 with HCM (OR = 2.04 (1.52–2.73), p=1.85 × 10−6).
Table 1.
Summary of Cohorts
| Tufts | Mayo Clinic | |
|---|---|---|
| Number of Subjects | 174 | 1012 |
| Age | 46.2 (15.5) | 44.68 (18.62) |
| Sex (% Men) | 103 (59%) | 602 (59%) |
| Mean Septal Thickness (mm) | 19.8 (5.91) | 20.98 (5.91) |
| Mutation Identified | 45 (27%) | 378 (37%) |
| LVOT Pressure Gradient (presence/absence) | 96 (55%) | 484 (48%) |
| Surgical Myectomy | 38 (22%) | 450 (46%) |
LVOT, left ventricular outflow tract. Data are presented as mean (SD) or n (%)
Table 2.
Significant Loci
| dbSNP rsID | Gene | Chr | BP (hg18) | P-Value | OR (95% CI) | SNP Location | |
|---|---|---|---|---|---|---|---|
| Unadjusted | Adjusted | ||||||
| rs516514 | FHOD3 | 18 | 32515046 | 1.25 × 10−7 | 0.04 | 2.45 (1.76–3.41) | Intronic |
| rs12341266 | RGS3 | 9 | 115396337 | 1.32 × 10−7 | 0.04 | 4.06 (2.40–6.80) | Non- Synonymous Coding |
Logistic regression analyses were performed with correction for the following co-variates: gender, presence of known HCM mutations, and age. P-values before and after adjustment for multiple hypothesis testing (Bonferroni protocol) are shown. Chr, chromosome; BP, chromosomal base pair; OR, odds ratio.
Figure 1.

Chromosome 18 locus that includes FHOD3 is associated with HCM. A. Graph shows the − log(p-value) and the location of each SNP contained on the 370CNV array that is within the chromosome 18 locus identified by the HCM GWA study. The SNP with the strongest association with HCM is shown in blue. SNPs in LD with rs516514 are shown from red (strong LD) to yellow (weak LD). Relative sites of recombination are shown in blue lines. B. Quantitative real time PCR demonstrating expression of FHOD3 RNA in mouse tissues. The strongest expression was found in the heart. C. Alignment of FHOD3 peptide showing conservation of FHOD3-V1151 (red box) in mammals. Amino acids conserved across all species shown are marked by asterisk.
Next, we searched the Gene Expression Omnibus (GEO) to determine whether any gene near rs516514 is expressed in the heart. Review of several GEO gene expression datasets, a representative example is GDS1096, demonstrated that FHOD3 is strongly expressed in the heart compared with other organs. We confirmed strong preferential expression of FHOD3 in the heart compared with other organs by quantitative real-time PCR analysis of a panel of mouse organ total RNA (Figure 1B). Further support for FHOD3 as opposed to other genes near rs516514 was derived from the observation that FHOD3 belongs to a family of proteins that include an FH2 domain, whose function is to promote actin filament formation. Finally, FHOD3 was reported to be required for assembly of the neonatal cardiomyocyte cardiac contractile apparatus34.
Independent replication of rs516514 association with HCM
We sought to confirm the association of rs516514 with HCM by testing the Mayo Clinic HCM (Table 1) and control cohorts. The Mayo Clinic HCM Cohort includes 1,012 FHOD3 genotyped cases. The Mayo Clinic control cohort included 1,326 subjects available from the eMERGE (Electronic Medical Records and Genomics Network) not known to have HCM and who all have a normal electrocardiogram. Our a priori threshold for significance in this replication study was p < 0.01. Applying an additive model to rs516514 genotypes in the Mayo Clinic case and control samples revealed a significant replication of the Tufts GWA HCM association (OR = 1.26 (1.12–1.41) p = 0.0001). This finding confirmed the association of rs516514 with HCM identified by our GWA study and suggested that a genetic variant within FHOD3 increased the risk of clinically apparent HCM.
FHOD3-V1151I shows a stronger association with HCM compared with rs516514
Approximately 50% of all clinically diagnosed HCM and 80% of reverse curvature-HCM is caused by mutations involving genes that encode key sarcomeric contractile proteins (myofilaments)35, 36. Consequently, we speculated whether the associated FHOD3 intronic polymorphism might be in linkage disequilibrium with a FHOD3 peptide variant that has the potential to biologically impact the physiology of the cardiac sarcomere. We queried the Exome Variant Server (http://evs.gs.washington.edu/EVS/) to ask whether a FHOD3 non-synonymous FHOD3 SNP, that changed the peptide sequence, was in linkage disequilibrium (LD) with rs516514. This survey identified 114 FHOD3 non-synonymous variants, and only FHOD3-V1151I was found to be in partial LD with rs516514 (rs2303510; D′=0.903, r2=0.287). FHOD3-V1151 is highly conserved among mammals and other species; nearby amino acids are completely conserved (Figure 1C). Using an additive model, we found that FHOD3-V1151I was more strongly associated with HCM (OR = 1.36 (1.20–1.54), p<0.0001) in the Mayo Clinic Cohort compared to the intronic rs516154 variant. Similarly, the p-value for association with HCM of the FHOD3-V1151I polymorphism was lower in the Tufts HCM Cohort (additive model OR = 2.01 (95% CI 1.64 –2.64), p=1.76 × 10−9) consistent with a stronger association of rs2303510 compared with rs516514. When rs514516 was added as a covariate in our logistic regression model the association of rs2303510 with HCM became non-significant indicating that the association of both SNPs with HCM is related to their being in partial linkage disequilibrium and less likely to be caused by their representing more than one HCM association signal. Finally, we found no significant statistical interaction between FHOD3 rs2303510 genotype status and contractile gene mutation. The discovery of a common variant that changes the amino acid sequence of FHOD3 associated with HCM suggests that the association of FHOD3 with HCM is based upon an effect mediated within the cardiac sarcomere as opposed to differences in expression level.
FHOD3 variants are not associated with differences in LV phenotypes
Next, we asked whether carriers of the FHOD3 polymorphisms demonstrated differences in LV phenotypes. We tested this question in the Mayo Clinic HCM Cohort because its large number of subjects offered greater power to detect a significant difference for quantitative traits. Patients homozygous for both the rs516514 and the FHOD3-V1151I minor alleles exhibited similar HCM phenotypes when compared with the reference genotypes (Table 3). These studies demonstrate that while we find evidence that FHOD3-V1151I is associated with HCM, FHOD3-V1151I is not associated with a particular LV phenotype including LV wall thickness or outflow tract obstruction.
Table 3.
Analysis of HCM Disease Expression by Genotype
| Genotype | FHOD3-V1151I | ||
|---|---|---|---|
| CC+CT | TT | P-value | |
| Number | 849 | 146 | |
| Septal Thickness | 21.0 (0.2) | 20.8 (0.5) | 0.87 |
| Rest Gradient | 43.9 (1.6) | 46.5 (3.6) | 0.50 |
| Obstructive (%) | 47 | 51 | 0.32 |
| Age at Dx | 44.8 (0.6) | 44.4 (1.5) | 0.87 |
| FHx HCM (%) | 31 | 33 | 0.63 |
| FHx Sudden Death (%) | 19 | 20 | 0.71 |
| Myectomy (%) | 46 | 41 | 0.36 |
| ICD (%) | 18 | 17 | 0.59 |
Data are presented as mean (SEM) or percentage
Both alleles of FHOD3-V1151I are expressed in HCM heart tissue
Next, we analyzed FHOD3 expression in heart tissue taken from patients with HCM at the time of septal myectomy, an operation in which a portion of the left ventricular septum is surgically removed to alleviate outflow tract obstruction. These studies confirmed expression of FHOD3 transcripts in the HCM heart. FHOD3 transcript abundance was increased in the HCM heart 1.67-fold (p=0.012) by quantitative PCR (Figure 2A). Western blotting revealed 2.05-fold (p=0.03) greater FHOD3 protein in HCM heart tissue versus control hearts (Figure 2B). Finally, we sought to determine whether transcripts encoding both alleles of the FHOD3-V1151I variant were expressed in the HCM tissue. Sanger sequencing of cDNA produced from HCM myectomy heart RNA identified the expression of both FHOD3-V1151I alleles in three cases heterozygous for the variant (Figure 2C). Increased expression of FHOD3 transcript and protein in HCM heart tissue, as well as expression of both alleles of FHOD3-V1151I, suggests that FHOD3 may have an important role in heart function.
Figure 2.
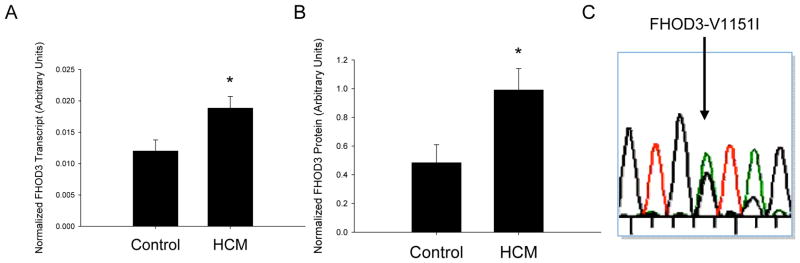
FHOD3 expression in heart and HCM tissue. A. Quantitative PCR analysis demonstrates significantly increased expression of FHOD3 transcript in heart tissue from 19 HCM case hearts and 17 controls. B. Quantitative Western blot analysis demonstrates significantly increased expression of FHOD3 protein in heart tissue from six HCM cases compared with six controls. C. Sequence analysis of the FHOD3 transcript in human myectomy samples demonstrates the presence of both nucleotide alleles of rs2303510 (green and black peaks at the position marked by arrow) indicating that both peptide forms of FHOD3-V1151I may be expressed. Asterisk, p<0.05.
Fhos is required for normal heart contractile function
We next sought to further define whether FHOD3 is plausibly related to myocardial phenotypes by defining whether it is required for normal adult heart function. Animal models provide well-recognized approaches to analyze genes whose human disease mutations are associated with disease. Drosophila has emerged as a tractable genetic model to determine whether genes associated with human disease may have a plausible role in heart function29, 30, 37–39. Therefore, to determine whether FHOD3 is required for contractile function in the fully formed adult heart, we analyzed flies genetically engineered for RNA interference-mediated knockdown (RNAi) of fhos, the fly homolog of the mammalian FHOD genes. Overexpression of fhos-specific RNAi (achieved using the Act5C-Gal4 driver line) produced a significant reduction in the fhos transcript when compared with control (Supplementary Figure). Heart-specific expression of fhos RNAi (achieved using the TinC-Gal4 driver line29) produced a significant decrease in fly heart fractional shortening with increases in the end-systolic and the end-diastolic dimensions (Figure 3A–D), indicative of significantly impaired contractile function. These results in flies and published work in mammalian cardiomyocytes34 are consistent with the requirement of FHOD3/fhos for normal heart contractile function and support a plausible role for FHOD3 variants being associated with HCM. This demonstration in Drosophila support future studies that will define the role of FHOD3 amino acid variants on the function of the mammalian heart.
Figure 3.
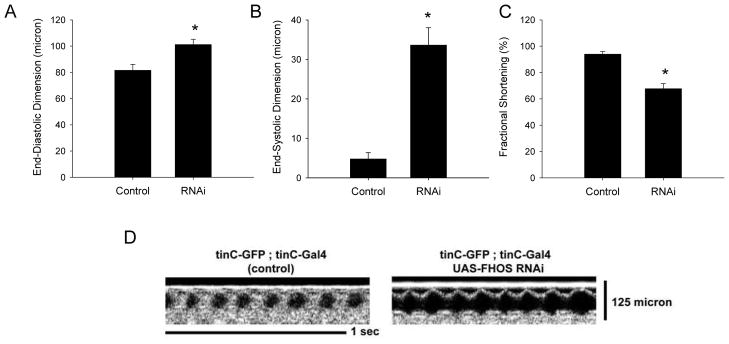
Decrease in fhos expression produces reduced fly heart contraction. Fly heart contractile function in flies expressing the fhos RNAi in the heart was examined by optical coherence tomography (OCT). Compared with control hearts (n=21) flies expressing the fhos RNAi in the heart (n=21) showed increased A. end-diastolic dimension, B. end-systolic dimension and C. reduced fractional shortening. D. representative OCT images from control and fhos RNAi flies. Asterisk, p<0.05.
Discussion
To our knowledge this is the first GWA study to identify a common gene variant associated with HCM, a disease caused by rare single gene mutations. In this GWA study, we identified an intronic SNP in FHOD3, a gene required for formation of the contractile apparatus, to be associated with HCM in two independent case-control cohorts of unrelated patients with HCM. This finding, together with the modestly increased FHOD3 expression both at the transcript and protein level in human HCM myectomy tissue, is consistent with an important role for FHOD3 in the HCM. However, these FHOD3 polymorphisms can not and should not be considered as HCM-causative variants because their minor allele frequencies are higher than the prevalence of HCM itself. The over-representation of FHOD3 alleles in the setting of HCM Our finding of a common genetic variant affecting a gene with a plausible role in heart phenotypes in HCM should encourage the application of GWA testing to other mono-genetic disorders to identify common gene variants associated with trait.
Our findings are consistent with carriers of the FHOD3-V1151I having an altered risk of manifesting clinically apparent HCM that would lead them to seek care in a dedicated HCM treatment center. Considering that several laboratories have demonstrated that patients carrying more than one contractile gene mutation have more severe HCM phenotypes11 we tested whether FHOD3-V1151I was also associated with differences in HCM phenotypes. We found no evidence that FHOD3-V1151I is associated with differences in LV wall thickness, LV outflow tract obstruction or other HCM clinical endpoints. We also speculated that by affecting sarcomere formation FHOD3-V1151I may alter remodeling of the cardiomyocyte in response to a contractile gene mutation, however we found no evidence to suggest that FHOD3-V1151I had a selective effect in patients found to carry a contractile gene mutation. While we found no evidence that FHOD3-V1151I affects HCM disease expression we acknowledge that our analyses may have lacked sufficient power to detect small phenotypic effects. Our results complement recent studies that have identified common polymorphisms associated with dilated non-ischemic cardiomyopathy40, further supporting a role for common genetic variants in cardiomyopathies otherwise caused by rare variants. We acknowledge that because the Tufts and the Mayo Clinic HCM cohorts do not have sufficient numbers of family members that include HCM-causing mutation positive subjects that do not have HCM we are not able to formally test whether FHOD3-V1151I affects penetrance of HCM. Our results should encourage future studies, perhaps based upon testing families that include members carrying an HCM mutation but lack phenotypic expression of HCM, to determine whether FHOD3 genotype status is associated with penetrance of HCM.
We find it compelling that a FHOD3 variant is associated with HCM because two reports have demonstrated that FHOD3 has a vital role in actin filament formation and maintenance34, 41. Mutations of genes encoding components of the thin filament8, including alpha-cardiac actin42, have been previously implicated as HCM disease associated genes. We found increased FHOD3 protein levels in patients with HCM, while patients with a dilated cardiomyopathy have reduced FHOD3 protein levels41. Our demonstration that reducing fhos fly heart transcript levels produces cardiac contractile dysfunction is consistent with an important role for FHOD3 in cardiomyopathy. Future studies will define the role of FHOD3 overexpression and peptide variant expression on cardiomyocyte and heart phenotypes.
The FHOD3 protein includes a formin homology-2 (FH2) domain and an FH1 domain. FH2 domains promote extension of actin filaments by blocking capping proteins from binding the actin filament end, which would terminate filament extension. The FH2 crystal structure indicates two states of the FH2 domain: one that binds to actin and the other that releases actin, both while protecting the filament from capping proteins43. Alanine mutants of two FHOD3 FH2 domain residues conserved with critical residues of the yeast protein Bni1p FH2 domain are unable to promote actin-nucleating activity and sarcomere formation in rat neonatal cardiomyocytes34. Intriguingly, we identified the FHOD3-V1151I peptide variant, which is conserved in the FH2 domain, to be associated with HCM. We speculate that the V1151I variant may alter the kinetics of FHOD3 actin binding resulting in either a permissive or a restrictive effect on actin filament formation.
We acknowledge several limitations to our study. First, our discovery cohort is smaller than most GWA study cohorts. Despite this limitation, the association of rs516514 found in the Tufts HCM Cohort was independently replicated in the larger Mayo Clinic HCM Cohort. Second, since our study has been performed in subjects of European background, our results may not be generalizable to HCM patients from different racial or ethnic backgrounds. Despite this limitation, the minor allele frequency of FHOD3 rs516514 is similar in Caucasians, African Americans, and Asians, which should motivate testing the association of FHOD3-V1151I with HCM in non-Europeans. The effects of population stratification were controlled by the inclusion of ancestry informative marker-derived covariates in our GWA study analysis. A significant proportion of the patients included in the Tufts and Mayo Clinic HCM cohorts were treated with septal reduction therapy (myectomy or alcohol septal ablation) suggesting that our results may not be generalizable to HCM patients with purely non-obstructive disease. Finally, our unbiased GWA study did not identify gene variants in the renin-angiotensinogen-aldosterone pathway that modify HCM44–46, nor did we identify polymorphisms previously associated with LV mass in the absence of HCM20 or incident heart failure47. Failure to find these associations in our GWA study may reflect associations that could not overcome the strict correction for multiple hypothesis testing that is necessary as part of our GWA study discovery strategy, or, perhaps, that the array platform did not adequately profile those alleles.
Our results support a model by which a common FHOD3 genetic variant alters the HCM disease phenotype in such a manner that increases the risk of clinically apparent disease. The requirement of fhos for fly heart contractility suggests that effects of FHOD3 on mammalian heart contractility may be responsible for its association with HCM. Future investigations will explore the molecular mechanisms supporting the association of FHOD3 variants with HCM.
Supplementary Material
Hypertrophic Cardiomyopathy (HCM) is characterized by excessive thickening and enlargement of the heart muscle causing heart failure, syncope and death. HCM is caused by rare mutations that change the peptide sequence of proteins in the cardiac sarcomere. We asked whether genetic polymorphisms common in the general population might also be found more frequently in HCM as has been recently shown for dilated cardiomyopathy. To address this question we performed a case-control genome wide association (GWA) study in the Tufts HCM Cohort and we validated our findings in the Mayo Clinic HCM cohort. These studies identified a peptide-altering variant in the FHOD3 gene associated with HCM. While HCM patients were more likely to carry the FHOD3 polymorphism carriers had similar HCM phenotypes as non-carriers. We found that the FHOD3 protein is expressed in the heart and its expression was increased in the setting of HCM. To answer whether this family of genes is important in muscle function, as might be expected of a gene associated with HCM, we studied the fruit fly whose homolog of FHOD3 is called fhos. Genetic knockdown of the fhos transcript specifically in the fly heart caused a decrease in heart contractile function. In summary, these studies have identified a new gene associated with HCM-one that is common in the general population and likely required for normal heart contractile function. Future studies will define the role of FHOD3 in cardiac function and cardiomyopathy.
Acknowledgments
The authors wish to thank Randy Kring, Westley Spiro, Michelle Arya, and Alessandra Alicea for their contribution to this manuscript.
Funding Sources: This project was supported by the following a grant from the John T. Babbitt Foundation (G.S.H.), the National Institutes of Health grant HL077378, the American Heart Association (E.C.W), and the Windland Smith Rice Comprehensive Sudden Cardiac Death Program and the National Institutes of Health grant P01HL 94291 (M.J.A.). The project described was supported by the National Center for Research Resources Grant Number UL1 RR025752 and the National Center for Advancing Translational Sciences, National Institutes of Health, Grant Number UL1 TR000073.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Seidman C. Genetic causes of inherited cardiac hypertrophy: Robert L. Frye Lecture. Mayo Clin Proc. 2002;77:1315–1319. doi: 10.4065/77.12.1315. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Casey SA, Hauser RG, Aeppli DM. Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol. 2003;42:882–888. doi: 10.1016/s0735-1097(03)00855-6. [DOI] [PubMed] [Google Scholar]
- 3.Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295–303. doi: 10.1056/NEJMoa021332. [DOI] [PubMed] [Google Scholar]
- 4.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 5.Spirito P, Autore C, Rapezzi C, Bernabo P, Badagliacca R, Maron MS, et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703–1710. doi: 10.1161/CIRCULATIONAHA.108.798314. [DOI] [PubMed] [Google Scholar]
- 6.Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–2218. doi: 10.1016/s0735-1097(00)01003-2. [DOI] [PubMed] [Google Scholar]
- 7.Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399–414. vi. doi: 10.1016/j.ccl.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–451. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 9.Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 10.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 12.Daw EW, Chen SN, Czernuszewicz G, Lombardi R, Lu Y, Ma J, et al. Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet. 2007;16:2463–2471. doi: 10.1093/hmg/ddm202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, et al. Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 2002;106:3085–3090. doi: 10.1161/01.cir.0000042675.59901.14. [DOI] [PubMed] [Google Scholar]
- 14.Semsarian C, Healey MJ, Fatkin D, Giewat M, Duffy C, Seidman CE, et al. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:2055–2060. doi: 10.1006/jmcc.2001.1466. [DOI] [PubMed] [Google Scholar]
- 15.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishi H, Kimura A, Harada H, Adachi K, Koga Y, Sasazuki T, et al. Possible gene dose effect of a mutant cardiac beta-myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 1994;200:549–556. doi: 10.1006/bbrc.1994.1483. [DOI] [PubMed] [Google Scholar]
- 17.Arnett DK, Hong Y, Bella JN, Oberman A, Kitzman DW, Hopkins PN, et al. Sibling correlation of left ventricular mass and geometry in hypertensive African Americans and whites: the HyperGEN study. Hypertension Genetic Epidemiology Network. Am J Hypertens. 2001;14:1226–1230. doi: 10.1016/s0895-7061(01)02200-2. [DOI] [PubMed] [Google Scholar]
- 18.Bella JN, MacCluer JW, Roman MJ, Almasy L, North KE, Best LG, et al. Heritability of left ventricular dimensions and mass in American Indians: The Strong Heart Study. J Hypertens. 2004;22:281–286. doi: 10.1097/00004872-200402000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Post WS, Larson MG, Myers RH, Galderisi M, Levy D. Heritability of left ventricular mass: the Framingham Heart Study. Hypertension. 1997;30:1025–1028. doi: 10.1161/01.hyp.30.5.1025. [DOI] [PubMed] [Google Scholar]
- 20.Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, et al. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. JAMA. 2009;302:168–178. doi: 10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 22.Pei YF, Zhang L, Li J, Deng HW. Analyses and comparison of imputation-based association methods. PLoS One. 2010;5(5):e10827. doi: 10.1371/journal.pone.0010827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Lee T, Lin N, Wolf MJ. Affecting Rhomboid-3 function causes a dilated heart in adult Drosophila. PLoS Genet. 2010;6:e1000969. doi: 10.1371/journal.pgen.1000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 28.Marcillac F, Bousquet F, Alabouvette J, Savarit F, Ferveur JF. A mutation with major effects on Drosophila melanogaster sex pheromones. Genetics. 2005;171:1617–1628. doi: 10.1534/genetics.104.033159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim IM, Wolf MJ. Serial examination of an inducible and reversible dilated cardiomyopathy in individual adult Drosophila. PLoS One. 2009;4:e7132. doi: 10.1371/journal.pone.0007132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maron BJ, Casey SA, Hurrell DG, Aeppli DM. Relation of left ventricular thickness to age and gender in hypertrophic cardiomyopathy. Am J Cardiol. 2003;91:1195–1198. doi: 10.1016/s0002-9149(03)00266-2. [DOI] [PubMed] [Google Scholar]
- 32.Andersen PS, Havndrup O, Hougs L, Sorensen KM, Jensen M, Larsen LA, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–370. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 33.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. Jama. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 34.Taniguchi K, Takeya R, Suetsugu S, Kan OM, Narusawa M, Shiose A, et al. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J Biol Chem. 2009;284:29873–29881. doi: 10.1074/jbc.M109.059303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binder J, Ommen SR, Gersh BJ, Van Driest SL, Tajik AJ, Nishimura RA, et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc. 2006;81:459–467. doi: 10.4065/81.4.459. [DOI] [PubMed] [Google Scholar]
- 36.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739–744. doi: 10.1016/S0025-6196(11)61527-9. [DOI] [PubMed] [Google Scholar]
- 37.Allikian MJ, Bhabha G, Dospoy P, Heydemann A, Ryder P, Earley JU, et al. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum Mol Genet. 2007;16:2933–2943. doi: 10.1093/hmg/ddm254. [DOI] [PubMed] [Google Scholar]
- 38.Wolf MJ, Rockman HA. Drosophila, genetic screens, and cardiac function. Circ Res. 2011;109:794–806. doi: 10.1161/CIRCRESAHA.111.244897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein JA, Kelly SM, LoPresti PP, Heydemann A, Earley JU, Ferguson EL, et al. SMAD signaling drives heart and muscle dysfunction in a Drosophila model of muscular dystrophy. Hum Mol Genet. 2010;20:894–904. doi: 10.1093/hmg/ddq528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32:1065–1076. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E. Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol. 2010;191:1159–1172. doi: 10.1083/jcb.201005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature. 2005;433:488–494. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 44.Kaufman BD, Auerbach S, Reddy S, Manlhiot C, Deng L, Prakash A, et al. RAAS gene polymorphisms influence progression of pediatric hypertrophic cardiomyopathy. Hum Genet. 2007;122:515–523. doi: 10.1007/s00439-007-0429-9. [DOI] [PubMed] [Google Scholar]
- 45.Perkins MJ, Van Driest SL, Ellsworth EG, Will ML, Gersh BJ, Ommen SR, et al. Gene-specific modifying effects of pro-LVH polymorphisms involving the renin-angiotensin-aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. Eur Heart J. 2005;26:2457–2462. doi: 10.1093/eurheartj/ehi438. [DOI] [PubMed] [Google Scholar]
- 46.van der Merwe L, Cloete R, Revera M, Heradien M, Goosen A, Corfield VA, et al. Genetic variation in angiotensin-converting enzyme 2 gene is associated with extent of left ventricular hypertrophy in hypertrophic cardiomyopathy. Hum Genet. 2008;124:57–61. doi: 10.1007/s00439-008-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–266. doi: 10.1161/CIRCGENETICS.109.895763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.