

Abstract
Background and Aims
Adiponectin is an adipose-secreted protein that has been linked to changes in insulin sensitivity, high-density lipoprotein cholesterol levels, and inflammatory patterns. Although fenofibrate therapy can raise adiponectin levels, treatment response is heterogeneous and heritable, suggesting a role for genetic mediators. This is the first genome-wide association study of fenofibrate effects on circulating adiponectin.
Methods and Results
Plasma adiponectin was measured in participants of the Genetics of Lipid Lowering Drugs and Diet Network (n=793) before and after a 3-week daily treatment with 160 mg of fenofibrate. Associations between variants on the Affymetrix Genome-Wide Human SNP Array 6.0 and adiponectin were assessed using mixed linear models, adjusted for age, sex, site, and family. We observed a statistically significant (P=5×10−8) association between rs2384207 in 12q24, a region previously linked to several metabolic traits, and the fenofibrate-induced change in circulating adiponectin. Additionally, our genome-wide analysis of baseline adiponectin levels replicated the previously reported association with CDH13 and suggested novel associations with markers near the PCK1, ZBP1, TMEM18, and SCUBE1 genes. The findings from the single marker tests were corroborated in gene-based analyses. Biological pathway analyses suggested a borderline significant association between the EGF receptor signaling pathway and baseline adiponectin levels.
Conclusions
We present preliminary evidence linking several biologically relevant genetic variants to adiponectin levels at baseline and in response to fenofibrate therapy. Our findings provide support for fine-mapping of the 12q24 region to investigate the shared biological mechanisms underlying levels of circulating adiponectin and susceptibility to metabolic disease.
Introduction
Adiponectin is a protein secreted by the adipose tissue that has been linked to improved insulin sensitivity, suppressed development of atherosclerosis, and altered inflammatory patterns (1). Several pharmaceutical agents, including PPAR-α agonists such as fenofibrate, have been proposed to target circulating adiponectin levels to prevent the onset and progression of cardiovascular disease (2). However, changes in adiponectin in response to fenofibrate therapy vary greatly between individuals, and the response trait is estimated to have moderate to high heritability (h2=0.38– 0.55), suggesting a role for pharmacogenetic determinants (3).
A linkage analysis from our group has identified 1p35.2 and 3q28 genomic regions as potential predictors of plasma adiponectin concentrations pre- and post-fenofibrate treatment in metabolically healthy individuals (3). However, these linkage scans were performed cross-sectionally and did not consider the change in adiponectin levels as an outcome variable. To our knowledge, no association study to date has comprehensively investigated genetic polymorphisms as potential predictors of adiponectin response to fenofibrate therapy. Additionally, most published studies on fenofibrate response have been conducted in individuals with impaired metabolic phenotypes, e.g. hypertriglyceridemia or central obesity, which could mediate the underlying biological mechanisms and limit the generalizability of the findings. In this study, we present the first genome-wide association analysis of fenofibrate effects on plasma adiponectin in a population with a comparatively low prevalence of dyslipidemia, as well as provide additional insights into the observed associations using gene-based tests and biological pathway methods.
Methods
Study Population
The GOLDN study participants were recruited from the Minneapolis and Salt Lake City sites of the National Heart, Lung, and Blood Institute Family Heart Study. The population, which consisted of self-reported European American pedigrees with at least two siblings, was described in detail in previous publications (4–6). Briefly, the purpose of the study was to identify pharmacogenetic determinants of response to an intervention which included daily treatment with 160 mg of micronized fenofibrate over the period of three weeks. Exclusion criteria included abnormal liver enzyme or creatinine tests, fasting triglycerides < 1500 mg/dL, pregnancy, insulin therapy for diabetes, as well as history of liver, kidney, pancreas, or gallbladder disease. All participants took fenofibrate solely for the purpose of this investigation rather than for a pressing medical concern, and compliance was assessed using pill counts at the end of the study as well as serum measurements of fenofibric acid; overall, 96% of participants were deemed compliant with the intervention. Participants were instructed to stop the use of other lipid-lowering drugs for at least four weeks, to fast for at least eight hours, and to abstain from alcohol for at least 24 hours prior to study visits.
The GOLDN study represents a uniquely appropriate population to study the genetic determinants of fenofibrate response for the following reasons: 1) the interventional nature of the study limits environmental heterogeneity, enhancing the power to detect genetic signals; 2) the large sample size of the study yields >90% statistical power at the 0.05 alpha level given an effect size as modest as 2%; 3) all participants were comprehensively phenotyped and genotyped (see below), resulting in an unbiased approach for investigating a variety of phenotypes, including changes in circulating adiponectin; and 4) the family-based design limits the bias due to confounding by population stratification. The study protocol was approved by Institutional Review Boards at the University of Minnesota, University of Utah, and Tufts University/New England Medical Center.
Adiponectin Measurements
Plasma circulating adiponectin was measured at baseline and after fenofibrate treatment using an ELISA kit from R&D Systems (Minneapolis, MN) (3). All samples were centrifuged at 2000 × g for 15 minutes at 4 degrees C within 20 minutes of collection, stored frozen at −70 degrees C, and analyzed at the same time for each participant to eliminate inter-assay variability (5). The reliability coefficient was estimated at 0.95 based on a comparison of 58 blind replicates embedded in study samples (3).
Genotyping
DNA extraction and purification as well as genotyping quality control have been described in detail in previous GOLDN study publications (5,6). Briefly, genotyping was done using Affymetrix Genome-Wide Human 6.0 array and genotypes were called using the Birdseed algorithm for a total of 906,600 single nucleotide polymorphisms (SNPs) (5). Participants with call rates <96% were removed from the analyses (n=16) (5). SNPs were removed from the analysis if they were monomorphic (55,530), had a call rate <96% (82,462), exceeded the previously defined thresholds for the number of families with Mendelian errors (9,592), or failed the Hardy-Weinberg equilibrium test at the 10−6 significance level (748) (5). After further excluding markers with minor allele frequency (MAF) < 1%, missing strand information, or discrepancies with the mlinfo file, untyped SNPs were imputed using MACH software (Version 1.0.16, University of Michigan, Ann Arbor, MI) and the Human Genome Build 36 as the reference (5). The final hybrid dataset included 2,543,887 SNPs, of which 584,029 were initially genotyped in the GOLDN population using the Affymetrix 6.0 chip (5).
Statistical Methods
Participants were excluded from the analysis if they were missing information on adiponectin levels or any of the relevant covariates, resulting in the final sample size of 793. If any participants were missing genotype information at specific loci, they were also excluded from the respective analyses. Phenotypes were defined as circulating adiponectin levels at baseline as well as ratios of post-fenofibrate treatment/baseline concentrations. As neither measure was normally distributed, they were subjected to log-transformations. A sensitivity analysis was conducted using an alternative definition of the response outcome as the difference in post-treatment and baseline adiponectin levels, adjusted for baseline values. Confounding due to population stratification was assessed using principal component analysis (EIGENSOFT 3.0, Harvard Medical School, Boston, MA) and found to be limited in the GOLDN data (5). The first 10 principal components were retained and tested for association with the adiponectin outcomes; no statistically significant associations were found and therefore the final models were not adjusted for ancestry.
Single SNP Genome-Wide Association Study
Linear mixed models were fit to assess the relationship between the additive genotypes and both baseline and response adiponectin outcomes (kinship package, R software, Vienna, Austria). The regression models were adjusted for sex, age, and center as fixed effects, and phenotypic dependence among family members as a function of their kinship as a random effect (5). Statistical adjustment for family relatedness further minimized the threat of confounding due to population substructure. Based on prior genome-wide association studies (GWAS) in populations of European descent, the genome-wide significance level was set at 5×0−8 (7). Deviations from the expected test statistic distribution were assessed using quantile-quantile (Q-Q) plots (Supplemental Figures 1 and 2). Genomic inflation factors (λ) were estimated at 1.14 (baseline) and 1.07 (fenofibrate response). The results were visualized using genome-wide Manhattan plots.
Figure 2.
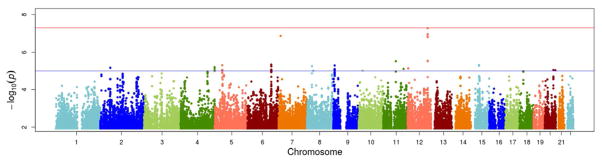
Manhattan plot of genome-wide results of testing for association between SNPs and adiponectin response to fenofibrate treatment. The X-axes display the chromosome on which the SNP is located, the Y-axes display −log10(P-value). The top horizontal line marks the genome-wide significance level (P-value= 5×10−8).
Gene-Based Association Tests
To supplement the single SNP tests, we used the VEGAS (Versatile Gene-Based Association Study) algorithm (8) to summarize evidence for association with adiponectin (baseline and response) on the gene level. VEGAS is a gene-based test, appropriate for analysis of family data, that incorporates information from all available markers within a gene and accounts for linkage disequilibrium between markers by using simulations from the multivariate normal distribution (8). For the gene-based test, statistical significance level was defined using a Bonferroni correction based on 17,787 autosomal genes, yielding α= 0.05/17,787= 2.8 × 10−6 (8). Our chosen approach to the multiple comparisons problem is likely to be conservative given the possibility of overlap between genes.
Biological Pathway Analysis
The computer program MAGENTA (Meta-Analysis Gene-set Enrichment of variaNT Association, Broad Institute, Boston, MA) was used to perform gene-set enrichment analysis. The method has been described in detail elsewhere (9). In brief, the method included four steps: first, mapping SNPs onto genes; second, scoring genes based on SNP association scores; third, correcting for six potential confounders (gene size, marker density, independent SNPs, recombination hotspots, genetic distance, linkage disequilibrium units) using the extended gene boundaries (+110 kb upstream and +40 kb downstream); fourth, gene-set enrichment analysis of GWAS data with false discovery rate < 0.05 correcting for multiple testing. In this analysis, 3,216 pathways or processes in the GO, PANTHER, INGENUITY, KEGG, REACTOME, and BIOCARTA databases were used focusing on gene-sets with at least 10 genes to obtain robustness of signals (10). At the end of this analysis, a revised false discovery rate (FDR) approach was used to flag statistical significance after correction for multiple testing in the presence of overlap of genes between gene sets and also non-independence of pathways or processes, genes, and SNPs (11).
Results
Table 1 lists the key demographic and clinical characteristics of the study population. All participants were of self-reported European ancestry and distributed evenly between the Minnesota and Utah study sites. The prevalence of dyslipidemia was low at baseline and further decreased after the fenofibrate treatment (5). Serum concentrations of circulating adiponectin also decreased from baseline to the end of the three-week follow-up period, although the decrease was not statistically significant, indicating considerable heterogeneity in fenofibrate response. Prior studies in cell culture suggest that the effect of fenofibric acid on adiponectin levels varies between individuals with and without mixed dyslipidemia, prompting additional sensitivity analyses (12). However, in our study, among subjects with baseline hypertriglyceridemia (serum triglycerides >200 mg/dL (n=156)), the levels of adiponectin were lower at baseline and also decreased from 6311 ng/mL to 6081 ng/mL following fenofibrate treatment (data not shown). The observed decrease in circulating adiponectin is likely explained by the fenofibrate-induced increase in the expression of adiponectin receptors, which in turn results in higher uptake and lower circulating levels of adiponectin (13–16); further large-scale studies are warranted to elucidate this relationship in normolipidemic individuals.
Table 1.
General characteristics of the study population (n=793).
| Variable | Mean or Median ± SD or % |
|---|---|
| Age, years | 48 ± 16 |
| Sex, % female | 50 |
| Field center, % from MN | 50 |
| Body mass index, kg/m2 | 28 ± 6 |
| Diabetes | |
| Diagnosed, % | 8 |
| Treated with medications, % | 65 |
| Hypertension | |
| Diagnosed, % | 26 |
| Treated with medications, % | 82 |
| Diagnosed with CHD, % | 5 |
| Adiponectin, ng/dL | |
| Baseline | 8123 ± 4807 |
| After fenofibrate treatment | 7770 ± 4424 |
| Glucose, mg/dL | |
| Baseline | 102 ± 20 |
| After fenofibrate treatment | 99 ± 19 |
| Insulin, mU/L | |
| Baseline | 14 ± 8 |
| After fenofibrate treatment | 13 ± 8 |
| Triglycerides, mg/dL | |
| Baseline | 141 ± 99 |
| After fenofibrate treatment | 91 ± 55 |
| HDL-C, mg/dL | |
| Baseline | 47 ± 13 |
| After fenofibrate treatment | 49 ± 13 |
| LDL-C, mg/dL | |
| Baseline | 123 ± 32 |
| After fenofibrate treatment | 105 ± 32 |
| Total cholesterol, mg/dL | |
| Baseline | 192 ± 39 |
| After fenofibrate treatment | 167 ± 35 |
CHD, coronary heart disease; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; MN, Minnesota; SD, standard deviation
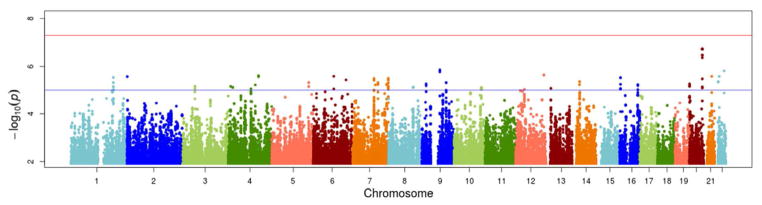
Manhattan plots summarizing the results of the genome-wide analyses for each outcome are shown in Figure 1 (baseline) and Figure 2 (response). The top loci associated with adiponectin levels at baseline and the corresponding fenofibrate response, as well as the direction and magnitude of the observed associations, are summarized in Table 2. No markers have reached genome-wide significance at baseline. However, 11 SNPs in a linkage disequilibrium block in an intergenic (closest genes: PMEPA1, PCK1, ZBP1) region of chromosome 20 showed suggestive associations with baseline adiponectin (P-values< 5 × 10−7). Other suggestive associations were observed with markers in the 12q24 region, we well as in PCSK5, SCUBE1, TMEM18, and CDH13 genes. Of those, CDH13 was found to be reproducibly associated with baseline adiponectin levels by previous GWAS studies (17–21).
Figure 1.


Manhattan plot of genome-wide results of testing for association between SNPs and baseline adiponectin levels. The X-axes display the chromosome on which the SNP is located, the Y-axes display −log10(P-value). The top horizontal line marks the genome-wide significance level (P-value= 5×10−8).
Table 2.
Top loci associated with circulating adiponectin at baseline and with the changes in adiponectin levels following fenofibrate treatment.
| SNP | Chr | Gene | β ± SE | MAFa | P-value |
|---|---|---|---|---|---|
| Baseline | |||||
| rs12481680 | 20 | Intergenic | 0.22±0.04 | 0.12 | 1.87 × 10−7 |
| rs10746997 | 9 | PCSK5 | −0.14±0.03 | 0.30 | 1.44 × 10−6 |
| rs5751452 | 22 | SCUBE1 | −0.31±0.06 | 0.06 | 1.56 × 10−6 |
| rs1716403 | 12 | Intergenic | −0.14±0.03 | 0.32 | 2.32× 10−6 |
| rs12999373 | 6 | TMEM18 | −0.14±0.03 | 0.24 | 2.71 × 10−6 |
| rs8053728 | 16 | CDH13* | −0.18±0.04 | 0.13 | 5.85 × 10−6 |
| Response | |||||
| rs2384207 | 12 | DDX54 | 0.06±0.01 | 0.18 | 5.00 × 10−8 |
| rs525304 | 11 | SHANK2 | 0.04±0.008 | 0.47 | 3.03 × 10−6 |
| rs12199015 | 6 | ENPP3 | 0.09±0.02 | 0.04 | 4.58 × 10−6 |
| rs950027 | 15 | SLC30A4 | −0.04±0.008 | 0.47 | 4.84 × 10−6 |
| rs7443270 | 5 | C6 | 0.04±0.009 | 0.31 | 4.95 × 10−6 |
| rs16925187 | 9 | KDM4C | 0.10±0.02 | 0.03 | 5.11 × 10−6 |
Within 200KB
Chr, chromosome; MAF, minor allele frequency; SNP, single-nucleotide polymorphism
The strongest evidence for association for the adiponectin response to fenofibrate was observed in the 12q24 genomic region. The rs2384207 polymorphism in that region (DDX54 gene) reached the threshold for genome-wide significance (P-value= 5 × 10−8), while 6 more variants in the adjacent TPCN1 gene all showed suggestive associations (P-values <3 × 10−6) but are not included as they are in linkage disequilibrium (r2>0.80) with the lead SNP. For the lead SNP, a likelihood-ratio based r2 measure (22) was estimated at 2.9% of outcome heterogeneity explained by variation at the rs2384207 locus. Additionally, a polymorphism on chromosome 7 (rs6954341) approached statistical significance but should be treated with extreme caution as it has a very low MAF (1%) and was not a part of a cluster of robustly associated variants; as such, it is excluded from the table. Other loci implicated in fenofibrate response were in the SHANK2, ENPP3, C6, and KDM4C genes; for the three latter genes, suggestive associations were observed with several SNPs comprising distinct linkage disequilibrium blocks.
The top findings of the gene-based test are shown in Table 3. The results were consistent with the single marker genome-wide association analysis. Baseline adiponectin was significantly associated (P-value <1.00 × 10−6) with more genetic predictors than fenofibrate response, although some of the signals overlapped (CDH13, CSMD1, RORA, A2BP1, CTNND2), likely due to the previously reported high correlations between pre- and post-fenofibrate treatment adiponectin levels (3). Additionally, the PTPRT gene, located in the same region on chromosome 20 as the top SNPs from the baseline single marker analysis, showed a significant (P-value= 2.00 × 10−6) association with the baseline outcome.
Table 3.
Top genes associated (P-value <1.00 × 10−6) with circulating adiponectin at baseline and with the changes in adiponectin levels following fenofibrate treatment.
| Chr | Gene | # SNPs |
|---|---|---|
| Baseline | ||
| 1 | BSND | 15 |
| 1 | IGSF21 | 30 |
| 1 | KIF26B | 16 |
| 1 | ZNF643 | 14 |
| 2 | LRP1B | 84 |
| 3 | CNTN4 | 21 |
| 4 | SORCS2 | 59 |
| 4 | WDR1 | 3 |
| 5 | CTNND2 | 64 |
| 7 | DYNC1I1 | 69 |
| 7 | LAMB1 | 24 |
| 7 | PTPRN2 | 27 |
| 8 | CSMD1 | 139 |
| 8 | RDH10 | 18 |
| 9 | ADAMTSL1 | 18 |
| 9 | PCSK5 | 34 |
| 10 | PRKG1 | 30 |
| 11 | NELL1 | 25 |
| 12 | IFLTD1 | 30 |
| 14 | NPAS3 | 67 |
| 15 | RORA | 26 |
| 15 | SLCO3A1 | 35 |
| 16 | A2BP1 | 64 |
| 16 | CDH13 | 112 |
| 16 | CMIP | 54 |
| 16 | KIAA0556 | 10 |
| 16 | WWOX | 77 |
| 17 | MSI2 | 35 |
| 22 | SCUBE1 | 21 |
| 22 | TTLL12 | 9 |
| Response | ||
| 1 | ESRRG | 53 |
| 3 | THRB | 45 |
| 5 | C6 | 60 |
| 5 | CTNND2 | 32 |
| 6 | ARHGAP18 | 14 |
| 6 | SYNE1 | 30 |
| 7 | DENND2A | 9 |
| 7 | MKRN1 | 9 |
| 8 | CSMD1 | 244 |
| 9 | GLIS3 | 17 |
| 11 | USP47 | 4 |
| 12 | TPCN1 | 46 |
| 15 | ALDH1A3 | 17 |
| 15 | BCL2L10 | 4 |
| 15 | LRRK1 | 4 |
| 15 | RORA | 44 |
| 16 | A2BP1 | 128 |
| 16 | CDH13 | 153 |
| 17 | SLC39A11 | 15 |
Chr, chromosome; SNP, single-nucleotide polymorphism
In the biological pathway analysis, the only borderline significant signal for the baseline outcome came from the PANTHER epidermal growth factor receptor (EGFR) pathway, comprised of 43 functionally related genes (gene set nominal P-value = 0.0009 < 0.006 after correction for multiple tests of pathways; FDR= 0.07). No significant gene enrichment was found for the response outcome (the strongest evidence was observed for the PANTHER RAS pathway, with the gene set nominal P-value of 0.0026 < 0.006 and FDR= 0.11).
Discussion
In this study, we present the first genome-wide investigation of fenofibrate effect on plasma adiponectin levels. Our analyses have identified a novel, statistically significant relationship between the change in circulating adiponectin following a three-week fenofibrate treatment and the rs2384207 variant, located in a linkage disequilibrium block in the 12q24 region. This GWAS adds to the body of evidence linking that genomic region with several metabolic phenotypes, specifically susceptibility to type 2 (23, 24) and autoimmune type 1 diabetes (25), abdominal adiposity (26, 27) and possibly circulating leptin levels (28). Furthermore, a recent meta-analysis of GWAS found robust associations of several SNPs in the 12q24.31 region and several metabolic traits including adiponectin (17). The signal in our study was observed in 12q24.13, a region spanning several genes including DDX54, TPCN1, SLC24A6, and OAS1/OAS2/OAS3. DDX54 encodes an RNA helicase that has been shown to contribute to differentiation and myelination of oligodendrocytes (29) while TPCN1 and SLC24A6 encode proteins implicated in calcium signaling (30, 31). Of utmost biological relevance, however, are the neighboring OAS1/OAS2/OAS3 genes, which code for enzymes involved in innate immunity (32) and could thus mediate the adiponectin/toll-like receptor 4 signaling pathway (33). However, previous studies reported evidence of long-range linkage disequilibrium between variants in the OAS cluster and type 1 diabetes susceptibility genes in a European population (34); it is possible that the linkage disequilibrium structure of 12q24.13 also has implications for our findings. Future studies including replications of the fenofibrate response analyses as well as fine-mapping of the 12q24 region will provide further insights in the biology of the observed association.
Although our analyses of adiponectin levels at baseline did not yield any statistically significant results, several findings that approached genome-wide significance are noteworthy. First, three loci (rs8058728, rs12921085, rs12929701) in or near the CDH13 gene showed a suggestive (P-value< 1×10−5) association with circulating adiponectin, replicating associations previously reported in several GWAS (12–16). Second, we observed suggestive signals from a linkage disequilibrium block on chromosome 20 (lead SNP: rs12481680), which contains PCK1, a gene encoding a gluconeogenic enzyme that is regulated by adiponectin signaling through the AMP-kinase pathway (35) and ZBP1, a gene coding for a DNA-binding protein shown to modulate innate immunity (36). Third, we saw suggestive associations with markers in the SCUBE1 and TMEM18 genes, which have been implicated in cardiovascular disease pathogenesis and obesity, respectively (37, 38). It is also notable that the associations with the top SNPs at baseline were characterized by greater magnitude and variability than for the response trait; also, most associations with baseline adiponectin levels were negative. Overall, these preliminary findings could inform future experiments aimed at functional validation of these biologically plausible loci.
In addition to the genome-wide signals from the single marker and gene-based studies, biological pathway analyses identified the EGFR pathway as borderline significantly associated with baseline adiponectin levels. This finding is highly biologically relevant, as prior experiments in mice show clear improvement in the metabolic profile, including increased serum adiponectin, following 14 days of administration of PD153035, an EGFR inhibitor (39). On the other hand, in vitro studies showed that adiponectin inhibits EGFR phosphorylation, heparin-binding EGF-stimulated protein synthesis, and heparin-binding EGF shedding, suggesting bidirectional cross-talk between adiponectin and EGFR signaling (40). To our knowledge, our study is the first to report this association in a human epidemiologic study; upon successful replication, these findings represent new insights into the etiologic role of adiponectin.
Several considerations are important to the interpretation of our findings. First, the GOLDN trial measured total rather than high molecular weight (HMW) adiponectin levels, which may represent the biologically active form of the protein (41). Nevertheless, prior reports have shown that measuring HMW adiponectin does not confer additional information over total adiponectin in studies of chronic disease, perhaps due to the high correlation between the phenotypes (42, 43). Second, although our baseline findings are consistent with other genome-wide studies, the fenofibrate response results have not been replicated. This challenge is common to pharmacogenetic studies, as very few cohorts are comparable on both drug intervention design, genotyping approach, and phenotype ascertainment (5). Furthermore, the Q-Q plots suggest slight genomic inflation (λ=1.07 for the fenofibrate response models), exacerbating the need for independent replication of our findings. As such, these results are to be considered preliminary, but informative of future studies of fenofibrate pharmacogenetics. Third, the homogeneity of our study population limits the generalizability of these findings, prompting the need for investigating genetic determinants of fenofibrate response in ethnically diverse samples in future studies. Fourth, the VEGAS gene-based test only estimates an empirical P-value without giving information on the direction of the observed association, and it is likely that the overall effect of a gene on a trait is determined by its constituent SNPs, some of which may have opposing effects. In light of this limitation, it is important to interpret our findings in context of all three statistical approaches, namely the single marker, gene-based, and biological pathway analyses. Finally, the presented results appear to be inconsistent with the moderate to high estimates of heritability of adiponectin response to fenofibrate (3). One possible explanation lies in the GWAS method itself, as it excludes such sources of “missing heritability” as rare functional variants, epigenetic traits, or gene-gene interactions. Another possibility, however, is that the estimate of heritability is too high as it erroneously includes variance due to shared environment, which could not be ascertained in the GOLDN study because household information was not available (3).
In conclusion, we have found preliminary evidence for association for several biologically relevant genetic loci with changes in plasma adiponectin following fenofibrate therapy. Upon successful validation, these findings will advance current understanding of biologic pathways underlying adipokine response to fenofibrate therapy.
Supplementary Material
Acknowledgments
source of funding: This work has been funded by the NHLBI grant U01HL072524-04.
Footnotes
Conflicts of interest: No authors declare conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem. 2004;50:1511–1525. doi: 10.1373/clinchem.2004.032482. [DOI] [PubMed] [Google Scholar]
- 2.Han SH, Quon MJ, Kim J, Koh KK. Adiponectin and cardiovascular disease: response to therapeutic interventions. J Am Coll Cardiol. 2007;49:531–538. doi: 10.1016/j.jacc.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 3.Rasmussen-Torvik LJ, Pankow JS, Peacock JM, Borecki IB, Hixson JE, Tsai MY, et al. Suggestion for linkage of chromosome 1p35. 2 and 3q28 to plasma adiponectin concentrations in the GOLDN study. BMC Med Genet. 2009;10:39. doi: 10.1186/1471-2350-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, et al. The -256T>C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem. 2007;53:1144–1152. doi: 10.1373/clinchem.2006.084863. [DOI] [PubMed] [Google Scholar]
- 5.Aslibekyan S, Kabagambe EK, Irvin MR, Straka RJ, Borecki IB, Tiwari HK, et al. A genome-wide association study of inflammatory biomarker changes in response to fenofibrate treatment in the Genetics of Lipid Lowering Drug and Diet Network. Pharmacogenet Genomics. 2012;22:191–197. doi: 10.1097/FPC.0b013e32834fdd41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irvin MR, Kabagambe EK, Tiwari HK, Parnell LD, Straka RJ, Tsai M, et al. Apolipoprotein E polymorphisms and postprandial triglyceridemia before and after fenofibrate treatment in the Genetics of Lipid Lowering Drug and Diet Network (GOLDN) Study. Circ Cardiovasc Genet. 2010;3:462–467. doi: 10.1161/CIRCGENETICS.110.950667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–359. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 8.Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. AMFS Investigators. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87 doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segre AV, Groop L, Mootha VK, Daly MJ, Altshuler D DIAGRAM Consortium, MAGIC investigators. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6:e1001058. doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–1188. [Google Scholar]
- 12.Labuzek K, Buldak L, Dulawa-Buldak A, Bielecka A, Krysiak R, Madej A, et al. Atorvastatin and fenofibric acid differentially affect the release of adipokines in the visceral and subcutaneous cultures of adipocytes that were obtained from patients with or without mixed dyslipidemia. Pharmacol Rep. 2011;63:1124–1136. doi: 10.1016/s1734-1140(11)70631-4. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes. 2005;54:3358–3370. doi: 10.2337/diabetes.54.12.3358. [DOI] [PubMed] [Google Scholar]
- 14.Civitarese AE, Jenkinson CP, Richardson D, Bajaj M, Cusi K, Kashyap S, et al. Adiponectin receptors gene expression and insulin sensitivity in non-diabetic Mexican Americans with or without a family history of Type 2 diabetes. Diabetologia. 2004;47:816–820. doi: 10.1007/s00125-004-1359-x. [DOI] [PubMed] [Google Scholar]
- 15.Debard C, Laville M, Berbe V, Loizon E, Guillet C, Morio-Liondore B, Boirie Y, Vidal H. Expression of key genes of fatty acid oxidation, including adiponectin receptors, in skeletal muscle of Type 2 diabetic patients. Diabetologia. 2004;47:917–925. doi: 10.1007/s00125-004-1394-7. [DOI] [PubMed] [Google Scholar]
- 16.Kaltenbach S, Staiger H, Weisser M, Haas C, Stumvoll M, Machicao F, et al. Adiponectin receptor gene expression in human skeletal muscle cells is not regulated by fibrates and thiazolidinediones. Int J Obes (Lond) 2005;29:760–765. doi: 10.1038/sj.ijo.0802957. [DOI] [PubMed] [Google Scholar]
- 17.Dastani Z, Hivert MF, Timpson N, Perry JRB, Yuan X, Scott RA, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 8(3):e1002607. doi: 10.1371/journal.pgen.1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morisaki H, Yamanaka I, Iwai N, Miyamoto Y, Kokubo Y, Okamura T, et al. CDH13 gene coding T-cadherin influences variations in plasma adiponectin levels in the Japanese population. Hum Mutat. 2012;33:402–410. doi: 10.1002/humu.21652. [DOI] [PubMed] [Google Scholar]
- 19.Chung CM, Lin TH, Chen JW, Leu HB, Yang HC, Ho HY, et al. A genome-wide association study reveals a quantitative trait locus of adiponectin on CDH13 that predicts cardiometabolic outcomes. Diabetes. 2011;60:2417–2423. doi: 10.2337/db10-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jee SH, Sull JW, Lee JE, Shin C, Park J, Kimm H, et al. Adiponectin concentrations: a genome-wide association study. Am J Hum Genet. 2010;87:545–552. doi: 10.1016/j.ajhg.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Li Y, Lange EM, Croteau-Chonka DC, Kuzawa CW, McDade TW, et al. Genome-wide association study for adiponectin levels in Filipino women identifies CDH13 and a novel uncommon haplotype at KNG1-ADIPOQ. Hum Mol Genet. 2010;19:4955–4964. doi: 10.1093/hmg/ddq423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun G, Zhu C, Kramer MH, Yang SS, Song W, Piepho HP, et al. Variation explained in mixed-model association mapping. Heredity. 2010;105:333–340. doi: 10.1038/hdy.2010.11. [DOI] [PubMed] [Google Scholar]
- 23.Ehm MG, Kkarnoub MC, Sakul H, Gottschalk K, Holt DC, Weber JL, et al. the American Diabetes Association GENNID Study Group. Genome-wide search for type 2 diabetes susceptibility genes in four American populations. Am J Hum Genet. 2000;66:1871–1881. doi: 10.1086/302950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindgren CM, Mahtani MM, Widen E, McCarthy MI, Daly MJ, Kirby A, et al. Genomewide search for type 2 diabetes mellitus susceptibility loci in Finnish families: the Botnia study. Am J Hum Genet. 2002;70:502–516. doi: 10.1086/338629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Genetics of Type 1 Diabetes in Finland, Wellcome Trust Case Control Consortium. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perusse L, Rice T, Chagnon YC, Despres JP, Lemieux S, Roy S, et al. A genome-wide scan for abdominal fat assessed by computer tomography in the Quebec Family Study. Diabetes. 2001;50:614–621. doi: 10.2337/diabetes.50.3.614. [DOI] [PubMed] [Google Scholar]
- 27.Wilson SG, Adam G, Langdown M, Reneland R, Braun R, Andrew T, et al. Linkage and potential association of obesity-related phenotypes with two genes on chromosome 12q24 in a female dizygous twin cohort. Eur J Hum Genet. 2006;14:340–348. doi: 10.1038/sj.ejhg.5201551. [DOI] [PubMed] [Google Scholar]
- 28.Dai F, Keighley ED, Sun G, Indugula SR, Roberts ST, Aberg K, et al. Genome-wide scan for adiposity-related phenotypes in adults from American Samoa. Int J Obes (Lond) 2007;31:1832–1842. doi: 10.1038/sj.ijo.0803675. [DOI] [PubMed] [Google Scholar]
- 29.Ueki T, Tsuruo Y, Yamamoto Y, Yoshimura K, Takanaga H, Seiwa C, et al. A new monoclonal antibody, 4F2, specific for the oligodendroglial cell lineage, recognizes ATP-dependent RNA helicase Ddx54: possible associations with myelin basic protein. J Neurosci Res. 2012;90:48–59. doi: 10.1002/jnr.22736. [DOI] [PubMed] [Google Scholar]
- 30.Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palty R, Ohana E, Herschfinkel M, Volokita M, Elgazar V, Beharier O, et al. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279:25234–25240. doi: 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- 32.Field LL, Bonnevie-Nielsen V, Pociot F, Lu S, Nielsen TB, Beck-Nielsen H. OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes. 2005;54:1588–1591. doi: 10.2337/diabetes.54.5.1588. [DOI] [PubMed] [Google Scholar]
- 33.Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011;286:13460–13469. doi: 10.1074/jbc.M110.204644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu HQ. Polychronakos C; Type 1 Diabetes Genetics Consortium. Reassessment of the type 1 diabetes association of the OAS1 locus. Genes Immun. 2009;(Suppl 1):S69–73. doi: 10.1038/gene.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadowaki T, Yamauchi T, Kubota N. The physiological and pathophysiological role of adiponectin and adiponectin receptors in the peripheral tissues and CNS. FEBS Lett. 2008;582:74–80. doi: 10.1016/j.febslet.2007.11.070. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type 1 interferon production. Immunity. 2011;35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu CF, Su YH, Huang YN, Tsai MT, Li LT, Chen YL, et al. Localization and characterization of a novel secreted protein SCUBE1 in human platelets. Cardiovasc Res. 2006;71:486–495. doi: 10.1016/j.cardiores.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Mei H, Chen W, Jiang F, He J, Srinivasan S, Smith EN, et al. Longitudinal replication studies of GWAS risk SNPs influencing body mass index over the course of childhood and adulthood. PLoS One. 2012;7:e31470. doi: 10.1371/journal.pone.0031470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prada PO, Ropelle ER, Mourao RH, de Souza CT, Pauli JR, Cintra DE, et al. EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet-fed mice. Diabetes. 2009;58:2910–2919. doi: 10.2337/db08-0506. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Liao Y, Xuan W, Zhao J, Bin J, Zhao H, Asakura M, et al. Antihypertrophic effects of adiponectin on cardiomyocytes are associated with the inhibition of heparin-binding epidermal growth factor signaling. Biochem Biophys Res Commun. 2010;393:519–525. doi: 10.1016/j.bbrc.2010.02.039. [DOI] [PubMed] [Google Scholar]
- 41.Tonelli J, Li W, Kishore P, Pajvani UB, Kwon E, Weaver C, et al. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes. 2004;53:1621–1629. doi: 10.2337/diabetes.53.6.1621. [DOI] [PubMed] [Google Scholar]
- 42.Glintborg D, Frystyk J, Hojlund K, Andersen KK, Henriksen JE, Hermann AP, et al. Total and high molecular weight (HMW) adiponectin levels and measures of glucose and lipid metabolism following pioglitazone treatment in a randomized placebo-controlled study in polycystic ovary syndrome. Clin Endocrinol (Oxf) 2008;68:165–174. doi: 10.1111/j.1365-2265.2007.03015.x. [DOI] [PubMed] [Google Scholar]
- 43.Almeda-Valdes P, Cuevas-Ramos D, Mehta R, Gomez-Perez FJ, Cruz-Bautista I, Arellano-Campos O, et al. Total and high molecular weight adiponectin have similar utility for the identification of insulin resistance. Cardiovasc Diabetol. 2010;9:26. doi: 10.1186/1475-2840-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.