

SUMMARY
Recent genome wide association studies have identified CLU, CR1, ABCA7 BIN1, PICALM and MS4A6A/MS4A6E in addition to the long established APOE, as loci for Alzheimer’s disease. We have systematically examined each of these loci to assess whether common coding variability contributes to the risk of disease. We have also assessed the regional expression of all the genes in the brain and whether there is evidence of an eQTL explaining the risk. In agreement with other studies we find that coding variability may explain the ABCA7 association, but common coding variability does not explain any of the other loci. We were not able to show that any of the loci had eQTLs within the power of this study. Furthermore the regional expression of each of the loci did not match the pattern of brain regional distribution in Alzheimer pathology.
Although these results are mainly negative, they allow us to start defining more realistic alternative approaches to determine the role of all the genetic loci involved in Alzheimer’s disease.
Keywords: Alzheimer’s disease, genetic risk, GWAS
INTRODUCTION
The recent application of genome wide association studies (GWAS) to the dissection of the risk for late onset Alzheimer’s disease (AD) has proved an outstanding success and has led to the identification of many new loci (CLU, PICALM, CR1, BIN1, MS4A6A/MS4A4E, CD33, CD2AP, ABCA7 and EPHA1) in addition to the long established apolipoprotein E locus (Harold et al., 2009, Lambert et al., 2009, Hollingworth et al., 2011, Naj et al., 2011). When such loci are identified, they simply appear as single nucleotide polymorphisms (SNPs), which have significantly different frequencies between cases and controls. It is not initially clear whether these risk SNPs are in linkage disequilibrium (LD) with coding changes or have an impact on gene expression. For all traits studied by GWAS only ~12% of the associated SNPs are located in, or occur in high LD with, protein coding regions of genes. The vast majority (~80%) of trait associated SNPs are located in intergenic regions or noncoding introns (Manolio, 2010). Alzheimer’s disease is no different: taking into account the 21 SNPs reported in the nine new loci by genome wide association studies assessing over 1500 cases and 1500 controls (see Supplementary Table 1 for details on the SNPs), 10 are located in intergenic regions; eight in intronic regions; one SNP is located in the 3’UTR of MS4A6A; and two SNPs are located in exons (one SNP is a non-synonymous variant in ABCA7 - Gly1527Ala and one synonymous variant was found as significant in PICALM). These findings clearly indicate that follow up studies should not only examine coding variability, but should also pay close attention to the potential roles of these intronic and intergenic regions in the regulation of gene expression (Hardy and Singleton, 2009, Manolio, 2010, Myers et al., 2007). In fact, for any disease associated SNP the true variant underlying the phenotype studied may be: 1) the GWAS hit itself; 2) a known common SNP in LD with the identified GWAS hit; 3) an unknown common SNP or rare single nucleotide variant tagged by a haplotype on which the hit occurs; or, 4) a linked copy number variant (Hindorff et al., 2009). In general, GWAS follow up studies rely on fine mapping of the associated locus or loci, deep resequencing of the associated region(s) in samples of interest (which allows the identification of all possible functional variants) and a variety of bioinformatic approaches to prioritize variants to be further studied (Stranger et al., 2011).
Confirmed functional variants underlying validated GWAS hits are still sparse in the literature, when considering all the diseases and traits studied, but each of these is extremely valuable to the respective research and clinical environments. For example, the IRF5 locus includes variants that disrupt intron splicing, decrease mRNA transcript stability, and delete part of the interferon regulating factor protein (Graham et al., 2007), explaining the independent associations of this locus with three different phenotypes: systemic lupus erythmatosis (Graham et al., 2006, Sigurdsson et al., 2005), inflammatory bowel disease (Dideberg et al., 2007), and rheumatoid arthritis (Stahl et al., 2010). Similarly, allele-specific chromatin remodelling affecting the expression of several genes in the ORMDL3 locus region (Verlaan et al., 2009) explains its association with asthma (Moffatt et al., 2007), Crohn’s disease (Barrett et al., 2008), and type 1 diabetes (Barrett et al., 2009). With this in mind we have undertaken an analysis of the recently identified AD risk loci with three components: (1) we have assessed by sequencing whether there is coding variability in linkage disequilibrium with the associated SNPs (2) we have assessed in a database of control human cerebral cortex samples whether the SNPs are associated with genetic variability in expression (3) we have assessed the regional distribution of expression and splicing of the genes at the risk loci to see whether this distribution is in any way consistent with the distribution of pathology in the disease.
MATERIALS and METHODS
GENOTYPING ANALYSIS
Samples
The 96 DNA samples selected for genotyping were previously used in a GWAS in AD (Corneveaux et al., 2010). These 96 Alzheimer disease samples were diagnosed according to the NINDS-ADRDA diagnostic criteria for Alzheimer disease, consisting of 67 females and 29 males with a mean age of 81 years (range 66–95) and mean age at onset of 71.9 years (ranging from 65 to 85 years).
SNPs studied
The GWAS SNPs studied were those found to be significantly associated with late onset Alzheimer’s disease (LOAD) by two recent studies: (Hollingworth et al., 2011, Corneveaux et al., 2010). For a complete list of SNPs analysed in the present study please refer to Table 1.
Table 1.
Features and minor allele frequencies of all SNPs studied here, including the GWAS hits.
| SNP | Gene/Locus | Chr | Chr position - Genome build 37.1 |
Chr position - Genome build 36.3 |
Category | mRNA Accession number |
SNP class | Ancestral allele |
MAF in our study |
1000 genomes MAF |
|---|---|---|---|---|---|---|---|---|---|---|
| rs4844600 | CR1 | 1 | 207679307 | 205745930 | Targeted | NM_000651.4 | Trialllelic SNP: cds-synonymous (Glu60Gu) or missense (Glu60Asp) | G | A=0.23 | A=0.209/263 |
| rs6656401 | CR1 | 1 | 207692049 | 205758672 | AD Risk - C | NM_000651.4 | Intronic | G | A=0.18 | A=0.107/135 |
| rs3737002 | CR1 | 1 | 207760773 | 205827396 | Targeted | NM_000651.4 | Missense (Thr1858Met) | C | T=0.28 | T=0.268/337 |
| rs17259045 | CR1 | 1 | 207782707 | 205849330 | Targeted | NM_000651.4 | Missense (Ans1990Ser) | A | G=0.09 | G=0.041/51 |
| rs17047661 | CR1 | 1 | 207782889 | 205849512 | Targeted | NM_000651.4 | Missense (Arg2051Gly) | A | G=0 | G=0.242/219 |
| rs4844609 | CR1 | 1 | 207782916 | 205849539 | Found | NM_000651.4 | Missense (Thr2060Ser) | T | A=0.02 | A=0.010/12 |
| rs6691117 | CR1 | 1 | 207782931 | 205849554 | Targeted | NM_000651.4 | Missense (Ile2065Val) | G | G=0.22 | G=0.382/481 |
| rs3818361 | CR1 | 1 | 207784968 | 205851591 | AD Risk - H | NM_000651.4 | Intronic | C | A=0.01 | A=0.301/379 |
| rs3811381 | CR1 | 1 | 207790088 | 205856711 | Targeted | NM_000651.4 | Missense (Pro2277Arg) | G | G=0.21 | G=0.165/208 |
| rs2296160 | CR1 | 1 | 207795320 | 205861943 | Targeted | NM_000651.4 | Triallelic SNP: missense Thr2419Ala or Thr2419Ser | C | A=0.2 | A=0.233/293 |
| rs76037557 | BIN1 | 2 | 127815174 | 127531644 | Targeted | NM_139351.1 | Intronic | G | 0 | NA |
| rs744373 | BIN1 | 2 | 127894615 | 127611085 | AD Risk - H | NA | 5' near BIN1 | T | G=0.38 | G=0.380/478 |
| rs79741566 | MS4A2 | 11 | 59857929 | 59614505 | Targeted | NM_001142303.1 | Missense (Trp103Gly) | T | 0 | NA |
| rs535630 | MS4A2 | 11 | 59861532 | 59618108 | Targeted | NM_000139.3 | Missense (Asn211Lys) | G | A=0 | A=0.086/30 |
| rs610932 | MS4A6A | 11 | 59939307 | 59695883 | AD Risk - H | NM_152852.1 | 3'UTR | A | T=0.39 | T=0.455/572 |
| rs7232 | MS4A6A | 11 | 59940599 | 59697175 | Targeted | NM_152852.1 | Missense (Thr185Ser) | T | A=0.33 | A=0.203/255 |
| rs670139 | MS4A4E | 11 | 59971795 | 59728371 | AD Risk - H | NA | 3' near MS4A4E region | C | T=0.46 | T=0.387/487 |
| rs10750931 | MS4A4A | 11 | 60059810 | 59816386 | Targeted | NM_148975.1 | Missense (Lys52Glu) | A | G=0 | G=0.139/303 |
| rs6591561 | MS4A4A | 11 | 60070176 | 59826752 | Targeted | NM_148975.1 | Missense (Met178Val) | A | G=0.28 | G=0.323/705 |
| rs74727972 | PICALM | 11 | 85723420 | 85401068 | Targeted | NM_007166.2 | Missense (Leu188Ile) | G | 0 | NA |
| rs541458 | PICALM | 11 | 85788351 | 85465999 | AD Risk - C | NA | 5' of PICALM region | T | C=0.29 | C=0.376/473 |
| rs3764645 | ABCA7 | 19 | 1042809 | 993809 | Targeted | NM_019112.3 | Missense (Glu188Gly) | A | G=0.46 | G=0.370/466 |
| rs72973581 | ABCA7 | 19 | 1043103 | 994103 | Targeted | NM_019112.3 | Missense (Gly215Ser) | G | A=0.04 | A=0.020/25 |
| rs3752232 | ABCA7 | 19 | 1043748 | 994748 | Targeted | NM_019112.3 | Missense (Thr319Ala) | G | G=0.01 | G=0.103/130 |
| rs4622634 | ABCA7 | 19 | 1043864 | 994864 | Found | NM_019112.3 | Intronic | G | C=0.47 | C=0.336/423 |
| rs3764647 | ABCA7 | 19 | 1044712 | 995712 | Targeted | NM_019112.3 | Missense (His395Arg) | G | G=0.03 | G=0.099/125 |
| rs3764648 | ABCA7 | 19 | 1044753 | 995753 | Found | NM_019112.3 | Intronic | C | T=0.35 | T=0.358/450 |
| rs3752233 | ABCA7 | 19 | 1045173 | 996173 | Targeted | NM_019112.3 | Missense (Arg463His) | G | A=0.03 | A=0.080/74 |
| rs3764650 | ABCA7 | 19 | 1046520 | 997520 | AD Risk - H | NM_019112.3 | Intronic | T | G=0.09 | G=0.207/261 |
| rs3745842 | ABCA7 | 19 | 1055191 | 1006191 | Targeted | NM_019112.3 | Missense (Arg1349Gln) | C | A=0.41 | A=0.401/504 |
| rs3752246 | ABCA7 | 19 | 1056492 | 1007492 | AD Risk - H | NM_019112.3 | Missense (Gly1527Ala) | C | G=0.19 | G=0.188/236 |
| rs4147918 | ABCA7 | 19 | 1058176 | 1009176 | Targeted | NM_019112.3 | Missense (Gln1686Arg) | A | G=0.03 | G=0.053/67 |
| rs4147934 | ABCA7 | 19 | 1065018 | 1016018 | Targeted | NM_019112.3 | Missense (Ala2045Ser) | G | G=0.27 | G=0.387/487 |
| rs4147935 | ABCA7 | 19 | 1065044 | 1016044 | Found | NM_019112.3 | Synonymous (Gly2053Gly) | C | T=0.36 | T=0.201/253 |
Chr: chromosome; AD Risk–C: this SNP was identified as a risk marker for AD in the GWAS study published by Corneveaux et al. (Corneveaux et al., 2010); Risk–H: this SNP was identified as a risk marker for AD in the GWAS study published by Hollingworth et al. (Hollingworth et al., 2011); Targeted: this SNP was identified as a common coding SNP in a gene containing an AD risk SNP from the previously mentioned GWASs; Found: this SNP was found when genotyping targeted SNPs because it was located near one of the targeted SNPs and showed up during the sequencing of the targeted SNP region.
Coding SNPs were chosen based upon their reported minor allele frequency (MAF) or heterozygosity in dbSNP. For this, publicly available data in dbSNP was used and SNPs were chosen based upon the fact that they induced a coding change in the resultant protein and that they had a MAF or heterozygosity greater than 0.05 in the general population. For CR1, SNPs were excluded if they were located in highly homologous exons in order to avoid genotyping errors.
Most of the SNPs studied conformed to these specifications, however there were some that did not and were included in the study because no better proxies were available (such as rs17259045, rs76037557, rs74727972, rs79741566, rs72973581).
DNA sequencing and data analysis
The genotypes of the coding SNPs used to establish the linkage disequilibrium structure were determined by Sanger sequencing. The exon in which the SNP is located was targeted for amplification, or, in the case of intronic GWAS SNPs, the sequence 150 bases upstream and 150 bases downstream was amplified. For the PCR reactions, AmpliTaq Gold ® 360 MasterMix (Applied Biosystems, Foster City, CA, USA) was used together with specific primers designed using ExonPrimer (http://ihg.gsf.de/ihg/ExonPrimer.html). For the SNPs rs3752239, rs4147934 and rs4147935, DMSO was included in the PCR protocol. Each purified PCR product was sequenced using Applied Biosystems BigDye terminator v3.1 sequencing chemistry and ran on an ABI3730xl (Applied Biosystems, CA) genetic analyzer as per manufacturer's instructions. The sequences were analyzed with Sequencher software, version 4.2 (Gene Codes Corporation, Ann Arbor, MI, USA).
DNA methylation and mRNA expression in the human brain
Tissue samples
Frozen samples from the frontal cerebral cortex and cerebellum were obtained from 387 Caucasian subjects without neurological disease in lifetime (Gibbs et al., 2010, Hernandez et al., 2011). Genomic DNA was extracted using phenol-chloroform and quantified on a Nanodrop1000 spectrophotometer before genotyping or bisulfite conversion for DNA methylation analysis.
CpG Methylation
Bisulfite conversion of genomic DNA was performed using Zymo EZ-96 DNA Methylation kits according to the manufacturers protocol, using 1 µg of DNA input. The CpG methylation status of DNA at >27,000 sites was determined using Illumina Infinium HumanMethylation27 BeadChips (Illumina Inc., San Diego, CA, USA). Samples were included in the analysis if the threshold call rate for inclusion of was >95% in the tissue. As a second quality control, we compared reported genders with methylation levels of CpG sites on the X chromosome. After these steps, 292 samples with data at 27,465 CpG sites in the frontal cortex tissue samples, and 27,419 sites in the cerebellum tissue samples were used for further analysis.
mRNA Expression
Messenger RNA (mRNA) expression was analyzed using Illumina HumanHT-12 v3 Expression Beadchips. Individual probes were excluded from analyses if the p-value for detection was > 0.01 and samples were excluded if <95% of probes were detected. Intensity values for each probe were normalized using cubic spline and transformed using log2 prior to statistical analyses. Probes were annotated using ReMOAT tool15 to exclude individual probes that are known to have problems in design or with ambiguous mapping. We also removed all probes that included any known SNP. After these quality control steps, data was available for 399 samples at 9814 probes from the frontal cortex, and 9587 probes in cerebellum.
Genotyping and Imputation of Control Brains in Epigenetic Analyses
The same tissue samples were genotyped using Illumina HumanHap550 v3, Human610-Quad v1 and Human660W-Quad v1 Infinium Beadchips and shared SNPs were extracted for each sample. We excluded samples where the reported sex did not match X chromosome heterogeneity from genotype data or if the per sample genome-wide call rate was less than 95%. Individual SNPs were excluded if there was a < 95% genotyping success rate per SNP, if minor allele frequency (MAF) < 0.01 or if Hardy-Weinberg equilibrium (HWE) p< 1×10−7. Multi-dimensional scaling was used to cluster samples after merging SNPs common to CEU, JPT, CHB and YRI samples from Phase II of HapMap. Outliers > 3 standard deviations from the mean component vector estimates for C1 or C2 for CEU samples were then removed, as were samples sharing greater than a proportion of 0.15 alleles. Genotypes for all European ancestry participants were imputed using MACHv1.0.16 with haplotypes derived from sequencing of 112 European ancestry samples present in the August 2009 release of phased data from the 1000 Genomes Project (available at http://www.sph.umich.edu/csg/abecasis/MACH/download/1000G-Sanger-0908.html). Data was imputed by first generating error and crossover maps as parameter estimates for the imputation on a randomly selected set of 200 samples over 100 iterations of the initial statistical model. These parameter estimates were then used to generate maximum likelihood allele dosages per SNP based on reference haplotypes for the entire study cohorts. We excluded SNPs with R2 quality estimates < 0.30, resulting in ~5.1 million SNPs available for analysis.
methQTL and eQTL mapping
SNPs with a fixed-effects p-value < 1×10−5 from the AD meta-analysis (Stage 1 + 2) were considered candidate quantitative trait loci (QTL) (Naj et al., 2011). For each SNP, CpG sites and expression probes within +/− 1MB were used for linear regression modelling using MACH2QTLv1.08. We estimated the association between the allelic dosage of each SNP against gene expression or methylation levels using linear regression models adjusted for covariates of gender and age at death, the first two component vectors from multi-dimensional scaling, post mortem interval (PMI), brain bank and batch in which preparation or hybridization were performed. SNPs with less than three minor homozygotes detected were excluded from analyses. We tested probes within 1 MB of each of 224 candidate probes in the expression datasets and 220 in the methylation datasets, resulting in 2542 associations for expression-QTLs and 11,522 associations for methylation-QTLs in the frontal cortex samples, and 2395 expression-QTLs and 11510 methylation-QTLs in the cerebellum. The resulting p-values were corrected for multiple testing using the Bonferroni method after removing SNPs having r2 > 0.5 with SNPs in adjacent sliding windows of 50 SNPs that moved two SNPs per iteration. After these filters, the analyses used 152 mRNA probes and 603 CpG sites
REGIONAL BRAIN EXPRESSION AND SPLICING ANALYSIS
Human post-mortem brain tissue collection and mRNA extraction
A detailed description of the samples used in the study, tissue processing and dissection is provided in Trabzuni et al. (2011). In brief, brain and CNS tissue originating from 137 control individuals was collected by the Medical Research Council (MRC) Sudden Death Brain and Tissue Bank, Edinburgh, UK (Millar et al., 2007), and the Sun Health Research Institute (SHRI) an affiliate of Sun Health Corporation, USA (Beach et al., 2008). All samples had fully informed consent for retrieval and were authorized for ethically approved scientific investigation (Research Ethics Committee number 10/H0716/3).
Total RNA was isolated from human post-mortem brain tissues using the miRNeasy 96 kit (Qiagen), processed with the Ambion® WT Expression Kit and Affymetrix GeneChip Whole Transcript Sense Target Labelling Assay, and hybridized to the Affymetrix Exon 1.0 ST Arrays following the manufacturers’ protocols. Hybridized arrays were scanned on an Affymetrix GeneChip® Scanner 3000 7G and visually inspected for hybridization artifacts.
Exon Array data analysis
All arrays were pre-processed using Robust Multi-array Averaging (RMA) (Irizarry et al., 2003) with quantile normalization and GC background correction in Partek’s Genomics Suite v6.6 (Partek Incorporated, St. Louis, MO, USA). In order to filter out low expression signals, detection above background (DABG) p-values of exon probe sets were calculated using Affymetrix Power Tools v1.14.3 (APT, http://www.affymetrix.com/partners_programs/programs/developer/tools/powertools.affx). After re-mapping the Affymetrix probe sets onto human genome build 19 (GRCh37) as documented in the Netaffx annotation file (HuEx-1_0-st-v2 Probeset Annotations, Release 31), we restricted analysis to 174,328 probe sets that had gene annotation, contained at least three probes with unique hybridization and had DABG p <0.001 in 50% of male or female individuals. We defined an expressed gene as any gene containing ≥1 exon with a median DABG p <0.001 in at least 50% of male or female individuals in at least one brain region. The gene-level expression was calculated for 19,597 genes by calculating the Winsorised mean value (winsorizing the data below 10% and above 90%) of all probe set signals annotated to a single gene. Region-specific expression and splicing was investigated using Partek’s mixed-model ANOVA and alternative splice ANOVA (Partek Genomics Suite v6.6). In all types of analysis, the date of array hybridization, brain bank and gender were included as co-factors. All p values were corrected for multiple comparisons using Bonferroni correction.
RESULTS AND DISCUSSION
GWAS are able to identify associations between phenotypes and genetic loci. Since only tagging SNPs and not all genetic variants are assessed in these studies it is not possible, solely from GWAS results, to accurately pinpoint the associated variant(s) or even gene(s). In this study we have chosen to study, for each associated region, the gene considered as the most likely to be associated by either having been reported as such in the original GWAS or because it was considered as the most interesting gene in the region from a functional perspective in regards to AD pathobiology.
ALZHEIMER’S DISEASE LOCI
CR1 - Complement component (3b/4b) receptor 1; chromosome 1q32
GWAS have consistently identified intronic SNPs in CR1 (rs6701713, rs1408077, rs3818361 and rs6656401) to be associated with an increased risk of AD onset (Fig. 1). The linkage disequilibrium between these intronic SNPs and common coding variants in the same gene was investigated.
Fig. 1.
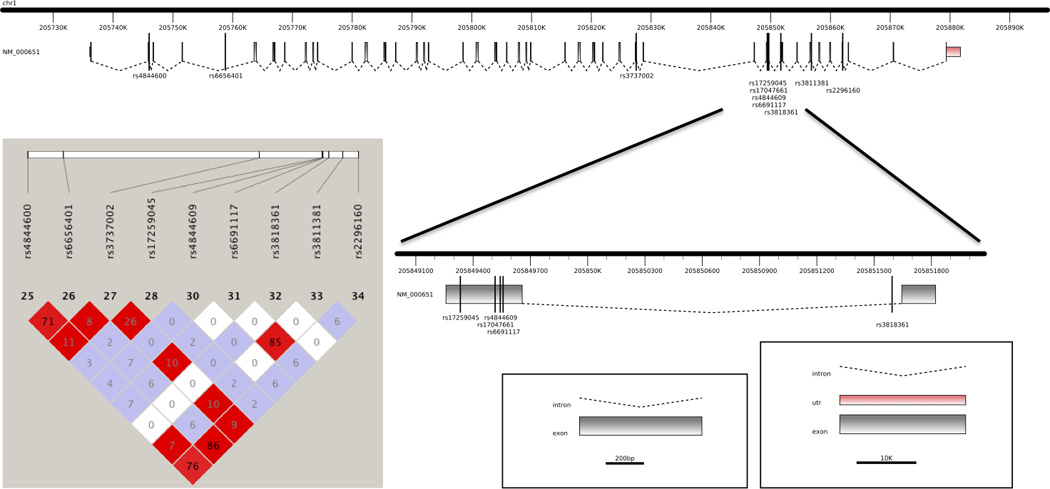
Representation of the CR1 gene with the localization of the SNPs found as significant by GWAS as well as the SNPs studied here. On the left, linkage disequilibrium plot for the CR1 SNPs found to be significant in GWAS and the common coding variants genotyped in this study.
The genotyping of eight common coding SNPs in CR1 in 96 pathologically confirmed late-onset AD cases revealed that the GWAS associated SNP rs6656401 was in LD with two coding SNPs: rs4844600 (p.Glu60Asp, NP_000642.3) and rs2296160 (p.Thr2419Ala, NP_000642.3), located in exons 2 and 44 respectively. The GWAS associated SNP rs3818361 was not in LD with any of the common coding SNPs studied here (Fig. 1).
The structure of the human CR1 gene is complex. The gene is composed of tandem long homologous repeating segments that encode binding sites for C3b or C4b. Four CR1 alleles differing in the total numbers of repeating segments are known and thought to have arisen through an homologous recombination with unequal crossover mechanism. The encoded protein is made up of four structurally significant domains. These are the signal peptide, extracellular, transmembrane and cytoplasmic domains (Wong, 1990). Three of these domains are homologous in each of the four allotypes but they differ in the lengths of the extracellular domain. This region is made up of short consensus repeats (SCRs) that are also known as complement-control-protein repeats (CCPs) (Klickstein et al., 1988). These repeats are highly conserved and are characterized by the presence of three cysteine residues and one tryptophan. In addition, there is a high degree of homology between every eighth SCR, thus grouping the SCRs into sevens. Each group of seven SCRs is termed a long homologous repeat (LHR) (Klickstein et al., 1988). The smallest allotype is rare and is termed either CR1-C or CR1-F’, it has three LHR (long homologous repeat) regions. The most common allotype is called CR1-A or CR1-F and has four LHR regions. The CR1-B or CR1-S allotype contains five LHR regions and the very rare CR1-D allotype contains six LHR regions (Holers et al., 1987).
The extracellular LHR regions contain the binding sites for the protein, including the binding sites for the complement fragments C3b and C4b, thus individuals with different CR1 alleles have different numbers of binding sites for these complement fragments and it is likely that this results in a different ability in the clearance of these fragments. There still remains some uncertainty about this process, mainly because it is currently poorly understood as to whether a decreased ability to clear complement fragments is beneficial or harmful in terms of AD aetiology. It had previously been thought that an increased ability to clear the complement fragments would be beneficial as it would decrease the activation of the complement system, generally thought of as pathogenic. However, it has been shown that individuals presenting CR1-B or CR1-S genotypes (the largest common allotype) actually have a raised risk of AD (Brouwers et al., 2011), indicating that an increased ability to clear the complement fragments C3b and C4b may be pathogenic in the situation of AD. Additionally, common variation at the CR1 locus, more specifically rs6656401 has been shown to have a broad impact on cognition. This effect was shown to be largely mediated by an individual's amyloid plaque burden (Chibnik et al., 2011).
Using microarrays, the only brain regions in which CR1 could be detected were white matter and cerebellum and the expression levels in both regions were low. This would suggest that the role of CR1 in AD may be related to its function at a systemic level or in relation to the brain vasculature. Taken together, our results suggest that CR1 genetic variability does not act through different splicing variants or through differences in expression. The most probable scenario is that the variants found to be significant in GWAS and the coding variants found here to be in LD with these GWAS hits are tagging the structural variants known to exist in the CR1 gene.
BIN1 - Bridging integrator 1; chromosome 2q14
Three SNPs in the BIN1 locus (rs744373 (Hollingworth et al., 2011, Hu et al., 2011), rs7561528 (Hu et al., 2011, Naj et al., 2011) and rs12989701 (Hu et al., 2011) have been identified by GWAS as associated with LOAD. These three SNPs lie in a noncoding region upstream of BIN1 and downstream of CYP27C1. In this locus, BIN1 (encoding the “bridging integrator 1” protein) appears to be the most likely functional candidate. Neither of the GWAS associated SNPs lie in regulatory regions, CpG islands or in microRNA target sites. Rs744373 is in a reported recombination hotspot (in HapMap) and all SNPs are in predicted transcription binding sites. There are only four non-synonymous SNPs published (NCBI SNP database, version 132) in BIN1 and for these, MAFs are described for rs112318500 and only for African populations. We attempted to genotype two SNPs in BIN1 in our cohort, rs76037557 and rs112318500, and were unable to identify the presence of the minor allele in any of the cases studied.
Although no significant eQTLs could be found for BIN1 in our samples, it is interesting to note the regional expression differences in its expression within the CNS. On the basis of the microarray results, BIN1 had the highest expression in white matter, with the mean gene expression being 5.1 times higher in white matter as compared to cerebellum (Bonferroni corrected p <1.0 × 10−30). In addition, we found evidence of alternative splicing by brain region with one or more of three isoforms (NM_139343, NM_139344 and NM_139345, Bonferroni corrected alternative splicing p value <1.0 × 10−30) being lower in white matter as compared to all other CNS regions (Fig. 2).
Fig. 2.
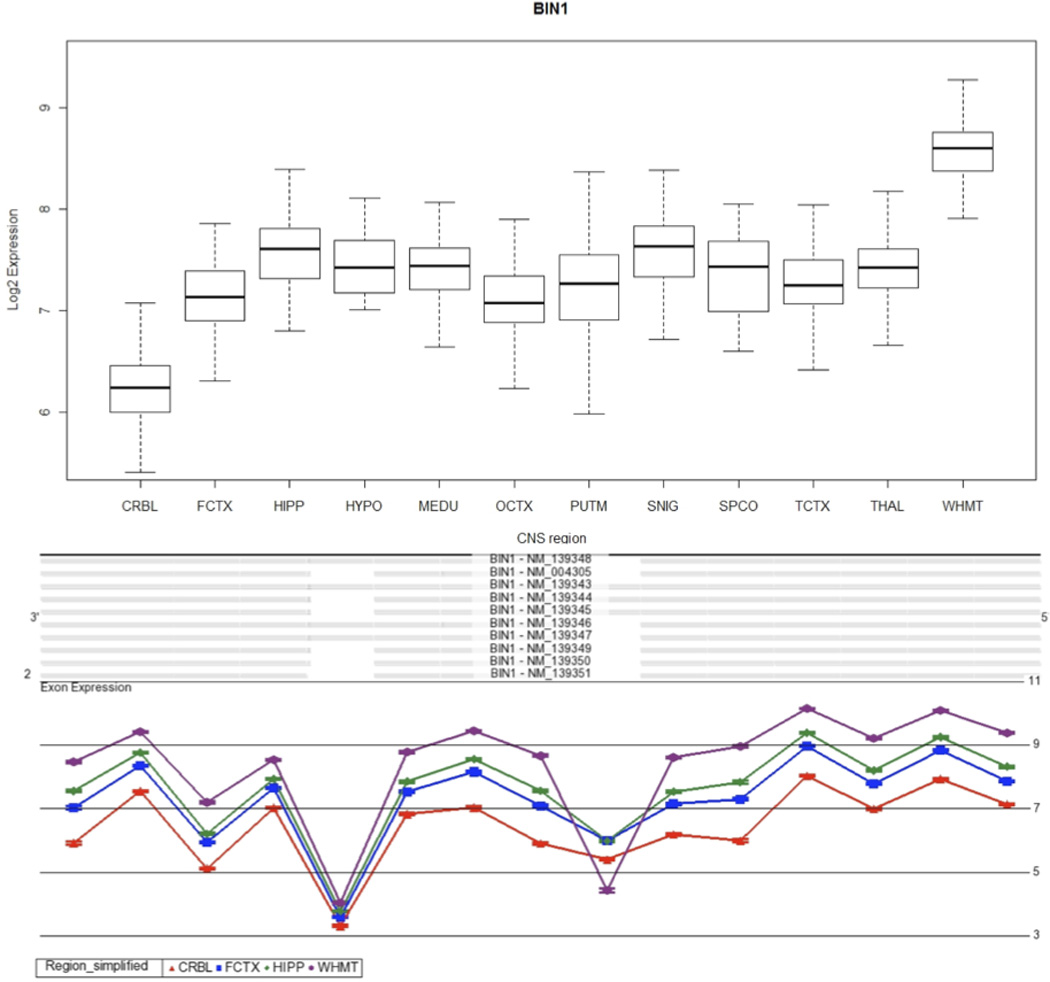
BIN1 expression and splicing across the human CNS. A, Boxplot showing BIN1 expression across 12 human CNS regions, as measured using Affymetrix exon arrays in 137 neuropathologically normal individuals. B, Plot of expression levels (y axis) for each probe set (x axis) for BIN1 in cerebellum, frontal cortex, hippocampus and white matter, showing a statistically significant interaction between probe set expression (“exon usage”) and brain region. Non-parallel probeset expression levels (highlighted in the boxed region) indicate region-dependent differential splicing of the corresponding exon. Plots are adapted from Partek Genomics suite auto-generated output.
Isoforms that are expressed in the central nervous system are thought to be involved in synaptic vesicle endocytosis and may interact with dynanim, synaptojanin, endophilin, and clathrin. More specifically, BIN1 has been shown to be involved in both dynamin- (Wigge and McMahon, 1998) and clathrin-mediated endocytosis (CME) (Pant et al., 2009). CME is thought to mediate the internalization of Amyloid Precursor Protein (APP; MIM 104760) from the cell surface (Nordstedt et al., 1993), after which the Aβ peptide can be cleaved from the APP. Presence of the Aβ peptide in turn inhibits CME (Kelly and Ferreira, 2007) and therefore stops excess APP from entering the cell by a mechanism of auto-inhibition. The BIN1 protein may also have an important endocytic role being involved in synaptic vesicle recycling at the synaptic terminal (Di Paolo et al., 2002) (Pant et al., 2009). Aberrant splice variants have been described to be expressed in tumor cell lines and misregulated alternative splicing of BIN1 has been shown to be associated with T tubule alterations and muscle weakness in myotonic dystrophy (Fugier et al., 2011), which leads us to hypothesize a similar situation for the CNS isoforms.
Both BIN1 and PICALM are part of endocytic pathways (Guerreiro and Hardy, 2011, Olgiati et al., 2011). The results we present here (Figs. 2 and 3) show both higher expression of BIN1 and a different complement of BIN1 isoforms in white matter. Along with the different balance of isoforms for PICALM in white matter (for more details see next section) we speculate that endocytic pathways function in a distinct manner in white matter as compared to other brain regions.
Fig. 3.
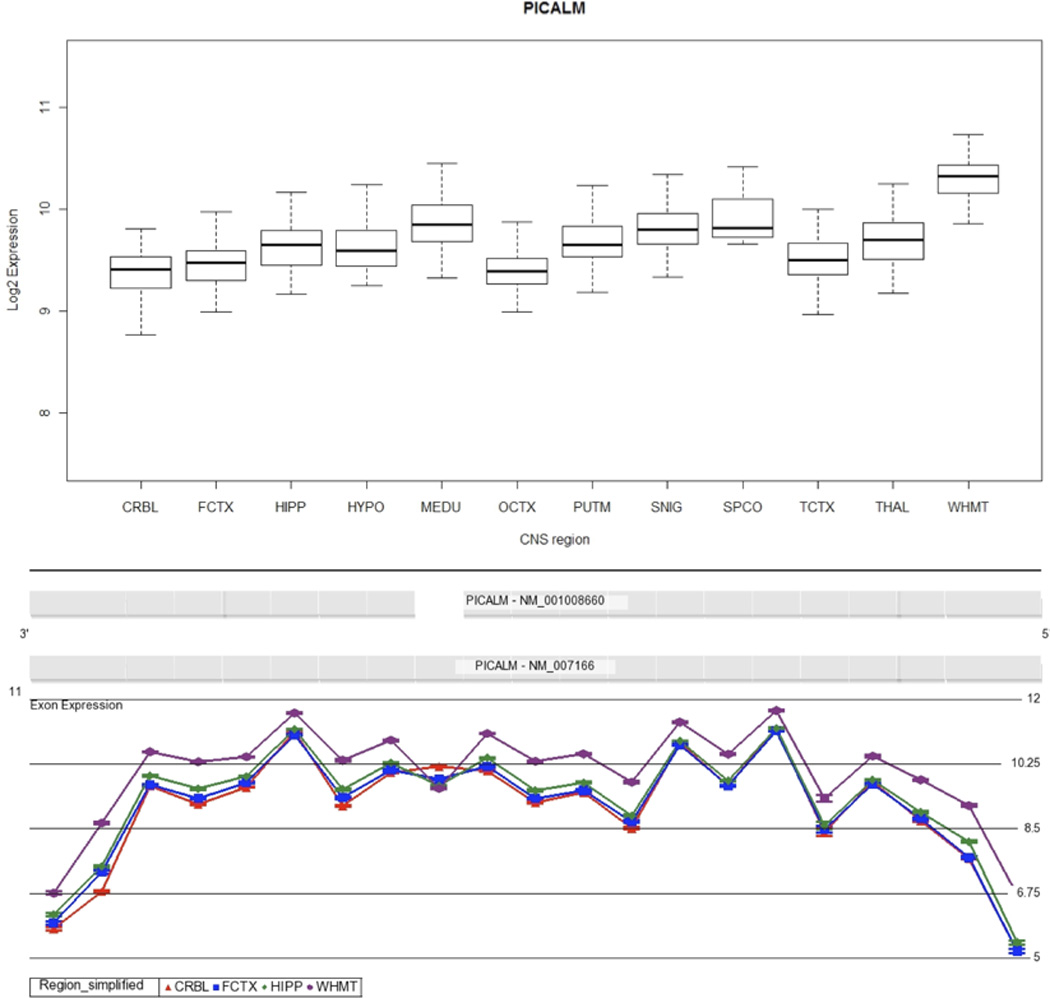
PICALM expression and splicing across the human CNS. A, Boxplot showing PICALM expression across 12 human CNS regions, as measured using Affymetrix exon arrays in 137 neuropathologically normal individuals. B, Plot of expression levels (y axis) for each probe set (x axis) for PICALM in cerebellum, frontal cortex, hippocampus and white matter, showing a statistically significant interaction between probe set expression (“exon usage”) and brain region. Non-parallel probeset expression levels (highlighted in the boxed region) indicate region-dependent differential splicing of the corresponding exon. Plots are adapted from Partek Genomics suite auto-generated output.
Alzheimer’s disease has historically been characterized by neuronal loss and gray matter atrophy. More recently the involvement of white matter in the disease has started to be considered (Hua et al., 2008). These results seem even more interesting in light of the association obtained by Braskie and colleagues between Clusterin rs11136000 and white matter microstructure in young adults (Braskie et al., 2011). Although no replication dataset was available in this study, white matter disturbances in AD seem to be a common factor worthy of further analyses.
PICALM - Phosphatidylinositol binding clathrin assembly protein; chromosome 11q14
There are four SNPs associated with the PICALM gene that have been identified in GWAS. These are rs592297 (Harold et al., 2009), rs561655 (Harold et al., 2009, Naj et al., 2011), rs541458 (Corneveaux et al., 2010, Harold et al., 2009, Lambert et al., 2009), and rs3851179 (Harold et al., 2009, Seshadri et al., 2010). The first of these SNPs is a synonymous variant (p.Gln174Gln) located in exon 5 of PICALM which may influence the activity of a sequence predicted to be an exon splicing enhancer. The other three SNPs are located upstream of the gene. rs561655 is found within a region that is thought to be a transcription factor binding site (Harold et al., 2009). It is possible that the change at this locus may increase or decrease the affinity of this region of the PICALM gene to transcription factors, thus changing the expression levels of the gene. rs541458 is located 8kb 5’ of PICALM and has been shown to be in LD with rs3851179, located 88.5kb 5’ of the gene (Harold et al., 2009). It is possible that the presence of both these SNPs in the 5’ region outside the gene may have an effect on the expression of the gene.
Eleven non-synonymous SNPs have been described in the PICALM gene (NCBI SNP database, version 132). From these, two were reported to have an established heterozygosity >0.05 (rs118027183 and rs74727972). Genotyping these SNPs in our cohort did not reveal any case with the minor allele, thus it is not likely that the association seen in the genome wide association studies is due to common coding variability in this gene.
Similarly to BIN1, PICALM is involved in CME (Dreyling et al., 1996, Tebar et al., 1999, Yao et al., 2005), and has been shown to have a particular influence on the activity of VAMP-2 (Harel et al., 2008), a SNARE protein responsible for directing neurotransmitter vesicles to the presynaptic membrane. This role of the protein, allied to the observation of a reduced synaptic density in the brains of AD patients, suggests that the activity of this protein is potentially important in disease aetiology. The mean gene expression of PICALM was found to be 1.9 times higher in white matter as compared to cerebellum (Bonferroni corrected p value <1.0 × 10−30). Two PICALM isoforms are detected by the exon array: full-length PICALM and a shorter form that lacks exon 13. Our results show that the short form of PICALM is expressed at lower levels in white matter as compared to the other CNS regions, in particular cerebellum (Figure 3, Bonferroni corrected alternative splicing p value <1.0 × 10−30).
Several methQTL associations were significant after multiple test correction. In cerebellum, these include associations with CpG sites in APOC1, BCL3 and CBLC on chromosome 19, as well as ME3, MGC34732 and MS4A6E on chromosome 11. In frontal cortex, six significant associations with a CpG site mapping to APOE, as well as one association at APOC2 were identified. From these, only the association observed between rs10751134 in PICALM and a CpG site in ME3 (a gene adjacent to PICALM, Fig. 4) is in moderate LD with the GWAS SNPs studied here (r2 between rs10751134 and rs561655, rs3851179, rs541458 in HapMap populations varies between 0.5 – 0.6) and may contribute to the genome-wide association signal found for PICALM.
Fig. 4.
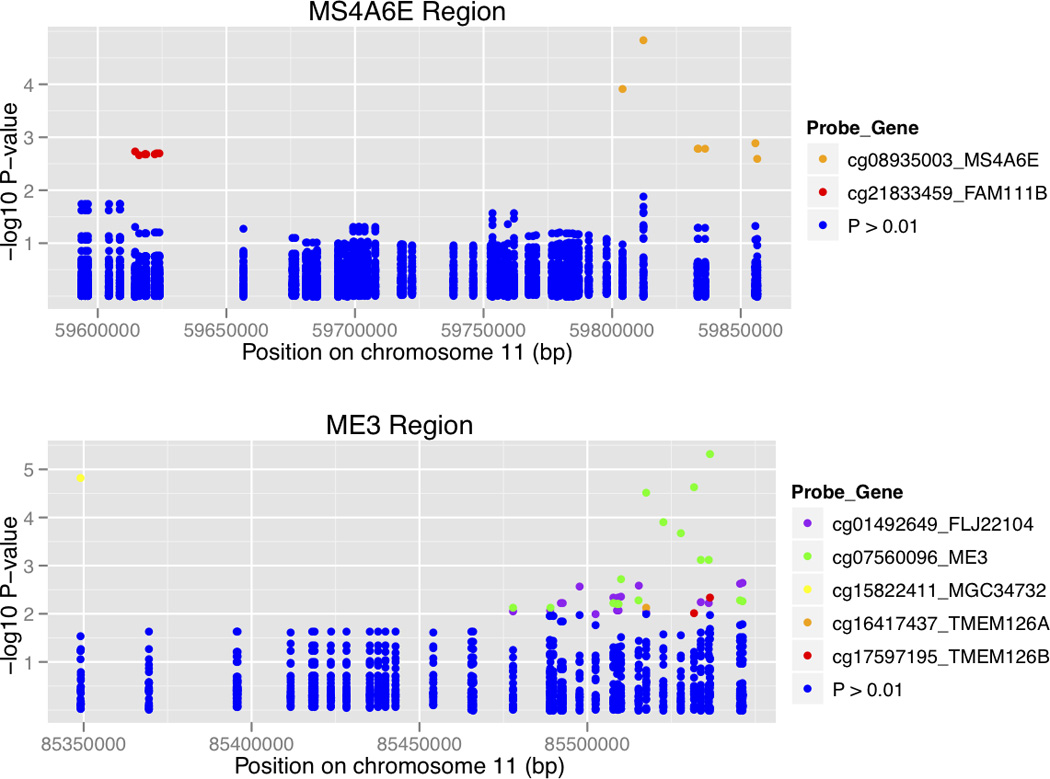
Association between genotypes and CpG sites in cerebellum. Here the results are shown as log-transformed P values color-coded to match the CpG probe in the loci. The regions associated in chromosome 11 are represented: in the top panel the MS4A6E locus and in the bottom panel the ME3 region. From these, only rs10897024 in PICALM (associated with a p-value of 4.65×10-6 with the probe cg07560096 in ME3, represented in green) is in moderate LD with the GWAS hits for PICALM (rs561655, rs3851179 and rs541458).
MS4A6A/MS4A4E - Membrane-spanning 4-domains, subfamily A, members 6A and 4E; chromosome 11q12.1
There are three SNPs in the MS4A gene cluster that have been associated with an increased risk of LOAD. These are rs4938933 in MS4A4A (Hollingworth et al., 2011, Naj et al., 2011), rs670139 in MS4A4E (Hollingworth et al., 2011, Naj et al., 2011) and rs610932 in MS4A6A (Hollingworth et al., 2011). Several SNPs are described in these genes. In order to have a comprehensive view of the locus we targeted non-synonymous coding SNPs in MS4A6A, MS4A4E and MS4A4A (Table 1). As expected the SNPs shown to be significantly associated with AD in the previous GWAS were in LD with each other. No significant LD was observed between these AD risk SNPs and the targeted coding SNPs studied here (highest r2=0.5 between rs610932 and rs7232), indicating that non-synonymous common genetic variability in the MS4A4 locus probably does not explain the associations established in the GWAS.
The role of the genes located in the MS4A cluster is so far poorly understood. The cluster is found on chromosome 11 and is made up of at least sixteen genes (Liang and Tedder, 2001). It has been suggested that the proteins encoded by the cluster may be ion channels or adaptor proteins (Liang et al., 2001, Zuccolo et al., 2010). It is likely that the genes in the cluster all have a similar role due to their high homology (Liang et al., 2001). However, until more is known of their function it is impossible to speculate as to the potential role of polymorphisms in these genes in AD aetiology. Whereas MS4A6A was detected in all brain regions, using microarrays, we were unable to confidently detect MS4A4A in cerebellum, frontal cortex or occipital cortex.
ABCA7 - ATP-binding cassette, sub-family A (ABC1), member 7; chromosome 19p13.3
Two SNPs in ABCA7 have been associated with LOAD: rs3752246 and rs3764650 (Hollingworth et al., 2011, Naj et al., 2011). rs3752246, in exon 32 of the gene, leads to a protein change (p.Gly1527Ala) which was the only coding non-synonymous change to be identified by GWAS. In an attempt to identify an associated functional variant at the ABCA7 locus, Hollingworth and colleagues chose to genotype rs3752246 in an additional cohort because this was a non-synonymous SNP with the highest LD with rs3764650 out of all HapMap ABCA7-coding variants based on r2 values (r2 = 0.36, D′ = 0.89) (Hollingworth et al., 2011). Although, in our study, we established a lower degree of LD (r2=0.1 between rs3764650 and rs3752246), this was also the highest LD found between rs3764650 and all the coding SNPs studied here (Figure 5). This suggests that the risk of AD conferred by the presence of rs3764650 is not due to the presence of multiple other common coding SNPs in the gene. rs3764650 SNP is found at position 115 of the intron between exons 13 and 14. There is no evidence to suggest that rs3764650 has an effect on the expression of the gene (Hollingworth et al., 2011). rs3752246 is predicted in silico to be a benign variant (Polyphen-2 score=0). This variant is in moderate LD (r2=0.6) with rs4147934, which is also predicted to be non-pathogenic. Nonetheless, rs3752246 was the only missense change to be identified by GWAS and functional studies of the real impact of these two variants at the protein level should further elucidate if any of these are the real risk associated variants in ABCA7.
Fig. 5.
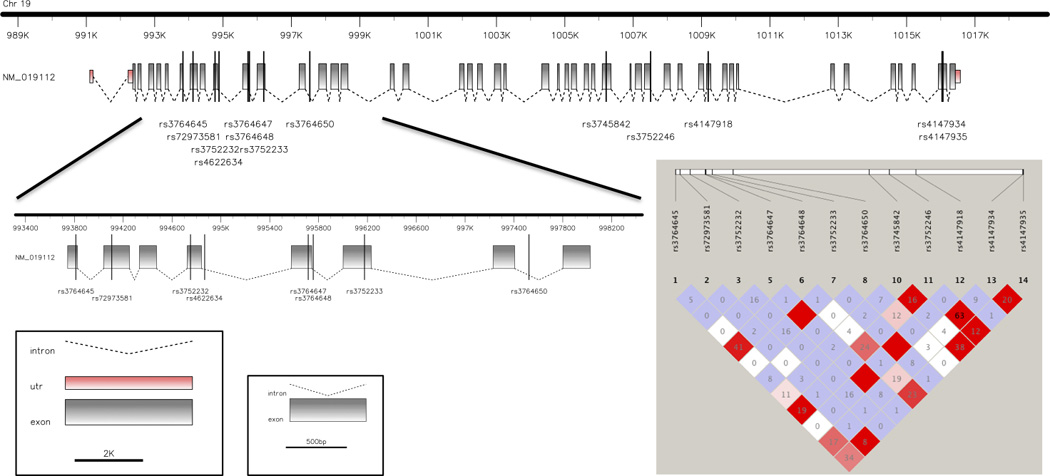
Representation of the ABCA7 gene with the localization of the SNPs found as significant by GWAS as well as the SNPs studied here. On the right, linkage disequilibrium plot for the ABCA7 SNPs found to be significant in GWAS and the common coding variants genotyped in this study.
ABCA7 had low, but detectable expression in all CNS regions with no striking regional differences in gene expression patterns (data not shown).
The ABCA7 gene is a member of a large family of ATP-binding cassette genes divided into the subfamilies A-G based upon sequence homology (Kim et al., 2008). The transporters of the A and G subfamilies are particularly responsible for the movement of lipids such as sterols, phospholipids and bile acids across membranes against the concentration gradient of the substrate (Schmitz et al., 2000, Schmitz and Kaminski, 2001, Kusuhara and Sugiyama, 2007). ABCA1 is known to have a role in the transport of cholesterol to lipid-free acceptors such as apoA-I and apoE. The high homology between ABCA1 and ABCA7 suggests that the two proteins should share a similar role. The high lipid content of the CNS means that lipid homeostasis is essential and so, changes in the ability to transport cholesterol, are potentially pathogenic. It has been shown that the levels of cholesterol influence the processing of the APP protein: during times of high intracellular content, the activity of α-secretase is inhibited, whilst the activity of β- and γ-secretases is enhanced (Bodovitz and Klein, 1996, Tun et al., 2002, Kalvodova et al., 2005, Vetrivel et al., 2005). The role of ABCA7 in this situation is currently unclear as there is conflicting evidence regarding its ability to transport cholesterol: work with ABCA7−/− mice has shown that cholesterol efflux is not dependent upon ABCA7 (Kim et al., 2005); however work with human embryonic kidney (HEK)293 cells has shown that transfection with ABCA7 cDNA leads to 1.7 to 2 times increase in cholesterol efflux (Chan et al., 2008). It is likely therefore that ABCA7 plays a role in cholesterol transport, but that this is a process that also involves other proteins.
CLU- Clusterin; chromosome 8p21-p12
The CLU gene encodes the clusterin or apolipoprotein J protein, expressed ubiquitously but with a known higher prevalence in the brain, ovary, testis and liver (de Silva et al., 1990). Levels of clusterin are shown to be elevated in the cortex and hippocampus areas of the brains of AD patients (Oda et al., 1994). Although we found high expression of CLU throughout the control CNS, we were unable to demonstrate that this gene was more highly expressed in cortex or hippocampus relative to other CNS regions (Fig. 6). Clusterin binds to Aβ plaques in the cerebrospinal fluid, forming a complex that is able to cross the blood-brain barrier (Zlokovic, 1996). Levels of clusterin in the plasma are positively correlated with the risk of AD (Schrijvers et al., 2011). It is therefore possible that the increased risk of AD induced by the SNPs described may be due to an increased level of expression. We sequenced the exonic regions of CLU in 495 AD cases and 330 healthy controls. In this study, a total of twenty-four variants were found in both cases and controls with similar frequencies, indicating that common coding variability in this gene does not underlie the association seen with the intronic SNPs (Guerreiro et al., 2010). In order to determine if common variants at the CLU locus effect expression of nearby (cis) mRNA transcripts, an eQTL analysis was also performed. No significant eQTL associations were observed for the SNPs previously associated with AD, which led us to conclude that the most likely mechanism underpinning the association is either small effects of genetic variability on resting gene expression, or effects on damage induced expression of the protein. These conclusions are also supported by the absence of significant differences in the expression of the gene between several brain regions and in the gene splicing (Fig. 6). More recently, rare coding variants in CLU have been associated with the risk for AD (Bettens et al., 2012). However, this variability was found to be independent of the common association signal identified by the GWAS. Small studies have also reported an association between rs9331888 and alternative splicing of CLU (Szymanski et al., 2011) and blood clusterin levels (Xing et al., 2012).
Fig. 6.
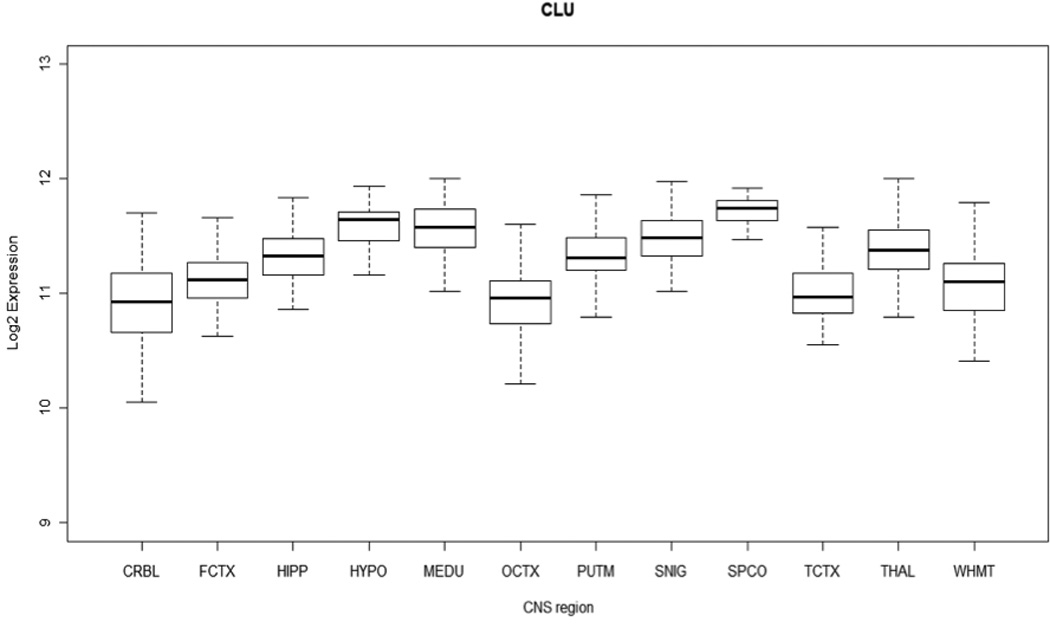
Boxplot showing CLU expression across 12 human CNS regions, as measured using Affymetrix exon arrays in 137 neuropathologically normal lindividuals.
CONCLUSION
Clearly, developing an understanding of the nature and mechanism of loci for Alzheimer’s disease (and other neurological diseases), which are identified by genome wide association studies and are not coding changes is going to be a considerable challenge (Table 2). This study shows that simple eQTL studies in control brain tissue may not identify effects in many cases. There remain several options which are not mutually exclusive: (1) the study is underpowered (although larger than most previous studies) (2) genetic variability in splicing is an important consideration (3) genetic variability in other RNA species at the locus is important besides the obvious mRNA (4) genetic variability in damage induced expression, and not in resting expression, is the important factor.
Table 2.
Initial loci identified by GWAS in AD and predicted type of causal variants in each locus.
| GWAS identified loci | |
|---|---|
| APOE* | Both amino acid and eQTL effects |
| CLU | Not common amino acid change / Possible alternative splicing |
| CR1 | Exon insertion polymorphism |
| BIN1 | Not common amino acid change |
| PICALM | Not common amino acid change / Possible methQTL |
| ABCA7 | Probably amino acid change |
| MS4A6A/MS4A4E | Not common amino acid change |
Locus not studied here
The first three possibilities can be gradually overcome by either more samples or by improvements (for example) in sequencing technologies, which would allow transcript QTLs to be assessed as well as QTLs for other RNA species. Understanding QTLs in damage induced expression is inherently difficult (Webster et al., 2009). Measuring changes in damage induced expression in tissue with changing cell populations and developing rigorous algorithms to interpret such data is problematic, but may be necessary, especially for understanding the etiology of late onset neurodegenerative diseases.
Supplementary Material
Acknowledgements
This work was supported in part by the Intramural Research Program of the National Institute of Health, National Institute on Aging (Z01-AG000950-10), and utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Genotyping of the TGEN2 cohort was supported by Kronos Science; The National Institute on Aging [R01 AG034504, R01 AG031581, P30 AG19610, Z01 AG000950-06, P30AG10161, R01AG15819]; The National Institute of Neurological Disorders and Stroke [R01 NS059873] The Banner Alzheimer's Foundation; The Johnnie B. Byrd Sr. Alzheimer’s Disease Institute; The Medical Research Council; and the state of Arizona. Many data and biomaterials were collected from several National Institute on Aging (NIA) and National Alzheimer’s Coordinating Center (NACC, grant #U01 AG016976) funded sites. This work was also partially supported by the MRC through the MRC Sudden Death Brain Bank. MR was supported by an MRC Training Fellowship and DT was supported by King Faisal Hospital.
We thank the brain donors and their families for their selfless donation to the fight against this disease. Amanda J. Myers, PhD (University of Miami, Department of Psychiatry) and John A. Hardy, PhD (Reta Lila Weston Institute, University College London) collected and prepared the series. Marcelle Morrison-Bogorad, PhD., Tony Phelps, PhD and Walter Kukull, PhD are thanked for helping to co-ordinate this collection. The directors, pathologist and technicians involved include: University of Michigan (NIH grant P50-AG08671): Dr. Roger Albin, Lisa Bain, Eszter Gombosi, The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek): Inge Huitinga, MD, Marleen Rademaker, Michiel Kooreman. We thank Colin Smith and Robert Walker at the Sudden Death Brain Bank for their help.
The National Institutes of Health, National Institute on Aging (NIH-NIA) supported the ADGC through the following grants: ADGC, U01 AG032984, RC2 AG036528; NACC, U01 AG016976; NCRAD, U24 AG021886; NIA LOAD, U24 AG026395, U24 AG026390; Banner Sun Health Research Institute P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01AG33193; Columbia University, P50 AG008702, R37 AG015473; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG06781, UO1 HG004610; Indiana University, P30 AG10133; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG016574; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, MO1RR00096, and UL1 RR029893; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG30146; TGen, R01 NS059873; University of Alabama at Birmingham, P50 AG016582, UL1RR02777; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573, P50, P50 AG016575, P50 AG016576, P50 AG016577; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991. The Kathleen Price Bryan Brain Bank at Duke University Medical Center is funded by NINDS grant # NS39764, NIMH MH60451 and by Glaxo Smith Kline. We thank Drs. D. Stephen Snyder and Marilyn Miller from NIA who are ex-officio ADGC members. Support was also from the Alzheimer’s Association (LAF, IIRG-08-89720; MP-V, IIRG-05-14147) and the US Department of Veterans Affairs Administration, Office of Research and Development, Biomedical Laboratory Research Program. P.S.G.-H. is supported by Wellcome Trust, Howard Hughes Medical Institute, and the Canadian Institute of Health Research.
ADNI Funding for ADNI is through the Northern California Institute for Research and Education by grants from Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., Alzheimer's Association, Alzheimer's Drug Discovery Foundation, the Dana Foundation, and by the National Institute of Biomedical Imaging and Bioengineering and NIA grants U01 AG024904, RC2 AG036535, K01 AG030514.
Abbreviations
- eQTL
expression quantitative trait loci
- GWAS
genome wide association studies
- AD
Alzheimer's disease
- SNPs
single nucleotide polymorphisms
- LD
linkage disequilibrium
- NINDS-ADRDA
National Institute for Neurological and Communicative Disorders and-Stroke-Alzheimer's Disease and Related Disorder Association
- LOAD
are onset Alzheimer's disease
- DMSO
Dimethyl sulfoxide
- PCR
polymerase chain reaction
- MAF
minor allele frequency
- HWE
Hardy–Weinberg equilibrium
- PMI
post mortem interval
- methQTL
methylation quantitative trait loci
- CNS
central nervous system
- RMA
Robust Multi-array Averaging
- CCPs
complement-control-proteins repeats
- SCRs
short consensus repeats
- LHR
a long homologous repeat
- ESR
erythrocyte sedimentation rate
- CME
clathrin-mediated endocytosis
- SNARE
Soluble N-ethylmaleimide-sensitive factor activating protein receptor
†Alzheimer’s Disease Genetics Consortium coauthors and affiliations
Liana G. Apostolova, MD1, Steven E. Arnold, MD2, Clinton T. Baldwin, PhD3, Robert Barber, PhD4, Michael M. Barmada, PhD5, Thomas G. Beach, MD PhD6, Gary W. Beecham, PhD7,8, Duane Beekly, BS9, David A. Bennett, MD10,11, Eileen H. Bigio, MD12, Thomas D. Bird, MD13, Deborah Blacker, MD14,15, Bradley F. Boeve, MD16, James D. Bowen, MD17, Adam Boxer, MD PhD18, James R. Burke, MD PhD19, Jacqueline Buros, BA3, Joseph D. Buxbaum, PhD20,21,22, Nigel J. Cairns, PhD FRCPath23, Laura B. Cantwell, MPH24, Chuanhai Cao, PhD25, Chris S. Carlson, PhD26, Regina M. Carney, MD27, Minerva M. Carrasquillo, PhD28, Steven L. Carroll, MD PhD29, Helena C. Chui, MD30, David G. Clark, MD31, Carl W. Cotman, PhD32, Paul K. Crane, MD MPH33, Elizabeth A. Crocco, MD34, Carlos Cruchaga, PhD35, Jeffrey L. Cummings, MD1, Philip L. De Jager, MD PhD36,37, Charles DeCarli, MD38, Steven T. DeKosky, MD39, F. Yesim Demirci, MD5, Ramon Diaz-Arrastia, MD PhD40, Malcolm Dick, PhD32, Dennis W. Dickson, MD28, Ranjan Duara, MD41, William G. Ellis, MD42, Nilufer Ertekin-Taner, MD PhD28,43, Denis Evans, MD44, Kelley M. Faber, MS45, Kenneth B. Fallon, MD29, Martin R. Farlow, MD46, Steven Ferris, PhD47, Tatiana M. Foroud, PhD45, Matthew P. Frosch, MD PhD48, Douglas R. Galasko, MD49, Mary Ganguli, MD50, Marla Gearing, PhD51,52, Daniel H. Geschwind, MD PhD53, Bernardino Ghetti, MD54, John R. Gilbert, PhD7,8, Sid Gilman, MD FRCP55, Bruno Giordani, PhD56, Jonathan D. Glass, MD57, Alison M. Goate, D.Phil35, Neill R. Graff-Radford, MD28,43, Robert C. Green, MD MPH58, John H. Growdon, MD59, Hakon Hakonarson, MD PhD60, Ronald L. Hamilton, MD61, Lindy E. Harrell, MD PhD31, Elizabeth Head, PhD62, Lawrence S. Honig, MD PhD63, Christine M. Hulette, MD64, Bradley T. Hyman, MD PhD59, Gail P. Jarvik, MD PhD65,66, Gregory A. Jicha, MD PhD67, Lee-Way Jin, MD PhD42, Gyungah Jun, PhD3,68,69, M. Ilyas Kamboh, PhD5,70, Jason Karlawish, MD71, Anna Karydas, BA18, John S.K. Kauwe, PhD72, Jeffrey A. Kaye, MD73,74, Ronald Kim, MD75, Edward H. Koo, MD49, Neil W. Kowall, MD76,77, Patricia Kramer, PhD78,73, Walter A. Kukull, PhD79,James J. Lah, MD PhD57, Eric B. Larson, MD MPH33,80, Allan I. Levey, MD PhD57, Andrew P. Lieberman, MD PhD81, Oscar L. Lopez, MD70, Kathryn L. Lunetta, PhD68, Wendy J. Mack, PhD82, Daniel C. Marson, JD PhD31, Eden R. Martin, PhD7,8, Frank Martiniuk, PhD83, Deborah C. Mash, PhD84, Eliezer Masliah, MD49,85, Wayne C. McCormick, MD MPH33, Susan M. McCurry, PhD86, Andrew N. McDavid, BA26, Ann C. McKee, MD76,77, Marsel Mesulam, MD87,88, Bruce L. Miller, MD18, Carol A. Miller, MD89, Joshua W. Miller, PhD42, Thomas J. Montine, MD PhD90, John C. Morris, MD23,91, Adam C. Naj, PhD7, Petra Nowotny, PhD35, Joseph E. Parisi, MD92,93, Elaine Peskind, MD94, Ronald C. Petersen, MD PhD16, Wayne W. Poon, PhD32, Huntington Potter, PhD25, Joseph F. Quinn, MD73, Ashok Raj, MD25, Ruchita A. Rajbhandary, MPH7, Murray Raskind, MD94, Barry Reisberg, MD47,95, Christiane Reitz, MD PhD63,96,97, John M. Ringman, MD1, Erik D. Roberson, MD PhD31, Ekaterina Rogaeva, PhD98, Roger N. Rosenberg, MD40, Mary Sano, PhD21, Andrew J. Saykin, PsyD45,99, Julie A. Schneider, MD100,10, Lon S. Schneider, MD30,101, William W. Seeley, MD18, Michael L. Shelanski, MD PhD102, Charles D. Smith, MD67, Joshua A. Sonnen, MD90, Salvatore Spina, MD54, Peter St George-Hyslop, MD FRCP98,103, Robert A. Stern, PhD76, Rudolph E. Tanzi, PhD59, John Q. Trojanowski, MD PhD24, Juan C. Troncoso, MD104, Debby W. Tsuang, MD94, Otto Valladares, MS24, Vivianna M. Van Deerlin, MD PhD24, Badri N. Vardarajan, MS3, Harry V. Vinters, MD1,105, Jean Paul Vonsattel, MD106, Li-San Wang, PhD24, Sandra Weintraub, PhD87,88, Kathleen A. Welsh-Bohmer, PhD19,107, Jennifer Williamson, MS63, Randall L. Woltjer, MD PhD108, Clinton B. Wright, MD MS109, Steven G. Younkin, MD PhD28,
1Department of Neurology, University of California Los Angeles, Los Angeles, California; 2Department of Psychiatry, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania; 3Department of Medicine (Genetics Program), Boston University, Boston, Massachusetts; 4Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, Texas; 5Department of Human Genetics, University of Pittsburgh, Pittsburgh, Pennsylvania; 6Civin Laboratory for Neuropathology, Banner Sun Health Research Institute, Phoenix, Arizona; 7The John P. Hussman Institute for Human Genomics, University of Miami, Miami, Florida; 8Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami, Miami, Florida; 9National Alzheimer's Coordinating Center, University of Washington, Seattle, Washington; 10Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois; 11Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago, Illinois; 12Department of Pathology, Northwestern University, Chicago, Illinois; 13Department of Neurology, University of Washington, Seattle, Washington; 14Department of Epidemiology, Harvard School of Public Health, Boston, Massachusetts; 15Department of Psychiatry, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts; 16Department of Neurology, Mayo Clinic, Rochester, Minnesota; 17Swedish Medical Center, Seattle, Washington; 18Department of Neurology, University of California San Francisco, San Fransisco, California; 19Department of Medicine, Duke University, Durham, North Carolina; 20Department of Neuroscience, Mount Sinai School of Medicine, New York, New York; 21Department of Psychiatry, Mount Sinai School of Medicine, New York, New York; 22Departments of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York; 23Department of Pathology and Immunology, Washington University, St. Louis, Missouri; 24Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania; 25Byrd Alzheimer Institute, University of Southern Florida Health, Tampa, Florida; 26Fred Hutchinson Cancer Research Center, Seattle, Washington; 27Department of Psychiatry, Vanderbilt University, Nashville, Tennessee; 28Department of Neuroscience, Mayo Clinic, Jacksonville, Florida; 29Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama; 30Department of Neurology, University of Southern California, Los Angeles, California; 31Department of Neurology, University of Alabama at Birmingham, Birmingham, Alabama; 32Institute for Memory Impairments and Neurological Disorders, University of California Irvine, Irvine, California; 33Department of Medicine, University of Washington, Seattle, Washington; 34Department of Psychiatry and Behavioral Sciences, Miller School of Medicine, University of Miami, Miami, Florida; 35Department of Psychiatry and Hope Center Program on Protein Aggregation and Neurodegeneration, Washington University School of Medicine, St. Louis, Missouri; 36Program in Translational NeuroPsychiatric Genomics, Institute for the Neurosciences, Department of Neurology & Psychiatry, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts; 37Program in Medical and Population Genetics, Broad Institute, Cambridge, Massachusetts; 38Department of Neurology, University of California Davis, Sacramento, California; 39University of Virginia School of Medicine, Charlottesville, Virginia; 40Department of Neurology, University of Texas Southwestern, Dallas, Texas; 41Wien Center for Alzheimer's Disease and Memory Disorders, Mount Sinai Medical Center, Miami Beach, Florida; 42Department of Pathology and Laboratory Medicine, University of California Davis, Sacramento, California; 43Department of Neurology, Mayo Clinic, Jacksonville, Florida; 44Rush Institute for Healthy Aging, Department of Internal Medicine, Rush University Medical Center, Chicago, Illinois; 45Department of Medical and Molecular Genetics, Indiana University, Indianapolis, Indiana; 46Department of Neurology, Indiana University, Indianapolis, Indiana; 47Department of Psychiatry, New York University, New York, New York; 48C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, Charlestown, Massachusetts; 49Department of Neurosciences, University of California San Diego, La Jolla, California; 50Department of Psychiatry, University of Pittsburgh, Pittsburgh, Pennsylvania; 51Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia; 52Emory Alzheimer's Disease Center, Emory University, Atlanta, Georgia; 53Neurogenetics Program, University of California Los Angeles, Los Angeles, California; 54Department of Pathology and Laboratory Medicine, Indiana University, Indianapolis, Indiana; 55Department of Neurology, University of Michigan, Ann Arbor, Michigan; 56Department of Psychiatry, University of Michigan, Ann Arbor, Michigan; 57Department of Neurology, Emory University, Atlanta, Georgia; 58Division of Genetics, Department of Medicine and Partners Center for Personalized Genetic Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts; 59Department of Neurology, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts; 60Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; 61Department of Pathology (Neuropathology), University of Pittsburgh, Pittsburgh, Pennsylvania; 62Sanders-Brown Center on Aging, Department of Molecular and Biomedical Pharmacology, University of Kentucky, Lexington, Kentucky; 63Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Neurology, Columbia University, New York, New York; 64Department of Pathology, Duke University, Durham, North Carolina; 65Department of Genome Sciences, University of Washington, Seattle, Washington; 66Department of Medicine (Medical Genetics), University of Washington, Seattle, Washington; 67Department of Neurology, University of Kentucky, Lexington, Kentucky; 68Department of Biostatistics, Boston University, Boston, Massachusetts; 69Department of Ophthalmology, Boston University, Boston, Massachusetts; 70University of Pittsburgh Alheimer's Disease Research Center, Pittsburgh, Pennsylvania; 71Department of Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania; 72Department of Biology, Brigham Young University, Provo, Utah; 73Department of Neurology, Oregon Health & Science University, Portland, Oregon; 74Department of Neurology, Portland Veterans Affairs Medical Center, Portland, Oregon; 75Department of Pathology and Laboratory Medicine, University of California Irvine, Irvine, California; 76Department of Neurology, Boston University, Boston, Massachusetts; 77Department of Pathology, Boston University, Boston, Massachusetts; 78Department of Molecular & Medical Genetics, Oregon Health & Science University, Portland, Oregon; 79Department of Epidemiology, University of Washington, Seattle, Washington; 80Group Health Research Institute, Group Health, Seattle, Washington; 81Department of Pathology, University of Michigan, Ann Arbor, Michigan; 82Department of Preventive Medicine, University of Southern California, Los Angeles, California; 83Department of Medicine - Pulmonary, New York University, New York, New York; 84Department of Neurology, University of Miami, Miami, Florida; 85Department of Pathology, University of California San Diego, La Jolla, California; 86School of Nursing Northwest Research Group on Aging, University of Washington, Seattle, Washington; 87Alzheimer's Disease Center, Northwestern University, Chicago, Illinois; 88Cognitive Neurology, Northwestern University, Chicago, Illinois; 89Department of Pathology, University of Southern California, Los Angeles, California; 90Department of Pathology, University of Washington, Seattle, Washington; 91Department of Neurology, Washington University, St. Louis, Missouri; 92Department of Anatomic Pathology, Mayo Clinic, Rochester, Minnesota; 93Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota; 94Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, Washington; 95Alzheimer's Disease Center, New York University, New York, New York; 96Gertrude H. Sergievsky Center, Columbia University, New York, New York; 97Department of Neurology, Columbia University, New York, New York; 98Tanz Centre for Research in Neurodegenerative Disease, University of Toronto, Toronto, Ontario; 99Department of Radiology and Imaging Sciences, Indiana University, Indianapolis, Indiana; 100Department of Pathology (Neuropathology), Rush University Medical Center, Chicago, Illinois; 101Department of Psychiatry, University of Southern California, Los Angeles, California; 102Department of Pathology, Columbia University, New York, New York; 103Cambridge Institute for Medical Research and Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom; 104Department of Pathology, Johns Hopkins University, Baltimore, Maryland; 105Department of Pathology & Laboratory Medicine, University of California Los Angeles, Los Angeles, California; 106Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Pathology, Columbia University, New York, New York; 107Department of Psychiatry & Behavioral Sciences, Duke University, Durham, North Carolina; 108Department of Pathology, Oregon Health & Science University, Portland, Oregon; 109Evelyn F. McKnight Brain Institute, Department of Neurology, Miller School of Medicine, University of Miami, Miami, Florida;
Footnotes
The authors have no conflicts of interest regarding the present study.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article:
Table S1 SNPs found associated with LOAD by GWAS assessing >1500 cases and >1500 controls
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organised for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
REFERENCES
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Engelborghs S, Lambert JC, Rogaeva E, Vandenberghe R, Le Bastard N, Pasquier F, Vermeulen S, van Dongen J, Mattheijssens M, Peeters K, Mayeux R, St George-Hyslop P, Amouyel P, de Deyn PP, Sleegers K, van Broeckhoven C. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener. 2012;7:3. doi: 10.1186/1750-1326-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- Braskie MN, Jahanshad N, Stein JL, Barysheva M, Mcmahon KL, de Zubicaray GI, Martin NG, Wright MJ, Ringman JM, Toga AW, Thompson PM. Common Alzheimer's disease risk variant within the CLU gene affects white matter microstructure in young adults. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:6764–6770. doi: 10.1523/JNEUROSCI.5794-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, Pasquier F, Montoya AG, Peeters K, Mattheijssens M, Vandenberghe R, de Deyn PP, Cruts M, Amouyel P, Sleegers K, van Broeckhoven C. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Kim WS, Kwok JB, Hill AF, Cappai R, Rye KA, Garner B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem. 2008;106:793–804. doi: 10.1111/j.1471-4159.2008.05433.x. [DOI] [PubMed] [Google Scholar]
- Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, Tran D, Aubin C, Buchman AS, Heward CB, Myers AJ, Hardy JA, Huentelman MJ, Corneveaux JJ, Reiman EM, Evans DA, Bennett DA, de Jager PL. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol. 2011;69:560–569. doi: 10.1002/ana.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, Nalls MA, Chen K, Lee W, Chewning K, Villa SE, Meechoovet HB, Gerber JD, Frost D, Benson HL, O'reilly S, Chibnik LB, Shulman JM, Singleton AB, Craig DW, van Keuren-Jensen KR, Dunckley T, Bennett DA, de Jager PL, Heward C, Hardy J, Reiman EM, Huentelman MJ. Association of CR1, CLU and PICALM with Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva HV, Harmony JA, Stuart WD, Gil CM, Robbins J. Apolipoprotein J: structure and tissue distribution. Biochemistry. 1990;29:5380–5389. doi: 10.1021/bi00474a025. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, de Camilli P. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron. 2002;33:789–804. doi: 10.1016/s0896-6273(02)00601-3. [DOI] [PubMed] [Google Scholar]
- Dideberg V, Kristjansdottir G, Milani L, Libioulle C, Sigurdsson S, Louis E, Wiman AC, Vermeire S, Rutgeerts P, Belaiche J, Franchimont D, van Gossum A, Bours V, Syvanen AC. An insertion-deletion polymorphism in the interferon regulatory Factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum Mol Genet. 2007;16:3008–3016. doi: 10.1093/hmg/ddm259. [DOI] [PubMed] [Google Scholar]
- Dreyling MH, Martinez-Climent JA, Zheng M, Mao J, Rowley JD, Bohlander SK. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci U S A. 1996;93:4804–4809. doi: 10.1073/pnas.93.10.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de La Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, de Munain AL, Sergeant N, Laquerriere A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1000952. e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martin J, Altshuler D, Behrens TW, Alarcon-Riquelme ME. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, Plenge RM, Koeuth T, Ortmann WA, Hom G, Bauer JW, Gillett C, Burtt N, Cunninghame Graham DS, Onofrio R, Petri M, Gunnarsson I, Svenungsson E, Ronnblom L, Nordmark G, Gregersen PK, Moser K, Gaffney PM, Criswell LA, Vyse TJ, Syvanen AC, Bohjanen PR, Daly MJ, Behrens TW, Altshuler D. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Beck J, Gibbs JR, Santana I, Rossor MN, Schott JM, Nalls MA, Ribeiro H, Santiago B, Fox NC, Oliveira C, Collinge J, Mead S, Singleton A, Hardy J. Genetic variability in CLU and its association with Alzheimer's disease. PLoS One. 2010;5:e9510. doi: 10.1371/journal.pone.0009510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Hardy J. Alzheimer's disease genetics: lessons to improve disease modelling. Biochem Soc Trans. 2011;39:910–916. doi: 10.1042/BST0390910. [DOI] [PubMed] [Google Scholar]
- Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 trafficking. Traffic. 2008;9:417–429. doi: 10.1111/j.1600-0854.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, Mcguinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, de Deyn PP, van Broeckhoven C, Livingston G, Bass NJ, Gurling H, Mcquillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O'donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ, Singleton AB. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holers VM, Chaplin DD, Leykam JF, Gruner BA, Kumar V, Atkinson JP. Human complement C3b/C4b receptor (CR1) mRNA polymorphism that correlates with the CR1 allelic molecular weight polymorphism. Proc Natl Acad Sci U S A. 1987;84:2459–2463. doi: 10.1073/pnas.84.8.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, de Deyn PP, van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alpérovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Björnsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossù P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O'Donovan M, Amouyel P, Williams J Alzheimer's Disease Neuroimaging Initiative; CHARGE consortium; EADI1 consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E, Dechairo B, John S, van Eerdewegh P, Soares H. Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer's disease. PLoS One. 2011;6:e16616. doi: 10.1371/journal.pone.0016616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Parikshak N, Lee S, Chiang MC, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer's disease: an MRI study of 676 AD, MCI, and normal subjects. NeuroImage. 2008;43:458–469. doi: 10.1016/j.neuroimage.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;147:60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, Fitzgerald ML, Kang K, Okuhira K, Bell SA, Manning JJ, Koehn SL, Lu N, Moore KJ, Freeman MW. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J Biol Chem. 2005;280:3989–3995. doi: 10.1074/jbc.M412602200. [DOI] [PubMed] [Google Scholar]
- Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–1166. doi: 10.1111/j.1471-4159.2007.05099.x. [DOI] [PubMed] [Google Scholar]
- Klickstein LB, Bartow TJ, Miletic V, Rabson LD, Smith JA, Fearon DT. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J Exp Med. 1988;168:1699–1717. doi: 10.1084/jem.168.5.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuhara H, Sugiyama Y. ATP-binding cassette, subfamily G (ABCG family) Pflugers Arch. 2007;453:735–744. doi: 10.1007/s00424-006-0134-x. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, de Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Liang Y, Buckley TR, Tu L, Langdon SD, Tedder TF. Structural organization of the human MS4A gene cluster on Chromosome 11q12. Immunogenetics. 2001;53:357–368. doi: 10.1007/s002510100339. [DOI] [PubMed] [Google Scholar]
- Liang Y, Tedder TF. Identification of a CD20-, FcepsilonRIbeta-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics. 2001;72:119–127. doi: 10.1006/geno.2000.6472. [DOI] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- Millar T, Walker R, Arango JC, Ironside JW, Harrison DJ, Macintyre DJ, Blackwood D, Smith C, Bell JE. Tissue and organ donation for research in forensic pathology: the MRC Sudden Death Brain and Tissue Bank. J Pathol. 2007;213:369–375. doi: 10.1002/path.2247. [DOI] [PubMed] [Google Scholar]
- Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, Zismann VL, Joshipura K, Huentelman MJ, Hu-Lince D, Coon KD, Craig DW, Pearson JV, Holmans P, Heward CB, Reiman EM, Stephan D, Hardy J. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, de Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, Mccormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE. Purification and characterization of brain clusterin. Biochem Biophys Res Commun. 1994;204:1131–1136. doi: 10.1006/bbrc.1994.2580. [DOI] [PubMed] [Google Scholar]
- Olgiati P, Politis AM, Papadimitriou GN, de Ronchi D, Serretti A. Genetics of late-onset Alzheimer's disease: update from the alzgene database and analysis of shared pathways. International journal of Alzheimer's disease. 2011;2011 doi: 10.4061/2011/832379. 832379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol. 2009;11:1399–1410. doi: 10.1038/ncb1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz G, Kaminski WE. ABC transporters and cholesterol metabolism. Front Biosci. 2001;6:D505–D514. doi: 10.2741/schmitz. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Kaminski WE, Orso E. ABC transporters in cellular lipid trafficking. Curr Opin Lipidol. 2000;11:493–501. doi: 10.1097/00041433-200010000-00007. [DOI] [PubMed] [Google Scholar]
- Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA : the journal of the American Medical Association. 2011;305:1322–1326. doi: 10.1001/jama.2011.381. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, Destefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, Jonsen A, Rantapaa-Dahlqvist S, Moller B, Kere J, Koskenmies S, Widen E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Ronnblom L, Syvanen AC. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJ, Costenbader KH, Criswell LA, Crusius JB, Cui J, de Bakker PI, de Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TR, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-Van Mil AH, van der Horst-Bruinsma IE, van der Schoot CE, van Riel PL, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Wijmenga C, Karlson EW, Toes RE, de Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski M, Wang R, Bassett SS, Avramopoulos D. Alzheimer's risk variants in the Clusterin gene are associated with alternative splicing. Transl Psychiatry. 2011;1 doi: 10.1038/tp.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999;10:2687–2702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, Weale ME, Hardy J. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem. 2011;119:275–282. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun H, Marlow L, Pinnix I, Kinsey R, Sambamurti K. Lipid rafts play an important role in A beta biogenesis by regulating the beta-secretase pathway. J Mol Neurosci. 2002;19:31–35. doi: 10.1007/s12031-002-0007-5. [DOI] [PubMed] [Google Scholar]
- Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Lariviere M, Moussette S, Grundberg E, Kwan T, Ouimet M, Ge B, Hoberman R, Swiatek M, Dias J, Lam KC, Koka V, Harmsen E, Soto-Quiros M, Avila L, Celedon JC, Weiss ST, Dewar K, Sinnett D, Laprise C, Raby BA, Pastinen T, Naumova AK. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet. 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–25900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, Rohrer K, Zhao A, Marlowe L, Kaleem M, McCorquodale DS, 3Jr, Cuello C, Leung D, Bryden L, Nath P, Zismann VL, Joshipura K, Huentelman MJ, Hu-Lince D, Coon KD, Craig DW, Pearson JV, Heward CB, Reiman EM, Stephan D, Hardy J, Myers AJ. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P, Mcmahon HT. The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci. 1998;21:339–344. doi: 10.1016/s0166-2236(98)01264-8. [DOI] [PubMed] [Google Scholar]
- Wong WW. Structural and functional correlation of the human complement receptor type 1. J Invest Dermatol. 1990;94:64S–67S. doi: 10.1111/1523-1747.ep12875150. [DOI] [PubMed] [Google Scholar]
- Xing YY, Yu JT, Cui WZ, Zhong XL, Wu ZC, Zhang Q, Tan L. Blood Clusterin Levels, rs9331888 Polymorphism, and the Risk of Alzheimer's Disease. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2011-111844. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Petralia RS, Bushlin I, Wang Y, Furukawa K. Synaptic distribution of the endocytic accessory proteins AP180 and CALM. J Comp Neurol. 2005;481:58–69. doi: 10.1002/cne.20362. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Cerebrovascular transport of Alzheimer's amyloid beta and apolipoproteins J and E: possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996;59:1483–1497. doi: 10.1016/0024-3205(96)00310-4. [DOI] [PubMed] [Google Scholar]
- Zuccolo J, Bau J, Childs SJ, Goss GG, Sensen CW, Deans JP. Phylogenetic analysis of the MS4A and TMEM176 gene families. PLoS One. 2010;5:e9369. doi: 10.1371/journal.pone.0009369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.