

Abstract
SHP-1 (PTPN6) is a member of the SHP sub-family of protein tyrosine phosphatases and plays a critical role in the regulation of the JAK/STAT signaling pathway. Previous studies suggested that SHP-1 contains a PTP1B-like second phosphotyrosine pocket that allows for binding of tandem phosphotyrosine residues, such as those found in the activation loop of JAK kinases. To discover the structural nature of the interaction between SHP-1 and the JAK family member, JAK1, we determined the 1.8 Å co-crystal structure of the SHP-1 catalytic domain and a JAK1-derived substrate peptide. This structure reveals electron density for only one bound phosphotyrosine residue. To investigate the role of the predicted second site pocket we determined the structures of SHP-1 in complex with phosphate and sulfate to 1.37 Å and 1.7 Å, respectively, and performed anomalous scattering experiments for a selenate-soaked crystal. These crystallographic data suggest that SHP-1 does not contain a PTP1B-like second site pocket. This conclusion is further supported by analysis of the relative dephosphorylation and binding affinities of mono-and tandem-phosphorylated peptide substrates. The crystal structures instead indicate that SHP-1 contains an extended C-terminal helix α2′ incompatible with the predicted second phosphotyrosine binding site. This study suggests that SHP-1 defines a new category of PTP1B-like protein tyrosine phosphatases with a hindered second phosphotyrosine pocket.
Keywords: phosphatase, cytokine signaling, protein-protein interaction, crystal structure, JAK-STAT, PTP1B
1. Introduction
Tyrosine phosphorylation occurs in response to extracellular stimuli and facilitates the rapid transmission of signals to manage cellular function. The protein tyrosine kinase and protein tyrosine phosphatase (PTP) families of enzymes mediate signal transmission by the addition or removal of phosphate to specific substrate tyrosine residues, respectively. Control of substrate specificity by these enzymes is mediated by multiple factors, including protein localization, protein-protein interactions, and specific sequences in the substrate target (Tonks and Neel, 2001). Therefore, understanding the exact molecular determinants of substrate specificity is critical to understanding the phosphotyrosine-driven networks that govern signal transduction.
Cytokines are a critical class of inter-cellular signaling molecules that transmit their messages to target cells by interaction with extracellular receptors. Cytokine receptor-ligand interaction, in turn, activates the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathway and consequent signaling events. The JAK kinase domains are well studied at the atomic level (Alicea-Velazquez and Boggon, 2011), but the molecular-level determinants of JAK kinase dephosphorylation are not understood as well. This aspect of JAK regulation is important because dephosphorylation of kinase activation loops is a key mechanism to downregulate activity. For cytokine signaling cascades one of the PTPs that performs this critical regulatory role is SHP-1.
SHP-1 (PTPN6) a member of the Src Homology 2 (SH2) domain containing protein tyrosine phosphatase (SHP) sub-family of PTPs and is involved in the regulation of cytokine, growth factor, and immunoreceptor signaling by dephosphorylation of cytoplasmic proteins (Tonks, 2006). SHP-1 can dephosphorylate all of the JAK kinase family members (David et al., 1995; Klingmuller, 1997; Migone et al., 1998; Yetter et al., 1995), and specifically regulates IL-2 and gp130 family cytokine signaling by dephosphorylating JAK1 (Migone et al., 1998). SHP-1 has also been implicated in myeloid cell adherence through interactions with paxillin, vimentin, and filamentous actin (Yeung et al., 1998), and it downregulates type I phosphatidylinositidol 3-kinase activity (Cuevas et al., 1999; Zhang et al., 2000). More recently, SHP-1 has been linked to phagolysosome maturation, where it plays a key role in recruitment of signaling proteins to the phagosome and ultimately regulates phagosomal acidification (Gomez et al., 2012). Here we conduct the first structural study of the interactions between SHP-1 and JAK1.
For the PTP family, primary substrate recognition takes place at the catalytic pocket via contacts with the incoming phosphotyrosine residue (Zhang et al., 1994), however, residues flanking the target phosphotyrosine can also contribute to substrate recognition (Vetter et al., 2000). Dephosphorylation site substrate selectivity has been very well-studied for PTP1B, a cytoplasmic PTP that plays a key role in regulation of insulin signaling by dephosphorylation of the insulin receptor kinase activation loop (IRKAL) (Byon et al., 1998), a role similar to that played by SHP-1 for the JAK-STAT pathway. Interestingly, PTP1B shows a preference for activity towards IRKAL that is phosphorylated on adjacent, or tandem, tyrosine residues (Salmeen et al., 2000). This is due to the presence of a second, non-catalytic, phosphotyrosine-binding pocket close to the active site of PTP1B (Salmeen et al., 2000). The structural determinants of this second pocket have recently been used to help categorize the PTP family into five classes, termed categories I to V (Barr et al., 2009). Category I PTP family members are most similar to PTP1B, and have been proposed to contain a similar second pocket that enables this category to preferentially recognize tandem-phosphorylated substrates. SHP-1 was proposed to be a member of the category I group of PTPs (Barr et al., 2009).
Here we investigate the structural basis for SHP-1 recognition of JAK1 activation loop (JAK1AL). We determine the co-crystal structure of SHP-1 with the JAK1AL to 1.8 Å resolution, measure the enzymatic activity of SHP-1 towards JAK1AL, and quantify the binding affinity of SHP-1 for different JAK1AL phosphorylation states. We also investigate the potential second pocket in SHP-1 and determine crystal structures of SHP-1 with phosphate or sulfate ions to 1.37 Å and 1.7 Å, respectively, and confirm our results by anomalous diffraction from selenate-soaked SHP-1 crystals. These studies suggest that the category I PTP group should be reclassified into two groups, one that recognizes tandem-phosphotyrosines via a PTP1B-like second pocket, and one that does not.
2. Materials and Methods
2.1 Protein expression and purification
The catalytic domain (residues 243–530) of human SHP-1 was subcloned into a modified pET-32 expression vector and expressed in Rosetta (DE3) Escherichia coli. SHP-1 was expressed as N-terminally hexahistidine (6×His) tagged protein by overnight induction with 0.5 mM isopropyl 1-thio-β-D-galactopyranoside at 16 °C. Cell pellets were resuspended in lysis buffer (20 mM Tris pH 8.5, 200 mM NaCl, 1.5 mg/mL lysozyme) supplemented with protease inhibitors, and lysed by repetitive freeze/thaw cycles and sonication. After centrifugation at 20,000 rpm for 1 hour at 4 °C, cleared lysate was loaded onto a Ni-NTA FF (GE) affinity column, eluted by an imidazole gradient and the tag removed by overnight incubation at 4 °C with TEV protease. Following a second affinity column to remove His-tagged TEV and uncut protein, SHP-1 was further purified by anion exchange chromatography using a Resource Q (GE) column. Purified SHP-1 catalytic domain was buffer exchanged to 20 mM Tris pH 8.0, 50 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (Sigma) (TCEP). 6×His-tagged SHP-1 was purified over a Ni-NTA FF affinity column, followed by anion exchange chromatography using a Resource Q column and was buffer exchanged to 20 mM Tris pH 8.0, 50 mM NaCl, 1 mM TCEP. SHP-1 variants (C453S, Q252A, Q252R, C453S/Q252A, and C453S/Q252R) were generated using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene).
2.2 Peptide preparation
Phosphopeptides corresponding to the JAK1 activation loop, JAK1AL, with sequence E1029TDKEpYYTVKD, E1029TDKEYpYTVKD, E1029TDKEpYpYTVKD, and K1032EpYpYTV were synthesized, HPLC purified and lyophilized by Tufts University Core Facility. The JAK1AL-derived phosphomimetic peptide, with sequence K1032E-[F2PMP]2-TV, was synthesized, HPLC purified and lyophilized by Cambridge Research Biochemicals (F2PMP indicates difluorophosphonomethyl phenylalanine). All peptides were resuspended in 100 mM Tris pH 8.0,100 mM NaCl buffer.
2.3 Crystallization
Crystallographic data were collected for four types of SHP-1 co-crystal, termed SHP-1:JAK1AL, SHP-1:PO4, SHP-1:SO4 and SHP-1:SeO4. For all crystals, sparse matrix and grid screens (Qiagen and Hampton Research) were set with a Matrix Hydra-II eDrop crystallization robot (Thermo Scientific) (Murthy, 2007) in order to identify initial crystallization conditions. Crystal quality was optimized by micro-seeding (Bergfors, 2003). SHP-1:JAK1AL. Purified 6×His SHP-1 wild type was buffer exchanged to 20 mM Tris pH 8.0, 50 mM NaCl, 1 mM TCEP and concentrated to 10 mg/mL. 6×His-SHP-1 wild type was incubated on ice for ~1hr with JAK1AL phosphomimetic peptide, K1032E-[F2PMP]2-TV at a 1:3 protein:peptide ratio prior to crystallization. Crystals were grown in 0.2 M Ca Acetate, 13% PEG-3350 by the hanging drop method. SHP-1:PO4. Purified SHP-1C453S was buffer exchanged to 20 mM Tris pH 8.5, 50 mM NaCl, 1 mM TCEP and concentrated to 10 mg/mL. SHP-1C453S was incubated with JAK1AL phosphomimetic peptide, K1032E-[F2PMP]-2-TV peptide at a 1:5 protein:peptide ratio prior to crystallization. Crystals were grown in 0.2 M AmPO4, 12% PEG-2000 by the hanging drop method. SHP-1:SO4. Purified SHP-1C453S was buffer exchanged to 100 mM Tris HCl pH 8.5, 5 mM EDTA, 5 mM DTT and concentrated to 15 mg/mL. SHP-1C453S was incubated on ice for ~1 hour with JAK1AL K1032EpYpYTV peptide at a 1:5 protein:peptide ratio prior to crystallization. Crystals were grown in 0.1 M MES pH 6.5, 1.5 M AmSO4, 3% PEG-400 by the hanging drop method. SHP-1:SeO4. Crystals were grown in ammonium sulfate conditions, as described for crystal form SHP-1:PO4, and optimized by micro-seeding. Single crystals were soaked in 2 M SeO4, 100 mM MES pH 6.5 and 30 % glycerol for 20 seconds prior to cryo-cooling.
2.4 Crystallography
X-ray diffraction data were collected on either beamline NECAT 24-ID-C at the Advanced Photon Source (APS) or beamline X6A of the National Synchrotron Light Source (NSLS) (Table 1). All data were processed using HKL2000 (Otwinowski, 1997). Initial phases were obtained for each structure by molecular replacement using Phaser (McCoy et al., 2007) with the SHP-1 phosphatase domain (PDB ID: 1FPR) (Yang et al., 2000) as the search model. Each structure was then autobuilt using ARP/wARP (Perrakis et al., 1999) to mitigate model bias. Standard refinement and model building were conducted using Phenix (Adams et al., 2010) and COOT (Emsley and Cowtan, 2004). The nal model was validated using MolProbity (Chen et al., 2010).
Table 1.
X-ray diffraction data reduction and structure refinement
| Crystal | SHP-1:Jak1AL | SHP-1:PO4 | SHP-1:SO4 | SHP-1:SeO4 |
|---|---|---|---|---|
| PDB ID | 4GS0 | 4GRZ | 4GRY | -- |
|
| ||||
|
Data collection
| ||||
| Space group | P31 | C2 | P21221 | P21221 |
| Cell | ||||
| a, b, c (Å) | 43.9, 43.9, 258.1 | 164.1, 45.4, 44.7 | 45.6, 57.3,110.9 | 45.6, 57.4, 111.5 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 91.2, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| X-ray source | APS NECAT | NSLS X6A | NSLS X6A | NSLS X6A |
| Wavelength (Å) | 0.9795 | 1.0781 | 0.9801 | 0.9784 |
| Resolution (Å)a | 50.0 – 1.8 (1.86 – 1.80) | 50.0 – 1.37 (1.42 – 1.37) | 30.0 – 1.7 (1.76 – 1.70) | 50.0 – 1.7 (1.76 – 1.70) |
| Total reflections | 200865 | 327790 | 174713 | 652259 |
| Unique reflections | 51289 | 68145 | 29703 | 62276 |
| Completeness (%)a | 98.6 (99.0) | 98.0 (88.6) | 90.5 (97.5) | 100. 0 (99.9) |
| Rmerge (%)a | 8.6 (66.9) | 4.9 (25.7) | 5.8 (40.0) | 10.0 (83.6) |
| <I/σ(I)>a | 13.5 (2.0) | 28.9 (4.8) | 19.5 (3.5) | 22.7 (2.8) |
| Multiplicitya | 3.9 (3.8) | 4.8 (3.5) | 5.9 (5.9) | 10.5 (9.1) |
| Wilson B factor (Å2) | 23.3 | 14.9 | 21.1 | 18.7 |
|
| ||||
|
Refinement statistics
| ||||
| Resolution (Å)a | 43.0 – 1.8 (1.83 – 1.80) | 39.6 – 1.37 (1.39 – 1.37) | 25.7 – 1.7 (1.75 – 1.70) | |
| R factor (%)a | 16.2 (26.1) | 16.9 (27.0) | 19.0 (24.9) | |
| Free R factor (%)a | 19.5 (33.1) | 18.9 (29.7) | 23.1 (28.5) | |
| Free R reflections (%) | 5.1 | 5.1 | 5.1 | |
| No of free R reflections | 2618 | 3445 | 1498 | |
| Molecules in AU | 2 | 1 | 1 | |
| Residue range built | A/ 260–523 B/ 243–527 |
A/ 243–524 | A/ 243–525 | |
| Ions built | PO4 | A/ SO4, SO4 | ||
| Peptide built | A/ A-A-[F2PMP]-A | |||
| No. of non-H atoms | 4734 | 2753 | 2468 | |
| No. of water molecules | 287 | 391 | 228 | |
|
| ||||
|
Model quality
| ||||
| R.m.s.d. bond lengths (Å) | 0.007 | 0.006 | 0.006 | |
| R.m.s.d. bond angles (°) | 1.062 | 1.109 | 0.994 | |
| Mean B factors (Å2) | ||||
| Overall | 41.8 | 16.5 | 31.4 | |
| Protein | A/36.5 B/33.0 | 18.8 | 25.4 | |
| Ligand/Water | 59.8 / 38.0 | 9.3 / 21.3 | 36.2 / 32.6 | |
| Ramachandran (%) | ||||
| Favored/Allowed/Disallowed | 97.5 / 2.2 / 0.4 | 97.3 / 2.3 / 0.3 | 98.5 / 1.5 / 0.0 | |
indicates high resolution shell.
In the SHP-1:JAK1AL co-crystal structure, the JAK1AL phosphomimetic peptide was built into unbiased positive electron density. We started by building a F2PMP residue into the active site because its electron density was unambiguous (Figure S2). Multiple rounds of refinement were conducted following building the F2PMP residue, and three more residues were built as alanines. Interestingly, however, no side-chain density was observed for any of these built alanines in either the Fobs-Fcalc or 2Fobs-Fcalc maps preventing us from unambiguously assigning the residues for JAK1AL. Restraints for the F2PMP phosphomimetic residue were generated using ELBOW (Moriarty et al., 2009). Re ned models are deposited in the protein data bank with accession codes: 4GRY, 4GRZ, 4GS0.
Selenate anomalous difference map. Data from SHP-1:SeO4 crystals were processed in HKL2000 (Otwinowski, 1997). Phases were obtained by molecular replacement using Phaser and the SHP-1:SO4 structure as a search model.
2.5 Malachite Green Assay
A Malachite Green Assay kit (Bioassay Systems) (Geladopoulos et al., 1991) was used to measure the dephosphorylation of JAK1AL phosphopeptides by SHP-1, SHP-1Q252R, or SHP-1Q252R. JAK1AL phosphopeptides (240 μM) were assayed with 15–30 nM SHP-1 in 20 mM Tris pH 8.0, 100 mM NaCl, 1 mM TCEP, at room temperature. The reaction was quenched and developed by addition of the Working Reagent. Absorbance was measured at 620 nm using a Safire microplate reader (Tecan). The amount of inorganic phosphate released was calculated from a phosphate standard curve.
2.6 Isothermal Titration Calorimetry (ITC)
SHP-1 and JAK1AL phosphopeptides were extensively dialyzed at 4°C against 20 mM Tris pH 8.0, 100 mM NaCl, 0.5 mM TCEP buffer. ITC experiments were performed at 25°C using a VP-ITC micro-calorimeter (GE). JAK1 phosphopeptides (200–300 μM) were titrated into the experimental cell, containing SHP-1C453S, SHP-1C453S/Q252A, or SHP-1C453S/Q252R (6–10 μM), over the course of 30–35 3 μL injections. In order to obtain the thermodynamic parameters of the SHP-1 / JAK1AL interaction, the raw data were integrated and the ITC isotherm was fitted to a single-site binding model using the Origin 5.0 software (MicroCal).
3. Results
PTPs have been well-studied structurally (Fig. 1) (Barr et al., 2009) and have been categorized into five groups (I to V) based on structural analysis of their mode of substrate recognition. Within this classification scheme the non-receptor PTP SHP-1 belongs to group I (Barr et al., 2009). This group contains a second aryl phosphate-binding site that binds a phosphotyrosine one residue C-terminal to the substrate phosphotyrosine (i.e. at position P+1). This type of interaction between a group I phosphatase and a tandem tyrosine phosphorylated substrate has previously been characterized between PTP1B and the tandem-phosphorylated activation loop of insulin receptor kinase (IRKAL) (Salmeen et al., 2000), however, the mode of binding differs from that previously observed for SHP-1 when bound to a mono-phosphorylated peptide derived from the Signal-Regulated Protein α-1 (SIRPα-1) (Yang et al., 2000). To further investigate the mode of substrate recognition utilized by SHP-1 for tandem-phosphorylated substrates, specifically those within the cytokine signaling pathway, we investigated the interactions of SHP-1 with the activation loop of the tyrosine kinase JAK1.
Figure 1. Conserved motifs within the catalytic domain of PTPs.
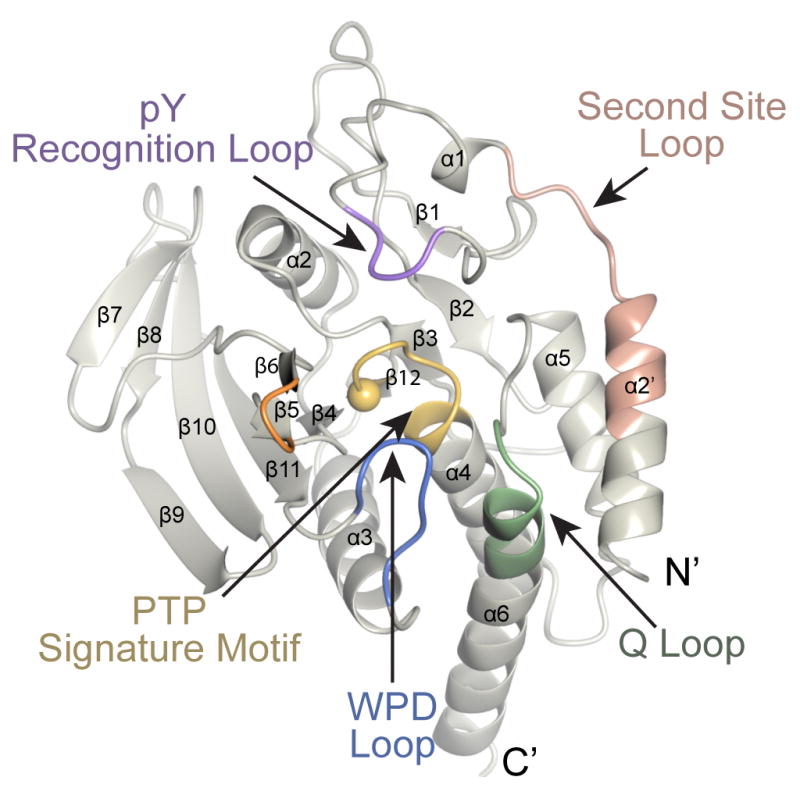
The key conserved motifs have been highlighted and are indicated by the arrows. The sphere within the PTP Signature Motif corresponds to the catalytic cysteine. The crystal structure SHP-1:PO4 is shown.
3.1 Co-crystal structure of SHP-1 with the JAK1 activation loop
We began by determining the 1.8 Å co-crystal structure of the catalytic domain of SHP-1 with a peptide derived from JAK1AL (denoted SHP-1:JAK1AL) (Table 1). The JAK1AL peptide, KE(F2PMP)2TV, contained two non-hydrolyzable phosphotyrosine mimetics (difluorophosphonomethyl phenylalanine, F2PMP) that have previously been successfully used to investigate tyrosine phosphatase-substrate interactions crystallographically (Ala et al., 2006; Bahta et al., 2011; Ivanov et al., 2005; Phan et al., 2003). In this crystal structure there are two copies of SHP-1 in the asymmetric unit that are structurally similar (RMSD 0.65 A over 257 Cα atoms) with a significant conformational difference observed for the phosphatase domain N-terminal helix, helix α2′. In chain B this helix is disordered but in chain A the helix is ordered and stabilized by crystal contacts (Supplementary Fig. S1). In this crystal structure the catalytic (WPD) loop is found in an open conformation, similar to that previously observed for SHP-1 in complex with a mono-phosphorylated SIRPα-1 peptide (Yang et al., 2000).
For both SHP-1 molecules very clear electron density is observed in the region of the active site, however we observed electron density for only one JAK1AL peptide that is bound to chain B. This density shows a bound phosphotyrosine mimetic F2PMP moiety (Fig. 2A–C and Supplementary Fig. S2). On building the F2PMP we expected to be able to trace the rest of the peptide and conclusively show a PTB1B-like tandem phosphotyrosine recognition interaction between SHP-1 and JAK1AL. We were, however, only able to build only the JAK1AL peptide backbone for four residues, and we could not establish the register of the peptide chain. Furthermore, we could not locate the second phosphotyrosine mimetic F2PMP moiety, even though it was directly adjacent to the clearly observed F2PMP bound in the active site. In our model we built the JAK1AL peptide as A-A-F2PMP-A.
Figure 2. Co-crystal structure of SHP-1 and JAK1 activation loop (AL) peptide in the bisphosphorylated state.

A) peptide (green) is shown as a space filling model. Ribbon rendition of SHP-1cat Chain B (purple) in complex with JAK1AL Zoomed-in view shows the interactions between JAK1AL and SHP-1. F2PMP engages in extensive hydrogen bonding contacts with the backbone of residues within the PTP Signature Motif and with the side chains of C453 and R459. The peptide backbone engages in interactions with the backbone of K277 and the side-chain of N278. B) Stereoview of the simulated annealing omit map of SHP-1:JAK1 contoured at 1.0σ. C) Superposition of the SHP-1:JAK1AL complex with PTP1B:IRKAL (Salmeen et al., 2000) (top row) and with SHP-1:Bzn-EJJ-amide (Asante-Appiah et al., 2002) (bottom row).
3.2 JAK1AL binds SHP-1 using a single aryl phosphate recognition site
We considered this lack of coherent side-chain register outside of the active site to be a curious finding. It potentially indicated lack of specificity for the bis-phosphorylated peptide, and little preference for binding either the N-terminal or C-terminal phosphotyrosine mimetic at the active site. Furthermore, our crystal structure potentially indicated an alternate binding mode for a group I PTP. We therefore compared the SHP-1:JAK1AL co-crystal structure with two modes of binding previously observed between PTP1B and tandem phosphorylated peptides; PTP1B in complex with the phosphorylated activation loop of insulin receptor kinase, IRKAL (Salmeen et al., 2000) (PDB ID: 1G1H) (denoted PTP1B:IRKAL), and PTP1B in complex with a bisphosphonate inhibitor, BzN-EJJ-amide, (Asante-Appiah et al., 2002) (PDB ID: 1LQF) that contains two tandem F2PMP moieties. On comparison of these structures the phosphotyrosine or phosphotyrosine mimetics that are bound in the active site (the P0 position) all adopt a very similar conformation (Fig. 2C). Furthermore, the substrate peptides all adopt a similar backbone conformation at positions P-1 and P-2. However, only PTP1B bound to IRKAL contains a phosphotyrosine bound in the second aryl phosphate site. For PTP1B in complex with BzN-EJJ-amide the second phosphotyrosine mimetic is observed at the P-1 position and is stabilized by a critical arginine residue (R47PTP1B). Since in SHP-1 the residue analogous to R47PTP1B is a lysine (K277), we suppose that this difference is the reason why SHP-1 does not bind tandem phosphorylated substrate in an identical manner to PTP1B in complex with BzN-EJJ-amide, however, it was not clear why SHP-1 did not bind a natural tandem-phosphorylated substrate using both of its previously suggested aryl phosphate recognition sites.
3.3 The predicted SHP-1 second pocket does not bind PO4, SO4 or SeO4
To investigate the reason for the lack of SHP-1 second pocket phosphotyrosine binding in our crystal structure we investigated whether crystal packing may influence the conformation. We found that in this crystal form both the active site and the second pocket are solvent exposed and accessible to bind the JAK1AL peptide. We therefore wondered whether the lack of second pocket binding for JAK1AL could indicate that SHP-1 is unable bind tandem-phosphorylated substrate via the second pocket. For PTP1B, the use of 1.9 M ammonium sulfate in the crystallization conditions results in sulfate ion binding both at the active site and at the second site (Ala et al., 2006; Li et al., 2005). Therefore, to investigate whether SHP-1 could bind a sulfate ion in the second pocket we conducted SHP-1 crystallization in a condition containing 1.5 M ammonium sulfate and determined the 1.8 Å crystal structure of the SHP-1 phosphatase domain bound to SO4 (denoted SHP-1:SO4) (Table 1).
On analysis of the SHP-1:SO4 crystal structure we observed good electron density for a sulfate ion in the catalytic site, coordinated by R459, S453, and the backbone of the PTP signature motif. We did not, however, observe any density that could be interpreted as a sulfate ion at the predicted site of the second pocket (Fig. 3) This was an unexpected result, as previous studies with PTP1B showed that sulfate could bind in the second pocket (Ala et al., 2006; Li et al., 2005). Therefore, to confirm the lack of ion binding to the second pocket we determined the crystal structure of SHP-1 grown using 0.2 M ammonium phosphate as the precipitant to 1.37 Å resolution (denoted SHP-1:PO4) (Table 1). We found that this structure also contained a phosphate ion at the catalytic site, but that there was no density for a PO4 ion at the second aryl phosphate binding site (Fig. 3) Finally, to confirm these results we soaked the SHP-1:SO4 crystals with 2 M SeO4 (denoted SHP-1:SeO4) and collected a X-ray diffraction data at the Se peak energy to investigate whether there was anomalous signal for a low occupancy ion bound in the second site. In the phased anomalous difference Fourier maps we observed a ~40 σ peak at the active site that corresponded well to the locations of the SO4 and PO4 ions. In contrast we did not observe any anomalous signal at the site of the predicted second pocket (Fig. 3D).
Figure 3. Analysis of the catalytic and second site pocket of SHP-1.
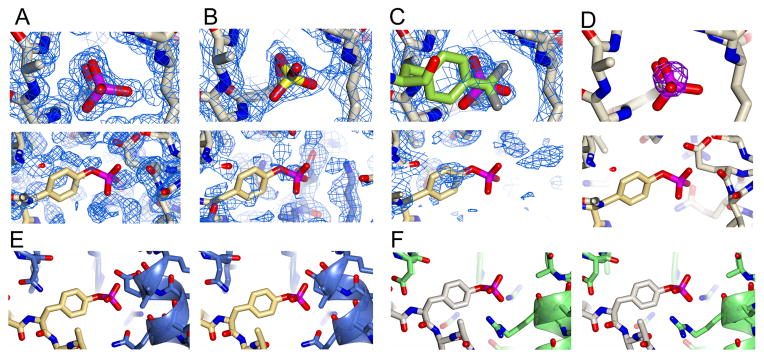
A–C) Top row shows the 2Fobs-Fcalc map contoured at 1.0σ at the catalytic pocket of SHP-1 in structures SHP-1:PO4, SHP-1:SO4, and SHP-1:F2PMP, respectively. The bottom row shows the 2Fobs-Fcalc map contoured at 1.0σ at the predicted second site pocket. For clarity the IRKAL from the structure of PTP1B:IRKAL (PDB ID: 1G1H) has been superposed to show the location of the predicted second phosphotyrosine binding site (shown in yellow). D) Top row shows selenate anomalous difference map at the catalytic pocket (with phosphate from SHP-1:PO4 superposed to for clarity). The bottom row shows the anomalous difference map at the predicted second phosphotyrosine binding site, with IRKAL from the structure of PTP1B:IRKAL superposed for clarity. The anomalous map is contoured at 10σ. E–F) Stereoviews of the predicted second site for SHP-1 and actual second site for PTP1B. Panel E shows SHP-1:PO4 with IRKAL from the structure of PTP1B:IRKAL (PDB ID: 1G1H) superposed to show the location of the predicted second phosphotyrosine binding site. Panel F shows the structure of PTP1B:IRKAL (PDB ID: 1G1H).
3.4 Surface electrostatic analysis indicates the lack of a second phosphotyrosine-binding pocket in SHP-1
We next analyzed the surface electrostatics of our SHP-1 crystal structures and compared them to that of PTP1B:IRKAL (Fig. 4) As expected, there is electropositive surface potential at the catalytic site for each of these structures, however, for the ion-bound SHP-1 crystal structures the predicted second aryl phosphate binding does not display a significant electropositive surface potential.
Figure 4. Comparison of SHP-1 surface electrostatics.

Surface electrostatics of the structures of PTP1B:IRK (top panel), SHP-1:PO4, SHP-1:SO4 and SHP-1:JAK1 (bottom panel) were rendered using CCP4mg. IRKAL and JAK1AL peptides are represented as yellow and green spheres, respectively. Arrows indicate the catalytic and predicted second phosphotyrosine binding site. The location of the predicted second phosphotyrosine binding pocket is highlighted in all structures by a green rectangle.
3.5 Structural determinants of the PTP second pocket
As surface electrostatic analysis was one of the determinants previously used to distinguish the presence of the second phosphotyrosine binding site in the class I PTPs (Barr et al., 2009) we wondered why the previous study classified SHP-1 with a positive electrostatic surface potential at the second site. We noted that the previous analysis had been conducted for the structure of SHP-1 in complex with the monophosphorylated SIRPα-1 peptide (Salmeen et al., 2000). In this structure, the carboxy-terminus of the SIRPα-1 peptide is inserted into the predicted second pocket site, inducing a conformational shift in the N-terminal helix α2′. This conformation is stabilized by crystal packing contacts.
We also noted that helix α2′ is conformationally flexible among our crystal structures, and that in the ion-bound structures, and in the full-length structure of SHP-1 (Wang et al., 2011), it contains one extra C-terminal turn when compared to PTP1B. This suggests the possibility of conformational divergence in the N-terminal helix of SHP-1 phosphatase domain, however in the context of the full-length SHP-1, and in our crystal structures of ion-bound SHP-1, the surface potential does not indicate a second pocket.
Our analysis of the N-terminal helix, helix α2′, also suggested that in the full-length enzyme in its active state, and in our ion-bound crystal structures, SHP-1 does not contain a cavity capable of binding a phosphotyrosine at the P+1 position (Fig. 3E–F). In SHP-1 the C-terminal helical turn of helix α2′, encompassing residues E254, V255, K256, and N257, creates a steric clash to the proposed position of the P+1 phosphotyrosine. In PTP1B, these residues are replaced by A27 and S28, thus shortening the helix by one turn and providing a cavity that allows phosphotyrosine binding. We therefore propose that the length of the N-terminal helix, helix α2′, is potentially a determinant of the presence of a second phosphotyrosine pocket, and thus that group I PTPs could be reclassified as group Ia (PTP1B-like) and group Ib (SHP-1-like). Based on previously determined crystal structures we propose that group Ia PTPs include PTP1B, TC-PTP, MEG2, BAS, DEP1, and GLEPP1; and group Ib phosphatases include SHP-1, SHP-2, LYP, and BDP1. We note, however, that the short side-chain residues S35 in LYP and S37 in BDP1 could potentially somewhat compensate for hindrance due to the extension in helix α2′, therefore the competence of these phosphatases to directly bind phosphotyrosine in the second pocket should be tested.
3.6 SHP-1 mutants in the predicted second pocket do not impact SHP-1 activity
Based on this analysis we confirmed the specificity of SHP-1 for phosphotyrosine substrates with the expectation that SHP-1 would display similar activities towards mono- and tandem-phosphorylated peptides. We therefore measured dephosphorylation of peptides corresponding to the phosphorylated JAK1 activation loop, E1029TDKEpYYTVKD (termed JAK1pYY), E1029TDKEYpYTVKD (termed JAK1YpY), and E1029TDKEpYpYTVKD (termed JAK1pYpY). We used a malachite green endpoint detection assay to measure release of inorganic phosphate (Pi) (Geladopoulos et al., 1991) following incubation of equimolar amounts of peptide with SHP-1. We found that SHP-1 displays increased catalytic activity towards JAK1pYpY when compared to equimolar amounts of the monophosphorylated peptides JAK1YpY and JAK1pYY (Fig. 5A). Because this increase is small, it could be largely attributed the effective doubling of phosphotyrosine substrate concentration for the tandem- versus mono-phosphorylated peptides, however, as PTP1B also displays an increase in activity towards tandem phosphorylated substrates (Salmeen et al., 2000), this result could also potentially indicate that SHP-1 might contain a second pocket. We therefore generated two mutated forms of SHP-1, Q252A and Q252R. Q252 is in the structural location analogous to PTP1B residue R24, and is one of the determinants of the second pocket in SHP-1 (Barr et al., 2009). These mutants should therefore either disrupt (Q252A) or potentiate (Q252R) phosphotyrosine binding to the second pocket. On phosphatase assay of the second pocket mutants, we found that dephosphorylation of JAK1pYpY by SHP-1Q252A or SHP-1Q252R did not differ significantly as compared with wild-type SHP-1 (Fig. 5B). These observations support the interpretation that the predicted second pocket does not impact dephosphorylation of tandem-phosphorylated substrate.
Figure 5. Biochemical assessment of SHP-1/JAK1AL interactions.
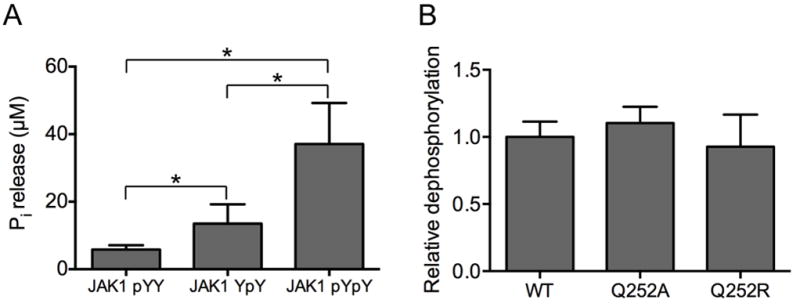
A) Dephosphorylation of JAK1AL phosphopeptides by SHP-1 assessed by Malachite Green Assay (Geladopoulos et al., 1991). Peptide dephosphorylation is quantified as the amount of released inorganic phosphate (Pi) per pmol of enzyme. * indicates P<0.05 by t-test. B) Dephosphorylation of JAK1AL in the tandem phosphorylated state by the different SHP-1 predicted second phosphotyrosine binding site variants Q252A and Q252R. Phosphate release was normalized to wild type SHP-1. The standard error of the mean is shown for both experiments.
3.7 SHP-1 preferentially binds tandem-phosphorylated peptide
Finally we asked whether the difference in activity towards substrate was due to a difference in SHP-1 binding affinity for JAK1pYpY, JAK1YpY or JAK1pYY. We used isothermal titration calorimetry (ITC) to measure the binding affinity of the mono- and tandem-phosphorylated JAK1 peptides to a catalytically inactive SHP-1 mutant, SHP-1C453S (Fig. 6A and Table 2). We find that SHP-1C453S binds JAK1pYpY approximately 20-fold tighter (Kd = 0.7 μM) than JAK1pYY (Kd= 13.4 μM), and approximately 8-fold tighter than JAK1YpY (Kd = 5.6 μM). The predicted second pocket mutants SHP-1C453S/Q252A or SHP-1C453S/Q252R, did not display a significant difference in binding affinity for JAK1pYpY (Kd = 0.6 and 0.5 μM, respectively) when compared with SHP-1C453S (Table 2). These results indicate that although the predicted second pocket does not impact binding between SHP-1 and tandem-phosphorylated substrate peptide, the protein displays increased affinity for tandem-phosphorylated substrate when compared to mono-phosphorylated peptide. Notably, the difference in binding affinity between the mono- and tandem-phosphorylated peptides is not as large as previously observed for PTP1B, which displays ~70-fold higher affinity for bis-phosphorylated over mono-phosphorylated IRKAL peptide (Salmeen et al., 2000).
Figure 6. Binding affinity measurements of the SHP-1/JAK1AL peptide interaction by ITC.
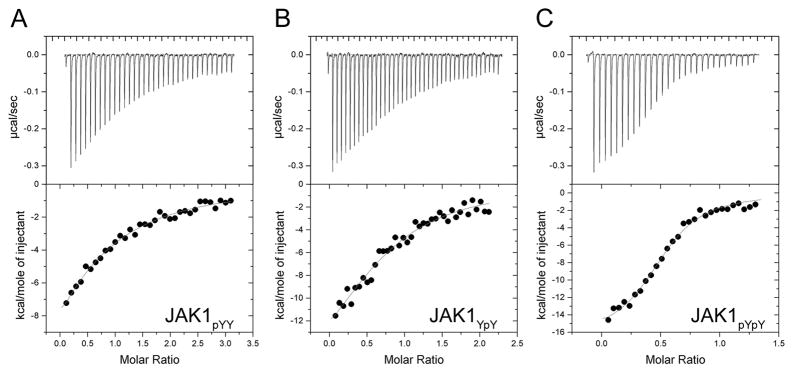
ITC was performed by titrating JAK1AL peptide (200–300 μM) into the experimental cell containing SHP-1C453S (6–10μM). The top panel shows the heat change upon ligand titration; the bottom panel shows the integrated data and ITC isotherm (solid line), fitted to a single-site binding model. A, B, and C show ITC data obtained from SHP-1C453S binding to JAK1pYY, JAK1YpY, and JAK1pYpY, respectively. Thermodynamic parameters for this data are presented in Table 2.
Table 2.
Thermodynamic parameters for the SHP-1/ JAK1AL peptide interaction measured by ITC.
| SHP-1 | JAK1 peptide | Kd (μM) | N | Enthalpy ΔH (kcal/mol) | Entropy TΔS (kcal/mol) |
|---|---|---|---|---|---|
| C453S | pY Y | 13.7 ± 1.4 | 0.7 | −28.7 ± 7.3 | −22.0 |
| C453S | Y pY | 5.6 ± 1.1 | 0.9 | −21.5 ± 3.5 | −14.4 |
| C453S | pY pY | 0.7 ± 0.1 | 0.8 | −16.6 ± 0.5 | −8.3 |
| C453S/Q252A | pY pY | 0.6 ± 0.1 | 0.9 | −16.6 ± 0.4 | −8.1 |
| C453S/Q252R | pY pY | 0.5 ± 0.1 | 0.9 | −17.4 ± 0.4 | −8.8 |
The NIHMS has received the file ‘mmc1.pdf’ as supplementary data. The file will not appear in this PDF Receipt, but it will be linked to the web version of your manuscript.
4. Discussion
The classification of PTP substrate recognition motifs has important implications for distinguishing the signaling pathways in which these enzymes function, and their mechanisms of action (Tiganis and Bennett, 2007). To better understand the mechanisms of substrate recognition utilized by the non-receptor tyrosine phosphatase, SHP-1, we conducted a structure-directed study to investigate recognition of its known substrate, the activation loop of the non-receptor tyrosine kinase JAK1. This kinase is a member of the JAK family and is responsible for downstream signaling from multiple hematopoietic cytokines via the IL-2 and gp130 receptor (Migone et al., 1998). One of the critical regulation mechanisms for JAK1, and consequently for cytokine signaling and the JAK/STAT pathway, is phosphorylation and dephosphorylation of the tandem tyrosines on the kinase activation loop. As the dephosphorylation event is mediated by SHP-1 (Shuai and Liu, 2003) it is important to understand how SHP-1 recognizes the JAK1 activation loop. Our phosphatase assays highlight a preference of SHP-1 for tandem- over mono-phosphorylated substrate and our measurements of binding affinity suggest that SHP-1 displays an increased affinity for the tandem-phosphorylated JAK1 activation loop when compared to the mono-phosphorylated form. The molecular basis for this preference, however, is not clear. Our crystal structure of SHP-1 in complex with JAK1 activation loop does not suggest an extended ordered conformation for the bound peptide, and the predicted second pocket mutants do not impact enzymatic activity or binding affinity. In addition, the structural data from SHP-1 in complex with high concentrations of phosphate or sulfate, and from SHP-1 crystals soaked with anomalously scattering selenate, do not indicate that a phosphate moiety can bind at the predicted second site. Furthermore, incorporation of mutations in the predicted second pocket that should directly impact either substrate binding or enzymatic activity towards tandem-phosphorylated substrate do not result in a measurable effect. This contrasts with previous analyses of the bona fide second site-containing phosphatase PTP1B, where mutation of the second site significantly impacts enzyme-substrate binding affinity and enzyme activity (Salmeen et al., 2000). We therefore conclude that the basis for SHP-1 preference for tandem-phosphorylated JAK1 is not due to the presence of a second pocket. Future studies will allow the determination of the mechanism of SHP1 preference for tandem-phosphorylated JAK1.
Our analysis also suggests further classification of the PTP family. A previous study had proposed that the catalytic domains of PTPs could be classified into five categories based on their structural features (Table 2) (Barr et al., 2009). SHP-1 was classified as a category I phosphatase because a previous SHP-1 crystal structure shared structural features with PTP1B suggesting the existence of a second aryl phosphate binding pocket. Our study conclusively shows that SHP-1 does not contain the predicted second phosphotyrosine-binding pocket. We propose that this difference is mainly attributed by two factors. First, the predicted second site of SHP-1 does not contain an electropositive surface that favors binding of the phosphotyrosine phosphoryl moiety. Second, the presence of an extra C-terminal turn in the N-terminal helix of SHP-1, helix α2′, that is not present in PTP1B. There is an insertion of two residues in SHP-1 that results in the extra helical turn, and consequently a steric block of the second pocket site (Fig. 7). This suggests that the category I phosphatases could potentially be further classified into categories Ia and Ib based on the presence of the N-terminal helix extension, with category Ia to include the PTP1B-like phosphatases which contain a non-hindered pocket and a shorter N-terminal helix, and category Ib, to include the SHP-1-like phosphatases whose second pocket is hindered because of the presence of an extended N-terminal helix. From the current crystallographic data available in the PDB category Ia PTPs could include PTP1B, TC-PTP, MEG2, BAS, DEP1, and GLEPP1 and category Ib PTPs could include SHP-1, SHP-2, LYP, and BDP1 (Fig. 7).
Figure 7. New categorization of PTPs.
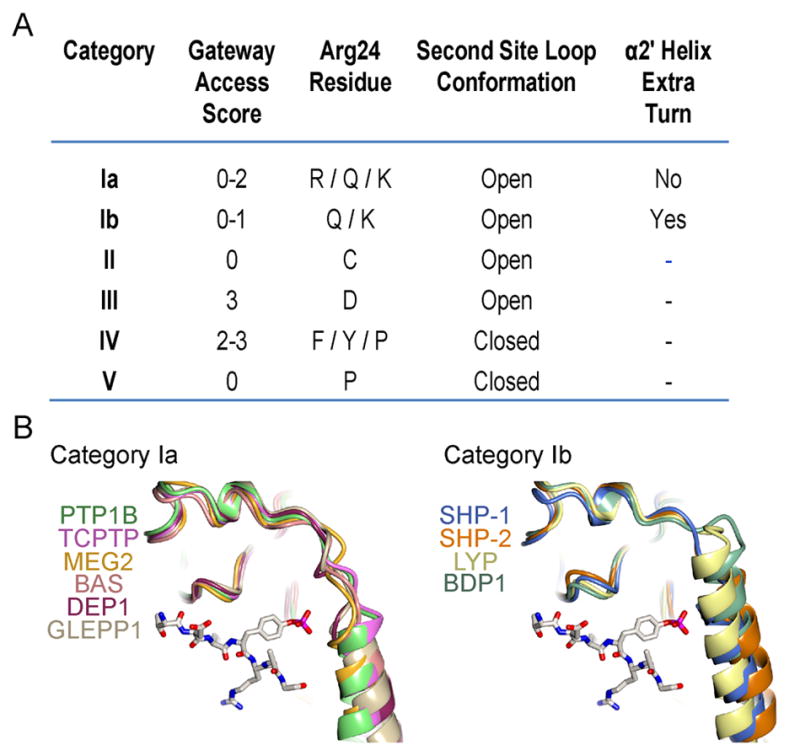
A) Summary of the structural features of the different categories of PTPs. Category Ib is the new category based on our structural findings. Adapted from (Barr et al., 2009). B) Comparison of the second phosphotyrosine binding pocket for PTP categories Ia and Ib. IRK tandem phosphorylated peptide is represented in cylinders as a reference for the second site pocket. The members of these two categories are listed. PDB IDs used: PTP1B, 1G1H; TCPTP, 1L8K; MEG2, 2PA5; BAS, 1WCH; DEP1, 2NZ6; GLEPP1, 2GJT; SHP-1, 4GRZ; SHP-2, 2SHP; LYP, 2P6X; BDP1, 2OC3.
Supplementary Material
Acknowledgments
We thank Ewa Folta-Stogniew, Amy Stiegler, Vivian Stojanoff, Anton Bennett, Elias Lolis, Ben Turk and Yang Deng. Beamline X6A at the National Synchrotron Light Source at the Brookhaven National Laboratory and the Northeastern Collaborative Access Team (NE-CAT) facility at the Advanced Photon Source at Argonne National Laboratory are thanked. Work funded by NIH grant R01 AI075133 (TJB) and Diversity Supplement R01 AI075133-S1.
Footnotes
Author contributions: Crystallography, NLAV, JJ and TJB; molecular biology, biochemistry, enzyme assays and ITC, NLAV; Study design and supervision, TJB; Manuscript preparation, NLAV, JJ and TJB.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala PJ, Gonneville L, Hillman MC, Becker-Pasha M, Wei M, et al. Structural basis for inhibition of protein-tyrosine phosphatase 1B by isothiazolidinone heterocyclic phosphonate mimetics. J Biol Chem. 2006;281:32784–32795. doi: 10.1074/jbc.M606873200. [DOI] [PubMed] [Google Scholar]
- Alicea-Velazquez NL, Boggon TJ. The use of structural biology in Janus kinase targeted drug discovery. Curr Drug Targets. 2011;12:546–555. doi: 10.2174/138945011794751528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante-Appiah E, Patel S, Dufresne C, Roy P, Wang Q, Patel V, et al. The structure of PTP-1B in complex with a peptide inhibitor reveals an alternative binding mode for bisphosphonates. Biochemistry. 2002;41:9043–9051. doi: 10.1021/bi0259554. [DOI] [PubMed] [Google Scholar]
- Bahta M, Lountos GT, Dyas B, Kim SE, Ulrich RG, et al. Utilization of nitrophenylphosphates and oxime-based ligation for the development of nanomolar affinity inhibitors of the Yersinia pestis outer protein H (YopH) phosphatase. Journal of medicinal chemistry. 2011;54:2933–2943. doi: 10.1021/jm200022g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergfors T. Seeds to crystals. J Struct Biol. 2003;142:66–76. doi: 10.1016/s1047-8477(03)00039-x. [DOI] [PubMed] [Google Scholar]
- Byon JC, Kusari AB, Kusari J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Molecular and cellular biochemistry. 1998;182:101–108. [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas B, Lu Y, Watt S, Kumar R, Zhang J, Siminovitch KA, et al. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem. 1999;274:27583–27589. doi: 10.1074/jbc.274.39.27583. [DOI] [PubMed] [Google Scholar]
- David M, Chen HE, Goelz S, Larner AC, Neel BG. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol. 1995;15:7050–7058. doi: 10.1128/mcb.15.12.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Analytical biochemistry. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- Gomez CP, Tiemi Shio M, Duplay P, Olivier M, Descoteaux A. The Protein Tyrosine Phosphatase Src Homology Region 2 Domain-Containing Phosphatase 1 Regulates Phagolysosome Biogenesis. J Immunol. 2012 doi: 10.4049/jimmunol.1103021. [DOI] [PubMed] [Google Scholar]
- Ivanov MI, Stuckey JA, Schubert HL, Saper MA, Bliska JB. Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol Microbiol. 2005;55:1346–1356. doi: 10.1111/j.1365-2958.2005.04477.x. [DOI] [PubMed] [Google Scholar]
- Klingmuller U. The role of tyrosine phosphorylation in proliferation and maturation of erythroid progenitor cells--signals emanating from the erythropoietin receptor. Eur J Biochem. 1997;249:637–647. doi: 10.1111/j.1432-1033.1997.t01-1-00637.x. [DOI] [PubMed] [Google Scholar]
- Li S, Depetris RS, Barford D, Chernoff J, Hubbard SR. Crystal structure of a complex between protein tyrosine phosphatase 1B and the insulin receptor tyrosine kinase. Structure. 2005;13:1643–1651. doi: 10.1016/j.str.2005.07.019. [DOI] [PubMed] [Google Scholar]
- McCoy A, Grosse-Kunstleve R, Adams P, Winn M, Storoni L, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci U S A. 1998;95:3845–3850. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta crystallographica Section D, Biological crystallography. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy T, Wang Y, Reynolds C, Boggon TJ. Automated Protein Crystallization Trials using the Thermo Scientific Matrix Hydra II eDrop. Journal of the Association for Laboratory Automation. 2007;12:213–218. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature structural biology. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Phan J, Lee K, Cherry S, Tropea JE, Burke TR, Jr, et al. High-resolution structure of the Yersinia pestis protein tyrosine phosphatase YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide. Biochemistry. 2003;42:13113–13121. doi: 10.1021/bi030156m. [DOI] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews Molecular cell biology. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- Vetter SW, Keng YF, Lawrence DS, Zhang ZY. Assessment of protein-tyrosine phosphatase 1B substrate specificity using “inverse alanine scanning”. J Biol Chem. 2000;275:2265–2268. doi: 10.1074/jbc.275.4.2265. [DOI] [PubMed] [Google Scholar]
- Wang W, Liu L, Song X, Mo Y, Komma C, et al. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J Cell Biochem. 2011;112:2062–2071. doi: 10.1002/jcb.23125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cheng Z, Niu T, Liang X, Zhao ZJ, et al. Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1. J Biol Chem. 2000;275:4066–4071. doi: 10.1074/jbc.275.6.4066. [DOI] [PubMed] [Google Scholar]
- Yetter A, Uddin S, Krolewski JJ, Jiao H, Yi T, et al. Association of the interferon-dependent tyrosine kinase Tyk-2 with the hematopoietic cell phosphatase. J Biol Chem. 1995;270:18179–18182. doi: 10.1074/jbc.270.31.18179. [DOI] [PubMed] [Google Scholar]
- Yeung YG, Wang Y, Einstein DB, Lee PS, Stanley ER. Colony-stimulating factor-1 stimulates the formation of multimeric cytosolic complexes of signaling proteins and cytoskeletal components in macrophages. J Biol Chem. 1998;273:17128–17137. doi: 10.1074/jbc.273.27.17128. [DOI] [PubMed] [Google Scholar]
- Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Seminars in immunology. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, et al. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.