

Abstract
Toll/IL-1R domain- (TIR-) containing adapter inducing IFN-β-(TRIF-) related adapter molecule (TRAM) serves as a bridging adapter that enables recruitment of TRIF to activated TLR4 and thereby mediates the induction of TRIF-dependent cytokines. A library of cell-permeating decoy peptides derived from TRAM TIR domain has been screened for the ability of individual peptides to inhibit TLR4 signaling in primary murine macrophages. Peptides derived from TRAM TIR BB loop (TM4) and C helix (TM6) inhibited the LPS-induced activation of MyD88-dependent and TRIF-dependent cytokines, as well as MAPK activation. TM4 and TM6 did not block macrophage activation induced by TLR2, TLR9, or RIG-I-like receptor agonists. Both TM4 and TM6 blocked co-immunoprecipitation of TRAM and TLR4 ectopically expressed in HEK293T cells. Both peptides also blocked the LPS-induced recruitment of MyD88 to TLR4 in primary murine macrophages. In vivo examination of TRAM-derived peptides demonstrated that all peptides that were inhibitory in vitro, profoundly suppressed systemic inflammatory response elicited in mice by a sublethal LPS dose, and protected mice against a lethal LPS challenge. This research identifies novel TLR inhibitors effective in vitro and in vivo and validates the approach taken in this study as a rational way for development of signaling inhibitors and lead therapeutics.
Introduction
Activation of a Toll-like Receptor (TLR) by a TLR agonist induces recruitment of Toll/IL1R domain- (TIR-) containing adapter proteins. Four adapter proteins participate in TLR4 signaling: MyD88 (1), TIRAP/Mal (2, 3); TRIF (4); and TRAM (5). A targeted mutation of MyD88 or TRIF results in activation of distinct set of genes in response to TLR4 stimulation (6, 7); whereas a knockout of MyD88 or TIRAP affects TLR4 responses similarly (7, 8). Similarly to the MyD88 and TIRAP adapter pair, targeted mutations of TRIF or TRAM produce a similar phenotype in mice (6, 9). Activated TLR4 dimerizes TIR domains of two receptor molecules and recruits one or two distinct pairs of adapter proteins, TIRAP and MyD88, or TRAM and TRIF. TIRAP and TRAM have been referred to as “sorting” or “bridging” adapters, as these adapters are directly engaged by the receptor. Recruitment of a bridging adapter stabilizes the receptor dimer and allows for recruitment of a signaling adapter, MyD88 or TRIF. MyD88 and TIRAP mediate rapid activation of NF-κB and mitogen-activated protein kinases (MAPKs) and induce MyD88-dependent cytokines, such as TNF-α and IL-1β (10). TRIF and TRAM activate a different signaling pathway that leads to activation of IFN regulatory factor 3 (IRF3) and IRF3-dependent genes, such as IFN-β or RANTES (5, 11). TRAM is required for recruitment of TRIF to endosomal TLR4 and activation of the TRIF-dependent TLR4 signaling (12).
The common structural feature of TLRs and TLR adapters is the TIR domain. The TIR domain is an interaction domain that mediates transient homotypic or heterotypic interactions of signaling proteins that contain TIR domains, thus enabling the formation of signaling complexes (13–15). Multiple interactions of TIR domains of TLRs and their adapters are pivotal in the early stages of TLR signaling as these interactions mediate adapter recruitment and thereby stabilize the receptor dimer (16–18). Although the critical role the TIR domains play in formation of initial signaling complex is commonly accepted, the architecture of the initial signaling complexes assembled after TLR activation remains to be clarified. It has been proposed that adapter recruitment is achieved through a cooperative interaction of several TIR domains in which the TIR of a recruited protein binds two (or more) TIR domains of an initial complex simultaneously (15–18). It also has been hypothesized that TLR4 activation leads to formation of several compositionally distinct complexes; e.g., Kagan et al. proposed that TLR4 engages MyD88 and TRIF sequentially and at distinct cellular locations (12), thus implying that one docking site in the TLR4 TIR might be sufficient for recruitment of several adapters. We and other groups hypothesized that TIRAP and TRAM share the same binding site at the TLR4 homodimer (15, 16, 19). However, it is still unclear which structural regions of TIRAP and TRAM mediate interaction with TLR4. A typical TIR domain consists of the central five-stranded parallel β-sheet (designated as βA-βE) surrounded by five α-helices (αA-αE). Available crystallographic and functional data suggest that TIR domains interact through topologically diverse structural regions (14–18). It has been proposed that TLR10 TIR domain dimerizes through interaction between BB loop and αC helix (20), whereas TLR4 TIR homodimerizes through interaction of BB loop with E helix (18, 21); while TLR1/TLR2 heterodimer is formed by interaction of TLR1 BB loop and TLR2 DD loop (22).
The mechanism by which a decoy peptide inhibits signaling is presumed to be that the peptide blocks the docking site of its prototype protein and thereby prevents a functional protein-protein interaction (17). Therefore, inhibition of signaling by a decoy peptide often indicates that the inhibitory peptide represents a functional protein interface. In this study we have screened a library of cell-permeable decoy peptides derived from the TRAM TIR for the ability of individual peptides to inhibit TLR4 signaling in vitro and identified two peptides that potently inhibit LPS signaling. One inhibitory peptide, TM4, represents the BB loop of TRAM TIR; the other, TM6, was derived from the third helical region of the TIR. Both peptides effectively inhibit induction of both MyD88-dependent and TRIF-dependent cytokines and activation of MAPKs by LPS. In accordance with these functional data, both peptides prevented co-immunoprecipitation of TLR4 with TRAM or MyD88. TM4 and TM6 and a truncated version of TM4, TM4-ΔC, effectively inhibit TLR4-driven inflammatory response in mice. All inhibitory TRAM peptides significantly diminished circulating cytokine levels induced by a sublethal dose of LPS, and dramatically improved survival of mice challenged with a lethal LPS dose.
Materials and Methods
Animals, Cell Culture and Treatment
C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, Maine). Harvesting, culturing, and stimulation of peritoneal macrophages were described in our previous publications (17–19). Escherichia coli K235 LPS (23) was used at the final concentration of 100 ng/ml. P3C and P2C (EMC microcollections GmbH, Tübingen, Germany) were used at 500 and 50 μg/ml, respectively. ODN 1668 and low molecular weight poly(I:C) in complex with LyoVec™ were obtained from InvivoGen; these agonists were used at 2.5 μM and 400 ng/ml, respectively.
Peptide Synthesis and Reconstitution
Peptides were obtained through the Biopolymer and Genomics Core Facility (UMB). All lyophilized peptides were of more than 95% purity. Peptide stocks were quantified by spectrophotometry (24) after reconstitution and stored at −80 C. Peptide sequences are shown in Table 1.
Table 1.
Sequences of decoy peptides.
| Peptide name | Peptide sequence | Structural region |
|---|---|---|
| TM1 | GAGAEEQDEEEFLK | βAa |
| TM2 | AEDDTDEALRVQDL | AA & αA |
| TM3 | QNDFGIRPG | αA, AB & BB |
| TM4b | IVFAEMPCGRLHLQ | BB, & αB |
| TM4-N | IVFAE | BB |
| TM4-M | EMPCG | BB |
| TM4-C | RLHLQ | αB |
| TM4-ΔN | MPCGRLHLQ | BB, & αB |
| TM4-ΔM | IVFARLHLQ | BB, & αB |
| TM4-ΔC | IVFAEMPCG | BB |
| TM4-E/A | IVFAAMPCGRLHLQ | BB, & αB |
| TM4-P/H | IVFAEMHCGRLHLQ | BB, & αB |
| TM4-C/H | IVFAEMPHGRLHLQ | BB, & αB |
| TM5 | NLDDAVNGSAWT | αB, BC, βC, & CC |
| TM6 | ENFLRDTWCNFQFY | αC & CD |
| TM7 | TSLMNSVSRQHKYNS | CD |
| TM8 | RPLNSPLPRE | CD, βD & DD |
| TM8/9 | PRERTPLALQTINA | DD, DE, & βE |
| TM9 | RTPLALQTINA | DD, DE, & βE |
| TM9/10 | QTINALEEES | βE & EE |
| TM10 | LEEESQGFSTQVE | βE, EE, & αE |
| TM11 | RIFRESVFERQQS | αE |
| CP | SLHGRGDPMEAFII | Randomized sequence |
- Structural regions of TIR domain are designated as follows: helices are designated by Greek α, f.e., αA – helix A; strands are indicated by Greek β; loops are indicated by two capital letters, f.e., AA – loop that connects strand A and helix A.
- This peptide was previously described by authors as TRAM-BP (19).
Expression vectors
The full length murine TRAM or TRIF TIR N-terminally tagged with hemagglutinin (HA) or Flag tag was cloned into the pEF-BOS vector. TLR4-Cer expression vectors was described earlier (18).
Cytokine ELISA
Two × 106 cells were plated in 12 well plates and treated with peptides for 30 min prior to LPS stimulation. TNF-α, IL-1β, IL-6, and IFN-β were measured using ELISA kits from Biolegend (San Diego, CA) as recommended by the manufacturer.
Immunoblotting and co-immunoprecipitation
HEK293T cells were transfected with indicated plasmids using Superfect Transfection Reagent (Qiagen, CA). Twenty four h post-transfection, the cells were lysed using the buffer containing 20 mM Hepes (pH 7.4), 150 mM NaCl, 10 mM NaF, 2 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 0.5% Triton X-100, 0.1 M DTT, 1 mM PMSF and protease inhibitor cocktail (Roche, Indianapolis, IN). Rabbit antibody against phospho-ERK, phospho-JNK, MyD88 (D80F5), STAT1-Y701, total STAT1, and GAPDH were purchased from Cell Signaling Technology (Belverly, MA). Rabbit anti-phospho-p38 was purchased from Promega (Madison, WI). Goat-anti-TLR4 (M-16), rabbit anti-TLR4 (H-80), anti-HA-HRP (F-7), and rabbit anti-β-actin were purchased from Santa Cruz Biotechnology. Mouse anti-Flag M2 and rabbit anti-HA antibody were from Sigma-Aldrich. GFP antibody (A1122) was from Invitrogen. Protein was quantified using Bio-rad protein quantification kit. Equal amount of protein was loaded and analyzed by 10% SDS-PAGE and immunoblotting. The cell extracts containing equal amount of protein were incubated with 1 μg of antibody for 2 hours, followed by 4 hour incubation with 25 μl protein G agarose beads (Roche), the beads were then washed 4 times with lysis buffer, boiled in 1x sample loading buffer, and analyzed by immunoblotting.
Quantitative real-time RT-PCR
Total RNA was isolated with Nucleospin RNA II kits (Macherey-Nagel, Inc. Bethlehem, PA) followed by DNase digestion. cDNA was synthesized from 1 μg of RNA using Goscript transcriptase (Promega), and subjected to real-time PCR with gene-specific primers for HPRT, TNF-α, IL-1β, IFN-β, RANTES on H7900 ABI system using Fast SYBR®Green master mix (Applied Biosystems, Foster City, CA).
Cell Viability analysis
Five ×104 mouse peritoneal macrophages were plated into 96-well tissue culture plates, incubated overnight, and treated with peptides for 3 h. After the treatment, cells were incubated with MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) at 0.5 mg/ml (Sigma-Aldrich) for 3 h. Fifty μl DMSO was added to cells before reading OD at 540 nm.
TRAM TIR model
The coordinate file for human TRAM TIR was kindly provided by Dr. Wei (25) (Shandong University, China). The model was visualized and analyzed using DeepView/Swiss-pdbViewer (Swiss Institute of Bioinformatics, Switzerland).
Animal experiments
Eight week old C57BL/6J mice were injected with 1 or 17.5 μg/g of animal weight of Escherichia coli K235 LPS i.p. Peptides reconstituted in PBS were administered i.p. or i.v. Control groups received equivalent volume of solvent (PBS). The blood samples were collected 1, 2, 4 and 8 h, or 2, 8, and 24 h after LPS challenge. The plasma samples were obtained and kept frozen until TNF-α and IL-6 were measured by ELISA. Survival of animals was monitored every 6 to 16 hours after LPS challenge. All animal experiments were carried out with institutional approval.
Results
Identification of inhibitory peptides
TRAM TIR decoy peptides were designed similarly to the libraries of TLR4- or TIRAP-derived peptides described previously (18, 26). Each TRAM peptide represents a structural element(s) that comprise a non-fragmented patch of TIR surface with all peptides collectively encompassing the TIR domain. The sequences of the TRAM-derived decoy peptides and the corresponding structural regions are provided in Table 1. The cell-permeating 16 amino acid-long sequence of Antennapedia homeodomain (RQIKIWFQNRRMKWKK) (27) was placed at the N terminus of each decoy sequence to render peptides cell-permeable. Two additional peptides, TM8/9 and TM9/10, that represent the border area between regions 8 and 9, and 9 and 10, respectively, were additionally tested.
To evaluate peptide inhibitory activities we first measured LPS-induced cytokine mRNA expression. Murine macrophages were pretreated with peptides (5 or 20 μM) for 30 min and then the cells were stimulated with LPS (100 ng/mL) for 1 hour. Two peptides, TM4 and TM6, potently inhibited the TLR4-mediated cytokine mRNA transcription (Fig. 1A, 1B). Decoy peptide TM4 is derived from the BB loop of TRAM TIR; we previously identified this peptide as an effective TLR4 inhibitor (19). TM4, even at the low dose of 5 μM, significantly decreased mRNA expression of all cytokines examined (Fig. 1A). Another TRAM-derived peptide that strongly inhibited mRNA transcription is TM6. TM6 effectively blocked induction of IFN-β and RANTES, two TRIF-dependent genes, at both concentrations used (Fig. 1A). However, the effect of TM6 at 5 μM on the MyD88-dependent cytokines, IL-1β and TNF-α, was not statistically significant. Both TM4 and TM6 potently inhibited expression of all cytokine genes at 20 μM (Fig. 1A).
FIGURE 1.

TRAM decoy peptides derived from BB loop and third helical region, TM4 and TM6, inhibit LPS-induced cytokine mRNA and activation of MAPKs and STAT1. Mouse peritoneal macrophages were pre-incubated with 5 or 20 μM (A, B) of control or decoy peptides for 30 min prior to stimulation with LPS (100 ng/ml). Cells were lysed 1 (panel A) or 5 h (panel B) after LPS challenge. Cytokine mRNA expression is normalized to the expression of the housekeeping gene, Hprt. Data represent the means ± s.e.m. of 4 independent experiments. Asterisks indicate data statistically different from the control group (p < 0.001). Cell lysates for immunoblotting was obtained 30 min (C) or 2 h (D) after LPS stimulation. GAPDH was used as a loading control. Panels C and D show a representative blot of 4 separate experiments.
To determine the duration of the inhibitory effect preliminary, we measured cytokine expression after 5 hour incubation. Two cytokines, RANTES and IL-6, have been selected for this test because these cytokines are strongly expressed at this time point, whereas expression of TNF-α and IFN-β is more transient and significantly decreased 5 hour post-stimulation (not shown). Pretreatment of cells with 20 μM of TM4 or TM6, but not with other peptides, profoundly suppressed expression of both cytokines, even at this late time point (Fig. 1B).
We next examined whether TRAM peptides inhibit the TLR4-mediated p38, ERK, and JNK MAPK activation. Murine macrophages pretreated with peptides for 30 minutes were challenged with LPS. Cell lysates were collected 30 minutes post-stimulation and analyzed for MAPK phosphorylation by Western analysis. Consistent with peptide effects on steady-state mRNA, both TM4 and TM6 inhibited activation of all three MAPKs (Fig. 1C). The inhibition of MAPKs by TM4 or TM6 was long-lasting; it persisted for at least 1 hour after LPS stimulation (Supplemental Fig. 1A). To characterize further the effects of TRAM peptides on MAPKs, murine macrophages were treated with TRAM peptides, but not challenged by LPS. Without LPS stimulation, TM3 weakly activated p38 and ERK MAPK, whereas effect of other peptides was insignificant compared to the LPS-induced activation (Supplemental Fig. 1B).
We previously demonstrated that LPS stimulation leads to STAT-1-Y701 phosphorylation through autocrine activation of type I IFN receptor by IFN-β (28). Figure 1D confirms this observation and shows that both TM4 and TM6 block LPS-induced STAT-1-Y701 phosphorylation. Inhibition of STAT1 phosphorylation is a consequence of the strong effect of both peptides on IFN-β transcription (Fig. 1A).
We next measured the effect of peptides on TNF-α, IL-6, IL-1β, IFN-γ, RANTES and IFN-β secretion. Macrophage supernatants were collected 24 h after LPS stimulation and cytokine contents measured by ELISA. Figure 2 shows that both TM4 and TM6 exert a strong and lasting inhibitory effect on secretion of all six cytokines examined (Fig. 2).
FIGURE 2.

TM4 and TM6 inhibit LPS-induced cytokine secretion. Mouse peritoneal macrophages were incubated in the presence of 5 or 20 μM of indicated TRAM peptide for 30 min prior to stimulation with LPS (100 ng/ml). Supernatants were collected 24 h later and analyzed for TNF-α (A), IL-6 (B), IL-1β (C), IFN-γ (D), RANTES (E) and IFN-β (F) contents. Data represent means ± s.e.m. of 3 independent experiments. * - p < 0.01. b.d. - below detection limit.
TM8, TM9, and TM10 peptides are derived from consecutive segments of TRAM primary sequence and represent adjacent areas on the TRAM TIR surface. To exclude the possibility of missing an inhibitory sequence due to arbitrary fragmentation of a peptide, we designed two additional peptides TM8/9 and TM9/10 that overlaps with TM8 and TM9, and TM9 and TM10, respectively (Table 1). Neither TM8/9 nor TM9/10 inhibited the TLR4 signaling significantly (Supplemental Fig. 1D).
TRAM is an adapter protein selectively involved in TLR4 signaling (5, 9). We next studied if the effects of TM4 and TM6 are specific and examined peptide effects on TLR2, TLR9, and RIG-I/MDA-5 pathway. Mouse macrophages were stimulated with 500 ng/ml of P3C or 50 ng/ml of P2C, agonists that activate signaling through TLR2/TLR1 or TLR2/TLR6 heterodimers, respectively. In sharp contrast with effect of these two decoy peptides on TLR4 signaling, TM4 and TM6 (20 μM) did not inhibit P3C- or P2C-induced p38, ERK, or JNK MAPK phosphorylation (Fig. 3A), or IL-1β and TNF-α mRNA induction (Fig. 3B). Neither peptide blocked cell activation induced by TLR9 or RIG-I/MDA-5 agonists, CpG oligonucleotide ODN 1668 or intracellularly delivered poly(I:C). The STAT1 Y701 residue was phosphorylated in macrophages in response to both agonists regardless the presence of TRAM peptides, albeit the STAT1 activation by poly(I:C) occurred at later time points (Fig. 3C). TRAM peptides did not affect the induction of IFN-β mRNA statistically significantly (Fig. 3D).
FIGURE 3.

TM4 and TM6 block TLR4, but not TLR2 (A and B), TLR9, or RIG-I-like receptor (C and D) signaling. Peptides derived from region 6 of TRAM and TIRAP TIR are strong inhibitors of TLR4-induced MAPKs activation and cytokine transcription (E, F). Peptides were used at 20 μM. P2C and P3C was used at 50 and 500 ng/ml respectively. Macrophages were treated with ODN 1668 at 2.5 μM or low molecular weight poly(I:C) in complex with LyoVec™ at 400 ng/ml. MAPK phosphorylation was measured in whole cell lysates collected 30 min after stimulation with LPS or P3C (A). To measure STAT1-Y701 phosphorylation cells were stimulated with ODN 1668 for 5 h or with poly(I:C) in complex with LyoVec™ for 16 h (C). IFN-β mRNA induction by TLR9 and RIG-I-like receptor agonists was measured 5 h after stimulation (D). Other experimental details are as in Figure 1. Data in panel A and E represent 3 independent experiments. Means ± s.e.m. of 3 independent experiments are shown in panels B, D, and F. * p < 0.01. (G) The residues that are conserved in TRAM and TIRAP peptides are underlined.
Peptide effects on cell viability were evaluated using the MTT test. Neither inhibitory peptide affected cell viability in a significant way, even after 3 h incubation in the presence of TM4 or TM6 (Supplemental Fig. 1C).
Inhibitory activities of peptides derived from the third helical region of TLR adapters
We observed previously that peptides derived from the BB loop of TLRs and TLR adapters differ markedly in ability to inhibit TLR4 (19, 29). Our new data show that, in addition to the TRAM BB loop peptide, peptide derived from the third helical region of TRAM, TM6, strongly inhibits TLR4 signaling (Figs. 1, 2). Interestingly, the sequence of the region represented by TM6, is highly similar to the sequence of the structurally homologous region of TIRAP/Mal ((26) and Fig. 3G). TIRAP/Mal peptide derived from this region, TR6, inhibited both TLR4 and TLR2 signaling (26). We next compared inhibitory efficiency of peptides derived from the homologous structural region of other TLR4 adapters. Both TRAM and TIRAP region 6 peptides, TM6 and TR6, potently inhibited the LPS-induced activation of p38 and ERK MAPKs (Fig. 3E). These peptides also blocked transcriptional activation of IL-1β and IFN-β genes (Fig. 3F). In contrast, the inhibitory effects of peptides derived from homologous region of TRIF and MyD88 TIR were less (Fig. 3E, 3F), especially on MAPK activation. Notably, region 6 in TRAM and TIRAP is the most similar region in these proteins. TIRAP and TRAM region 6 sequences are PGFLRDPWCKYQML and ENFLRDTWCNFQFY, respectively, and have 7 identical amino acids (underlined). This is by margin the highest degree of local sequence conservancy that has been found in all four TLR4 adapters. Such a high degree of local conservancy of the surface exposed residues strongly suggests that this region is functionally important. It is noteworthy that there is no notable similarity in the homologous regions of MyD88 and TRIF (Fig. 3G).
Inhibitory properties of truncated TM4 variants
TM4 is a very efficient overall inhibitor of TLR4 (Fig. 1 and our previous publications (17, 19)). We next sought to identify amino acids that are critical for TM4’s inhibitory effects. We used three groups of modified peptides to address this question. First, three short peptides each of which contained only 5 amino acids of TM4 were examined. TM4-N contains five N-terminal amino acids of TM4; TM4-M contains the TM4 middle 5 amino acid section; and TM4-C contains the 5 C-terminal amino acids (Sequences are shown in Table 1). The second group is comprised of three modified TM4 peptides that lack TM4-N, TM4-M, and TM4-C; these peptides are designated as TM4-ΔN, TM4-ΔM, and TM4-ΔC, respectively (Table 1). Finally, we tested three peptides, TM4-E/A, TM4-P/H, and TM4-C/H, in which a single amino acid has been replaced (sequences are shown in Table 1).
Among three short peptides, TM4-N was the strongest inhibitor. TM4-M demonstrated intermediate inhibition, whereas TM4-C was least active (Fig. 4A, 4B and Supplemental Fig. 2A). Consistently with these data, TM4-ΔN, peptide that lacks the N-terminus of TM4, was least inhibitory; whereas deletion of the TM4 C terminus did not affect peptide inhibitory potency significantly (Fig. 4C, 4D and Supplemental Fig. 2B). The inhibitory efficiency of TM4-ΔC is close to that of TM4. TM-ΔM also retains some inhibitory properties.
FIGURE 4.

Inhibitory activities of truncated TM4 variants. (A–D). The experimental details are as described in Figure 1. Peptide sequences are shown in Table 1. Data in panels a and c represent 4 independent experiments; panels B and D show means ± s.e.m. of duplicate samples obtained in 3 independent experiments. * p < 0.01.
All three single amino acid replacements affected TM4 properties weakly. TM4-E/A, TM4-P/H, and TM4-C/H inhibited TLR4-induced MAPKs activation (Fig. 4A) and cytokine expression (Fig. 4B), although the inhibitory efficacy of TM4-C/H was less than that observed for other two modified peptides. Collectively, these data demonstrate that the five N-terminal amino acid of TM4 peptide together with its middle segment are more important for TLR4 inhibition.
TM4 and TM6 block adapter recruitment to TLR4
We used co-immunoprecipitation assays to study peptide effects on TLR adapter recruitment. To study effect of peptides on TRAM recruitment, HEK293T cells were co-transfected with mouse TLR4 tagged with Cerulean fluorescent protein (TLR4-Cer) (18) and HA-tagged mouse TRAM (HA-TRAM). TM4 or TM6, but not a non-inhibitory TM3 peptide, blocked TLR4/TRAM co-immunoprecipitation (Fig. 5A). Interestingly, TR6, a TIRAP-derived inhibitory peptide that has sequence similar to TM6, also inhibited TLR4/TRAM association, although less efficiently than TM6 (Fig. 5A).
FIGURE 5.
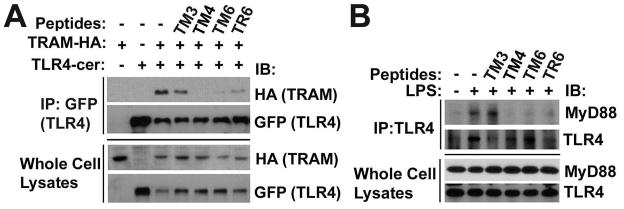
TM4, TM6 and TR6 block adapter recruitment to TLR4. (A) Six x 105 HEK293T cells were transfected with 1 μg pcDNA3.1-mTLR4-Cer and pEF-BOS-mTRAM-HA. Twenty four h post-transfection the cells were treated with 20 μM of each peptide for 1 hour. Cell lysates were immunoprecipitated with anti-GFP antibody and blotted with anti-HA-HRP. (B) Two × 106 mouse peritoneal macrophages were pretreated with 20 μM of each peptide for 30 min prior to stimulation with 100 ng/ml LPS. Cell extracts were obtained 30 min after LPS treatment, immunoprecipitated with goat anti-TLR4 Ab and blotted with rabbit anti-MyD88 or anti-TLR4 Ab. Data are representative of 3 independent experiments.
To study peptide effects on MyD88 recruitment, we used primary mouse peritoneal macrophages. In this cellular model, MyD88 co-immunoprecipitates with TLR4 in the agonist-dependent manner (30) (Fig. 5B). All inhibitory peptides tested, TM4, TM6, and TR6, efficiently prevented the LPS-induced TLR4/MyD88 association (Fig. 5B). TIRAP/Mal is necessary for MyD88 recruitment to TLR4 (3), whereas TRAM recruits TRIF and is not required for MyD88 recruitment and activation of MyD88-dependent genes (11). Yet, TIRAP- and TRAM-derived peptides cross-reacted in the sense that each peptide blocked the recruitment of both adapters, TRAM and MyD88. This observation agrees fully with the fact that the adapter-derived peptides are equally effective inhibitors of MyD88- or TRIF-dependent cytokines (Figs. 1, 2, and reference (26)). Collectively, these results suggest that TM4 and TM6 disrupt the assembly of TLR4 signaling complexes by blocking the TIR:TIR interactions required for adapter recruitment, possibly mediated by a common binding site for recruitment of TIRAP and TRAM to TLR4 TIR dimer.
Inhibitory peptides blunt LPS-induced cytokine response in mice
We next studied whether peptides identified as inhibitory in in vitro tests are effective in vivo. C57BL/6J mice were challenged i.p. with a sublethal LPS dose (1 μg/g of animal weight) and plasma TNF-α and IL-6 levels were monitored for 8 hours after the challenge. In the first series of experiments, we injected 10 nmol/g of TM4 or TM6 i.p. 1 hour before LPS challenge. The 10 nmol/g dose corresponds to 38, 41, and 32 mg/kg of TM4, TM6, and TM4-ΔC, respectively. Pretreatment of mice with either inhibitory peptide dramatically decreased systemic IL-6 and TNF-α levels measured 2 hours after LPS challenge; after the treatment, the plasma cytokine levels were only 5 - 15 % of cytokine levels induced in the PBS-treated group (Fig. 6A). The effect was lasting; TM4 and TM6 significantly decreased circulating levels of both cytokines at every time point throughout the observation period (Fig. 6B).
FIGURE 6.

TM4, TM6 and TM4-ΔC effectively suppress LPS-induced cytokine induction in vivo (A–E) and protect mice from lethal endotoxemia (F, G). (A–E) C57BL/6J mice were injected i.p. with purified Escherichia coli K235 LPS (1 μg/g) or PBS. Peptides (10 nmol/g of animal weight, i.e., 38, 41, 40, and 32 mg/kg for TM4, TM6, TR6, and TM4-ΔC, respectively) were injected i.p. (A, B, D, E) or i.v. (C) 1 h before (A, B, and C) or 30 min after (D, E) i.p. injection of LPS. Data in panels A and D show plasma IL-6 and TNF-α levels measured 2 h after LPS challenge. Peptide TM8/9 was used as a non-inhibitory control peptide. Data represent the means ± s.e.m. for 6–12 blood samples obtained in at least three independent experiments. * p < 0.01. (F, G) C57BL/6J mice were challenged i.p. with a lethal LPS dose (17.5 μg/g). Peptides (10 nmol/g) were injected i.p. 1 h before LPS challenge. Panel G also shows survival in the group treated with TM4-ΔC 3 h after LPS challenge (n=7). The statistical significance of changes in mortality and survival time was determined by the Mantel Cox log-rank test using GraphPad Prism software (Version 5.04). * p < 0.01.
Optimization of peptide dose
When used at a lower dose of 2.5 nmol/g, TM4 appears to affect the circulating TNF-α equally strongly as it does at 10 nmol/g (Supplemental Fig. 3A). However, the lower dose was less efficacious against the circulating IL-6 levels, especially at the later time points (Supplemental Fig. 3C). A similar decrease in the TM6 dose resulted in a less efficient inhibition of both cytokines (Supplemental Figs. 3B, 3D). Interestingly, these results, obtained in in vivo experiments, parallel very closely the effects of low peptide doses on macrophage expression of cytokine mRNA (Fig. 1A, 1B). Increasing the peptide dose to 25 nmol/g did not augment the efficiency of TM4 and TM4-ΔC with respect to circulating IL-6 at 4 or 8 hour time points (Supplemental Figs. 3C, 3C). Based on these observations, the dose of 10 nmol/g was chosen for all subsequent experiments.
Effect of injection route
Efficient cellular uptake of cell-permeating peptides is well established for monolayers of cells in culture; however, the tissue permeability of peptides is expected to be lower. Diminished peptide permeability through the tissues of the body might limit their inhibitory efficacy in vivo. Results of experiments shown in Figure 6B demonstrate that peptides effectively mitigate the TLR4-driven inflammatory symptoms when administered via the same route as the TLR agonist, i.e., i.p. To understand if peptides might be effective as systemic inhibitors of TLR4, we used separate routes of administration for the TLR4 agonist and TLR4 antagonists in the next series of experiments. Peptides (10 nmol/g) were injected i.v.; LPS was administered i.p. Intravenous injection of TM4 or TM4-ΔC significantly lowered the levels of circulating IL-6 and TNF-α throughout the 8 hour observation period following the LPS injection (Fig. 6C). However, the inhibitory effect of i.v. injection was less compared to that after i.p. administration of an inhibitory peptide (Fig. 6C, 6B, respectively). Although a statistically significant difference in cytokine levels after i.p. versus i.v. treatment was detected only for the effect of TM4 on IL-6 4 hours after LPS injection (p<0.01, not shown); for all other time points, the p values of the difference between the untreated and i.v.-treated group were lower than between the untreated and i.p.-treated animals for both TM4 and TM4-ΔC peptides (Fig. 6B, 6C, and data not shown). These data show that the i.v. injection of decoy peptide is less effective compared to i.p. administration in this model of inflammation; nevertheless, the results indicate that the peptides are efficient systemic TLR inhibitors.
Effects of timing of peptide treatment
TLR activation launches a complex chain of signaling events that ultimately results in production of multiple cytokines. Secreted cytokines act back on cells and activate a multitude of different signaling pathways, many of which help to sustain inflammation. In our previously described experiments, mice were pretreated with inhibitory peptides before activation of TLR signaling by LPS. We next wanted to determine if the peptides administered after LPS are still effective for mitigation of TLR4-driven inflammatory symptoms. In this series of experiment, mice were first challenged i.p. with LPS (1μg/g); peptides were injected at the dose of 10 nmol/g i.p. 30 minutes after LPS. The administration of inhibitory peptides 30 min after LPS did not affect levels of circulating TNF-α and IL-6 measured 1 h after LPS administration (i.e., 30 min after administration of peptides) (Fig. 6E). However, 2 hours after LPS challenge, i.e., 1.5 hour after injection of peptides, the plasma levels of both cytokines were markedly diminished in mice treated with any of the three inhibitory peptides tested, TM4, TM6, or TM-ΔC (Fig. 6D, 6E). The observed diminution in the cytokine levels at 2, 4, and 8 hours after LPS was less than that in the experiments when mice were pretreated with inhibitory peptides (Fig. 6A, 6B, 6D, 6E). TM8/9, peptide that did not inhibit macrophage signaling in vitro, did not affect the LPS-induced cytokines in vivo two hours after LPS challenge or thereafter (Fig. 6E). Our data suggest that both prophylactic and therapeutic administration of peptides mitigate the inflammatory response to LPS.
Inhibitory peptides protect mice from a lethal LPS challenge
TM4, TM6, and TM4-ΔC strongly suppress systemic cytokine response induced by a sublethal LPS dose. We next studied if inhibitory peptides protect mice from a lethal LPS challenge. TR6, a previously described inhibitory peptide derived from the third helical region of TIRAP (26), was also included in this study as an additional control. C57BL/6J mice were injected i.p. with 17.5μg/g of LPS. This dose induced 100% mortality in the control group (Fig. 6F). Pretreatment of mice with 10 nmol/g of TM4 or TM4-ΔC rescued all animals from the lethal LPS dose (Fig. 6F). TM6 also dramatically improves survival compared to the untreated group; however, three of 13 animals died after LPS challenge in this group. TR6, an inhibitory TIRAP peptide that share significant sequence similarity with TM6, improved animal survival comparably to TM6 (Fig. 6F). In an additional control series of experiments, we used peptide TM8/9 as this peptide did not inhibit TLR4 in in vitro tests. Although we had one survivor in the group of seven animals injected with 17.5 μg/g of LPS following pretreatment with TM8/9, the difference in survival rate was highly significant between each of the control groups and the TM4-, TM6-, or TM4-ΔC-treated group (p<0.0012, for all pairs). The peptide-treated survivors appeared active and healthy at the time the experiments were terminated, 7 days after injection of LPS.
We next studied if the TRAM decoy peptides are effective when administered therapeutically, i.e., in our model of the disease, after injection of LPS. TM4-ΔC was chosen for these tests. The peptide was administered i.p. 3 hours after i.p. injection of LPS. Five of seven animals survived the lethal LPS challenge in this group, whereas no animals survived in the untreated group (0/12) while all animals (10/10) survived in the group pretreated with TM4-ΔC (Fig. 6G). These data demonstrate that inhibitory peptides have strong potential as therapeutics, because the difference in survival rate between the untreated and the group treated with TM4-ΔC therapeutically is highly significant (p=0.009 as determined by the Gehan-Breslow-Wilcoxon test). The results presented strongly suggest that select decoy peptides effectively lessen the systemic inflammatory response induced by activation of TLR4 and, therefore, might be effective for treatment of septic shock.
Discussion
Homotypic or heterotypic interactions of TIR domains mediate the early stages of TLR signaling complex assembly: agonist-induced dimerization of receptor TIR domains followed by recruitment of TIR-containing adapters (13, 15, 16, 31). Blockage of any interface involved in the formation of the initial complex is predicted to abolish the TLR signaling because the molecular interactions that underlie the initial steps of TLR signaling complex assembly are highly cooperative (18). Therefore, molecular tools capable of blocking a specific TIR:TIR interaction potentially have a significant therapeutic value because excessive TLR signaling is a pathogenic mechanism in many inflammatory diseases. We previously screened libraries of decoy peptides derived from TIR domains of TLR4 and TIRAP/Mal (18, 26). This study expands our previous work and tests peptides derived from TRAM. Although previous studies have identified a number of TLR4 inhibitory peptides (18, 19, 26, 29, 32, 33), due to diversity of sequences that define the surface exposed area of TIR domains (13, 34), identification of novel peptides would be valuable as it may yield insight into mechanisms of TLR complex assembly and recognize novel TLR targets and TLR-targeting agents. The library of TRAM peptides was designed similarly to TLR4 and TIRAP sets; each TRAM peptide represents a non-fragmented patch of the TRAM TIR surface with all peptides of the library encompassing the domain. The approach taken in these 3 studies has been highly successful in that that a unique set of inhibitory peptides that block TLR4 signaling at low micromolar concentration has been identified in each TIR-specific library (18, 19, 26). In general, the sequences of identified inhibitory peptides are dissimilar, as are the surface-exposed residues of TIR domains. TIRAP and TRAM peptides derived from the third helical region, designated as TR6 (26) and TM6, were the most similar among identified inhibitors with 50% amino acid homology. In each set of the TIR peptides tested, the inhibitory peptides represent different structural regions of the TIRs, as illustrated by the observation that no TIR structural region would consistently produce an inhibitory decoy in all three TIR domains studied.
Two TRAM-derived peptides, TM4 and TM6, potently inhibit TLR4 signaling. TM4 is derived from the BB loop, while TM6 represents the short CC loop, together with the third helical region of the domain. TM4 and TM6 regions are located in close proximity on the TIR surface, although they are not juxtaposed (Fig. 7). The sufficient separation between the areas, especially if the C terminal part of TM4 is excluded, together with significant curvature of this part of the TIR (Fig. 7A), suggests that these regions interact with different TIR domains in the receptor/adapter complex.
FIGURE 7.
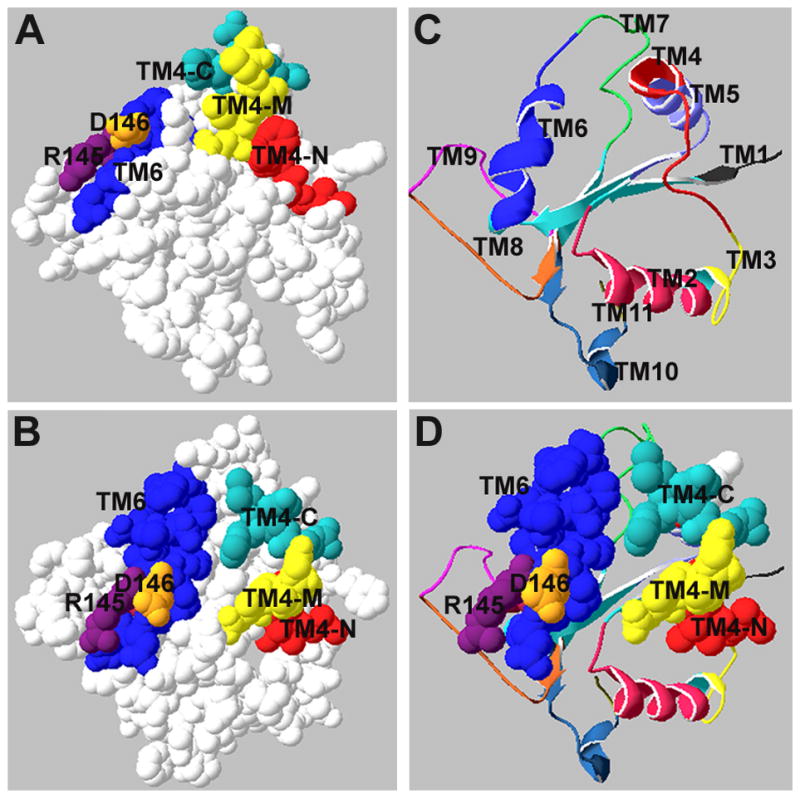
Relative positions of TRAM TIR surface regions represented by TM4 and TM6. Regions represented by TRAM peptides that potently inhibit TLR4 signaling are highlighted in color (A and B). (C) Ribbon representation of TRAM backbone demonstrates relative positions of 11 TRAM decoy peptides. Images were produced using the DeepView viewer.
Both peptides potently inhibit all manifestations of TLR4 activation we examined in primary macrophages: the cytokine gene expression and secretion and activation of MAPKs and STAT-1 were all potently inhibited. Importantly, the TRAM peptides block not only the induction of cytokines traditionally associated with the TRIF-dependent signaling pathway, e.g., IFN-β, RANTES, and IL-6 (9), but also the MyD88-dependent cytokines, e.g., TNF-α and IL-1β. This finding parallels and expands the result of screenings of TLR4- and TIRAP-derived peptides, that also have not identified an inhibitory peptide that would selectively block the expression of MyD88-dependent or TRIF-dependent cytokines (18, 26). This consistent pattern of specificity likely indicates that all inhibitory peptides identified in these screenings target the very early, cooperative stages of the assembly of multi-TIR complex and that the adapter recruitment interfaces overlap significantly. Co-immunoprecipitation assays further confirmed that the inhibitory peptides block recruitment of adapters to TLR4. Importantly, each peptide tested, including TR6, a TIRAP-derived peptide that is similar to TM6, blocked recruitment of both adapters, TRAM and MyD88. This observation is also consistent with the absence of specificity of any of these peptides with respect to blockage of MyD88-dependent or TRIF-dependent cytokines. Interestingly, VIPER, a TLR4 inhibitory peptide derived from viral protein A46 (33), demonstrates similar pattern of specificity as it blocks all TLR4 signaling by disrupting both TLR4:TIRAP/Mal and TLR4:TRAM interactions (35). Although effects of VIPER on TLR4 are similar to the effect of TRAM peptides, sequences of these peptides are dissimilar. Further studies are required to understand whether VIPER and TRAM peptides affect TLR4 via similar mechanism.
The TRAM peptides exerted a lasting inhibitory effect. A significant decrease in cytokine content was detected in macrophage supernatants collected 24 hours post-stimulation for every cytokine measured (Fig. 2). This level of stability is sufficient for both tested peptides to mitigate efficiently the systemic LPS-induced inflammatory response in vivo after a single-dose treatment.
While TM4 and TM6 failed to inhibit MyD88-dependent and TRIF-dependent pathways selectively, they exhibited specificity for TLR4 as neither peptide blocked TLR2-, TLR9-, and RIG-I-like receptor-mediated activation of cytokine transcription and protein phosphorylation (Fig. 3A–D). This finding demonstrates that TRAM peptides are specific inhibitors of TLR4. However, considering that TR6, a TIRAP-derived peptide that is similar to TM6, inhibits both TLR4 and TLR2 (26), it is difficult to interpret the observed pattern of TM6 specificity at this time. Small differences in the affinity of TR6 and TM6 peptides to their specific TIR target(s), together with differences in unspecific peptide binding to the entire cell proteome, could account for this difference in specificity.
It has been previously reported that the replacement of P116 or C117 by histidine negatively affects the function of the full length TRAM (5, 11). Interestingly, replacement of same residues in the context of the TM4 peptide affects the peptide inhibitory activity weakly. Both TM4-P/H and TM4-C/H inhibit LPS-induced cytokines and MAPKs quite potently (Fig. 4A, 4B). This observation suggests that while introduction of a larger residue into an interface of a folded protein is likely to prevent binding by causing a steric hindrance, a similar replacement introduced into a peptide may not be as critical for a peptide/protein binding due to the higher conformational flexibility of the peptide. The fact that all 3 single amino acid replacements did not strongly affect the TM4 inhibitory efficiency also suggests that a sequence of several amino acids determines the binding behavior of a peptide, while the contribution of a single amino acid is often minor. Unlike a single amino acid replacement, deletion of 5 N-terminal amino acids markedly diminished inhibition with TM4 peptide, whereas deletion of 5 C-terminal amino acids had a minimal impact on inhibitory efficiency. Both TM4 and its abridged version, TM4-ΔC, potently inhibited the TLR4-driven response in vitro and in vivo. The shorter peptide would be predicted to have a higher permeability. Yet, even in vivo tests did not reveal a statistically significant difference in the efficiency of these peptides, although slightly better inhibition of IL-6 at later time points was observed in the experiments when the peptides and LPS were injected to animals via different routes (Fig. 6C, upper panel).
This study investigated the inhibitory properties of TIR-derived decoy peptides in vivo in much more detail than our previous study (26). The most exciting result of the in vivo examination of TRAM peptides is that the inhibitory peptides are very good candidate therapeutics for mitigation of TLR4-elicited systemic inflammation. Each TLR4 inhibitory peptide tested effectively lowered the systemic cytokine levels over a relatively long period of time after a single injection. The peptides were effective systemic TLR inhibitors; this is demonstrated by the observation that the significant inhibition of TLR4 signaling was achieved even when the peptides and LPS were injected to animals via different routes. All TLR4 inhibitory TRAM peptides performed very well in survival tests; select peptides rescued 100% of animals injected with an LPS dose that caused 100% mortality in the untreated group.
In summary, this study identifies TRAM-derived decoy peptides that effectively block TLR4 signaling in vitro and in vivo. The results of peptide screening also suggest that TRAM BB loop and the N-terminal half of the helical region between the third and fourth β-strand of TRAM TIR are protein interfaces important for binding of TRAM to TLR4 signaling complex and activation of the receptor. Finally, this work validates the approach taken in this and our two prior studies (18, 26) as a rational way to develop novel signaling inhibitors and lead therapeutics.
Supplementary Material
Acknowledgments
Authors thank Wendy Lai for ELISA measurement of IFN-β. Patent pending.
This work was supported by NIH grants AI-082299 (VYT) and AI-018797 (SNV).
Abbreviations used in this paper
- CP
control peptide
- ODN
oligonucleotide
- P2C
S-[2,3-bis(palmitoyloxy)-(2-RS)-propyl]-[R]-Cys-Ser-Lys4-OH
- P3C
2,3-bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-Ser-Lys4-OH
- poly(I:C)
polyinosinic-polycytidylic acid
- RIG-I
retinoic acid-inducible gene 1
- TIR
Toll/Interleukin-1 Receptor domain
- TIRAP/Mal or TIRAP
TIR domain-containing adapter protein, also known as MyD88-adapter-like
- TRAM
TRIF-related adapter molecule, also known as TICAM-2
- TRIF
TIR domain-containing adapter-inducing interferon-β
- VIPER
viral inhibitory peptide of TLR4
References
- 1.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Molecular cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 2.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O’Neill LA. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 8.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 10.Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J Biol Chem. 2003;278:49751–49762. doi: 10.1074/jbc.M305820200. [DOI] [PubMed] [Google Scholar]
- 12.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 14.Dunne A, Ejdeback M, Ludidi PL, O’Neill LA, Gay NJ. Structural complementarity of Toll/interleukin-1 receptor domains in Toll-like receptors and the adaptors Mal and MyD88. J Biol Chem. 2003;278:41443–41451. doi: 10.1074/jbc.M301742200. [DOI] [PubMed] [Google Scholar]
- 15.Nunez Miguel R, Wong J, Westoll JF, Brooks HJ, O’Neill LA, Gay NJ, Bryant CE, Monie TP. A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE. 2007;2:e788. doi: 10.1371/journal.pone.0000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monie TP, Moncrieffe MC, Gay NJ. Structure and regulation of cytoplasmic adapter proteins involved in innate immune signaling. Immunol Rev. 2009;227:161–175. doi: 10.1111/j.1600-065X.2008.00735.x. [DOI] [PubMed] [Google Scholar]
- 17.Toshchakov VY, Vogel SN. Cell-penetrating TIR BB loop decoy peptides a novel class of TLR signaling inhibitors and a tool to study topology of TIR-TIR interactions. Expert Opin Biol Ther. 2007;7:1035–1050. doi: 10.1517/14712598.7.7.1035. [DOI] [PubMed] [Google Scholar]
- 18.Toshchakov VY, Szmacinski H, Couture LA, Lakowicz JR, Vogel SN. Targeting TLR4 signaling by TLR4 Toll/IL-1 receptor domain-derived decoy peptides: identification of the TLR4 Toll/IL-1 receptor domain dimerization interface. J Immunol. 2011;186:4819–4827. doi: 10.4049/jimmunol.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toshchakov VU, Basu S, Fenton MJ, Vogel SN. Differential involvement of BB loops of toll-IL-1 resistance (TIR) domain-containing adapter proteins in TLR4- versus TLR2-mediated signal transduction. J Immunol. 2005;175:494–500. doi: 10.4049/jimmunol.175.1.494. [DOI] [PubMed] [Google Scholar]
- 20.Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund P. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283:11861–11865. doi: 10.1074/jbc.C800001200. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Z, Georgel P, Li C, Choe J, Crozat K, Rutschmann S, Du X, Bigby T, Mudd S, Sovath S, Wilson IA, Olson A, Beutler B. Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc Natl Acad Sci U S A. 2006;103:10961–10966. doi: 10.1073/pnas.0603804103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gautam JK, Ashish, Comeau LD, Krueger JK, Smith MF., Jr Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J Biol Chem. 2006;281:30132–30142. doi: 10.1074/jbc.M602057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu B, Xin B, Jin M, Wei T, Bai Z. Comparative and phylogenetic analyses of three TIR domain-containing adaptors in metazoans: implications for evolution of TLR signaling pathways. Developmental and comparative immunology. 2011;35:764–773. doi: 10.1016/j.dci.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Couture LA, Piao W, Ru LW, Vogel SN, Toshchakov VY. Targeting Toll-like Receptor (TLR) Signaling by Toll/Interleukin-1 Receptor (TIR) Domain-containing Adapter Protein/MyD88 Adapter-like (TIRAP/Mal)-derived Decoy Peptides. J Biol Chem. 2012;287:24641–24648. doi: 10.1074/jbc.M112.360925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 28.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 29.Toshchakov VY, Fenton MJ, Vogel SN. Cutting Edge: Differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J Immunol. 2007;178:2655–2660. doi: 10.4049/jimmunol.178.5.2655. [DOI] [PubMed] [Google Scholar]
- 30.Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, Wahl LM, Fenton MJ, Vogel SN. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci U S A. 2009;106:10260–10265. doi: 10.1073/pnas.0812956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy SL, Kurtz SE, Macarthur CJ, Trune DR, Hefeneider SH. Identification of a peptide derived from vaccinia virus A52R protein that inhibits cytokine secretion in response to TLR-dependent signaling and reduces in vivo bacterial-induced inflammation. J Immunol. 2005;174:3006–3014. doi: 10.4049/jimmunol.174.5.3006. [DOI] [PubMed] [Google Scholar]
- 33.Lysakova-Devine T, Keogh B, Harrington B, Nagpal K, Halle A, Golenbock DT, Monie T, Bowie AG. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J Immunol. 2010;185:4261–4271. doi: 10.4049/jimmunol.1002013. [DOI] [PubMed] [Google Scholar]
- 34.Slack JL, Schooley K, Bonnert TP, Mitcham JL, Qwarnstrom EE, Sims JE, Dower SK. Identification of two major sites in the type I interleukin-1 receptor cytoplasmic region responsible for coupling to pro-inflammatory signaling pathways. J Biol Chem. 2000;275:4670–4678. doi: 10.1074/jbc.275.7.4670. [DOI] [PubMed] [Google Scholar]
- 35.Stack J, Bowie AG. Poxviral protein A46 antagonizes Toll-like receptor 4 signaling by targeting BB loop motifs in Toll-IL-1 receptor adaptor proteins to disrupt receptor:adaptor interactions. J Biol Chem. 2012;287:22672–22682. doi: 10.1074/jbc.M112.349225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.