

Abstract
Interleukin 17 (IL-17) cytokines play a crucial role in a variety of inflammatory and autoimmune diseases. They signal through heterodimeric receptor complexes consisting of members of IL-17 receptor (IL-17R) family. A unique intracellular signaling domain was identified within all IL-17Rs, termed SEFIR [SEF (similar expression to fibroblast growth factor genes) and IL-17R]. SEFIR is also found in nuclear factor κB (NF-κB) activator 1 (Act1), an E3 ubiquitin ligase, and mediates its recruitment to IL-17Rs. Here we report the structure of the first SEFIR domain from IL-17RB at 1.8Å resolution. SEFIR displays a five-stranded parallel β-sheet that is wrapped by six helices. Site-directed mutagenesis on IL-17RB identified helix αC as being critical for its interaction with Act1 and IL-25 (IL-17E) signaling. Using the current SEFIR structure as a template, the key functional residues in Act1 are also mapped as part of helix αC, which is conserved in IL-17RA and RC, suggesting this helix as a common structural signature for heterotypic SEFIR-SERIR association. On the other hand, helix αB′ is important for homo-dimerization of Act1, implicating a dual ligand-binding model for SEFIR domain, with distinct structural motifs participating in either homotypic or heterotypic interactions. Furthermore, although IL-17RB-SEFIR structure resembles closest to the Toll/Interleukin-1 receptor (TIR) domain of TLR10 with low sequence homology, substantial differences were observed at helices αC, αD and DD′ loop. This study provides the first structural view of the IL-17 receptor intracellular signaling, unraveling the mechanism for the specificity of SEFIR versus TIR domain in their respective signaling pathways.
Introduction
Members of IL-17 cytokine family (IL-17A to IL-17F) are important regulators of both innate and adaptive immune responses. In particular, IL-17E (also referred as IL-25), produced by mucosal epithelial cells and many other immune cell types, plays a critical role in the initiation and propagation of the TH2-type immune response (1–4). IL-17E (IL-25) signals through heterodimeric receptor complex composed of IL-17RA and IL-17RB, members of IL-17 receptor (IL-17R) family. The IL-17R family members are single trans-membrane proteins containing fibronectin type-III (FnIII) domains within extracellular portion and a unique structural motif within cytoplasmic tails termed as SEFIR [SEF (similar expression to fibroblast growth factor genes) and IL-17R] domain (5). SEFIR domains share limited homology with Toll/IL-1 receptor (TIR) domains of Toll-like receptors (TLRs) and IL-1R (5). Proteins containing SEFIR and TIR domains together constitute the STIR (SEFIR/TIR)-domain superfamily. NF-κB activator 1 (Act1), also a member of the SEFIR protein family, is an essential component in signaling of several IL-17 cytokines (6–9). Upon ligand stimulation, Act1 is recruited to IL-17R complexes in a SEFIR-dependent manner, suggesting the crucial role of SEFIR-mediated protein-protein interactions in IL-17R signaling.
Although SEFIR domains are analogous to the TIR domains, the critical structural elements from the SEFIR domains seem specific for IL-17 receptor signaling. In particular, a key functional moiety called BB′ loop that is critical for TIR-TIR interaction in TLRs and IL-1R signaling (10) was found dispensable for SEFIR-SEFIR interaction between Act1 and IL-17R (11). Instead, our recent report revealed a CC′ loop structural motif as critical for this interaction, which could be a signature for SEFIR-mediated IL-17 signaling. In addition, although the SEFIR domains are relatively conserved among IL-17Rs, it was suggested that IL-17RB is unique in that it directly interacts with TRAF6 through a putative binding site within its SEFIR domain (12). However the molecular basis for the specific association of IL-17RB and TRAF6 is unknown.
Given the importance of SEFIR-SEFIR interactions in IL-17 cytokine signaling, a better mechanistic understanding of these interactions are crucial for the development of new and improved therapeutics for the treatment of IL-17 cytokine-mediated inflammatory diseases. Here we report the first crystal structure of SEFIR domain from IL-17RB at 1.8Å resolution. We identified distinct structural elements in SEFIR domain that are key to either homotypic or heterotypic interactions, suggesting a dual ligand-binding mode for SEFIR domains. Our structural and functional analyses suggest a unique signaling model on how SEFIR-SEFIR interactions take place, providing structural insights into the molecular mechanisms of IL-17 cytokine signaling.
Materials and Methods
Protein Expression and Purification
The mouse IL-17RB-SEFIR (amino acid residues 314 to 486) was cloned into a modified pET28 vector, as a SUMO fusion protein with a N-terminal 6 × His tag. The IL-17RB-SEFIR domain was expressed and purified using similar protocol as previously described (13), however using protease Ulp1 for removing the His-tagged SUMO moiety. The purified protein failed in our crystallization trials. Limited proteolysis by chymotrypsin and subsequent peptide sequencing was used in aiding the design of a new stable construct (325–486). Residues K364, K367, K368 and K369 were additionally mutated to alanines to reduce surface entropy (14). This mutant protein crystallized successfully. Mouse Act1-SEFIR protein was purified as described (11). The TRAF domain of mouse TRAF6 (residues 341–512) was cloned in a modified pET28 vector with a N-terminal 6×His tag followed by a tobacco etch virus (TEV) protease recognition site (ENLYFQG). The expression and purification of the TRAF domain protein were essentially the same as IL-17RB-SEFIR, while using TEV for removing the N-terminal 6×His tag.
Crystallization, data collection and structure determination
IL-17RB-SEFIR crystallized in a condition containing 0.2 M sodium acetate, pH 5.0, 1.6 M Ammonium citrate dibasic. 20% glycerol was added to the mother liquid as cryoprotectant. A set of data was collected on a SeMet substituted crystal at beamline 19-ID at the Advanced Photon Source, Argonne National Laboratory. The structure was solved by Single Wavelength Anomalous Dispersion method using HKL3000 (15). The SeMet phased model was used to solve the native structure by molecular replacement method using phaser (16). PHENIX program (17) was used for refinement and Coot (18) was used for iterative manual model building. Translation, libration and screw-rotation displacement (TLS) groups used in the refinement were defined by the TLMSD server (19). The current model is of good geometry and refinement statistics (Table 1). The final structure was refined to 1.8Å resolution and crystallographic Rwork and Rfree are 18.0% and 22.3%, respectively. The final protein model has 97.0% of all residues residing in the most-favored region of the Ramachandran plot and 3.0% in additionally allowed region, as calculated by Molprobity (20). All molecular graphic figures were generated with PYMOL (21).
Surface plasmon resonance (SPR) analysis
Binding between analytes (mouse Act1-SEFIR) and ligands [wild type IL-17RB-SEFIR (WT-RB-SEFIR), the crystallized mutant IL-17RB-SEFIR (RB-SEFIR-M)] was conducted on a Biacore 3000. The ligands were immobilized on a CM5 sensor chip in 10 mM acetic acid (pH 4.5 or pH 5.0). The analytes were allowed to flow over the chip in a buffer of 10 mM Hepes (pH 7.4), 150 mM NaCl, and 0.005% surfactant P20. The chips were regenerated with 1 M NaCl or 0.05% SDS. Binding KD values were determined by Biaevaluation software.
Cell culture, plasmids and antibodies
MEFs and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin G (100 μg/ml), and streptomycin (100 μg/ml). MEFs expressing IL-17RB were established by retroviral infection with pMSCV-IL-17RB-IRES-GFP. Complementary DNA (cDNA) encoding Myc-tagged mouse IL-17RB and its point mutants were subcloned into the plasmid pMSCV-IRES-GFP. cDNA encoding Myc-tagged mouse IL-17RB was also subcloned into pMSCV-IRES-GFP. cDNA encoding V5-tagged mouse Act1 (mAct1) were subcloned into the vector pcDNA3.1. cDNA encoding FLAG-tagged mouse Act1 and its mutants, D-BB′(residues 425–432 deleted) and point mutant H472D were subcloned into the plasmid pMSCV-IRES-GFP.
Transfection, retroviral infection and coimmunoprecipation
All transfections were conducted with FuGENE according to the manufacturer’s instructions. For reconstitution assays in MEFs, cells were infected by retroviral supernatant as described previously (8). For coimmunoprecipations, cell extracts were incubated with antibody (1 μg) and protein A beads (20 μl). After overnight incubation, beads were washed four times with lysis buffer, resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed by Western blotting according to standard procedures.
Real-time polymerase chain reaction assays
Total RNA was extracted with Trizol reagent according to the manufacturer’s instructions. The cDNAs were synthesized and real-time reverse transcription polymerase chain reaction (RT-PCR) assays were performed as described previously (8).
Results
The structure of IL-17RB-SEFIR
The IL-17RB-SEFIR structure, representing the first structure of any SEFIRs, displays a compact globular architecture comprised of a five-stranded (βA to βE) parallel β-sheet that is wrapped by six helices (αA, αB′, αB, αC, αD and αE, Fig. 1A) with loops of various lengths connecting the secondary structures (the individual loop that connects the β-strand and its next neighboring helix is termed after the name of the secondary structures, i.e., the DD′ loop connects strand βD and helix αD). The structure is well defined except for a 20 amino acids (A.A.) region connecting strand βC and helix αC (residues 396–416, we named as CC′ segment), the electron density of which is not observable. To evaluate if the missing density was due to proteolysis during crystallization, we analyzed the crystals used for data collection and found by SDS-PAGE that the protein in the crystal was intact, suggesting a conformational disorder instead of degradation of the material (Supplemental Fig. 1A). There are two topological possibilities for connecting residues 396 to 416. One scenario is to crossover the DD′ loop from its outer surface, forming a knot topology. Alternatively, residues 396 to 416 could thread inwards and through the DD′ loop from the inner side toward the protein core (Supplemental Fig. 1B). However, after carefully analyzing the structure, we found the latter alternative to be not feasible. In particular, the side chains of the residues located in the DD′ loop pack tightly, leaving no space for accommodating a peptide chain to go through the DD′ loop (Fig. 1B, Supplemental Fig. 1C). Specifically, L393, P394, F420, L450, Y454 and L457 are forming a tightly packed hydrophobic core with close non-bonding atom-to-atom distance for van der Waals interactions (i.e., the distance of L450 Cδ1 to Y454 Cδ2 is 4.5Å). Therefore, the enclosed space within DD′ loop is fully occupied, prohibiting the CC′ segment to go through. Using Falc-loop Modeling Server (22), the missing CC′ segment was modeled containing two portions, a helix insertion (CC′-ins) and a loop (CC′-loop) (Supplemental Fig. 1D), which is consistent with the secondary structures defined in our previous homology-modeled IL-17RB-SEFIR based on a TIR template (11).
Fig. 1. Structure of IL-17RB-SEFIR.
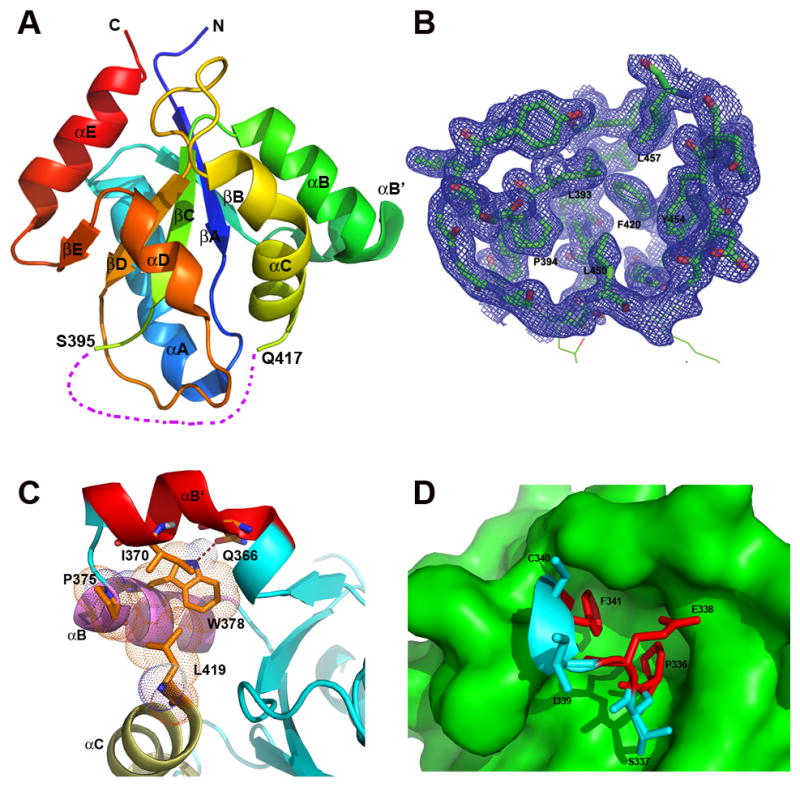
(A) Overall structure. The secondary structures are labeled and shown in rainbow colors, with N-terminus colored in blue and C-terminus in red. The CC′ segment (residues 396–416) is disordered and not visible from the current structure, as shown by a purple dashed line. (B) The 2mFo–DFc electron density map (30) covering a section of the structure containing the DD′ loop (contoured at 1.0 σ level). The DD′ loop is well defined, which embraces a space filled with hydrophobic residues that are well defined from the electron density map. (C) Helix αB′ of IL-17RB-SEFIR is tethered to helices αB and αC via hydrophobic interactions and hydrogen bond. (D) IL-17RB-SEFIR doesn’t contain a true TRAF6 binding motif. The two residues P336 and F341 in the putative TRAF6 binding motif (VYPSEICF, shown in sticks with key residues colored in red) are completely buried inside the SEFIR structure (the surface of the rest of the structure is shown in green), not possible for interacting with other proteins. The proline and phenylalanine residues at corresponding positions in the canonical TRAF6 motif are critical for binding.
In the current structure, there is a short helix αB′ (residue 363–373) in the linkage between helixαB and strand βB, which is nearly perpendicular to helix αB (Fig. 1). Helix αB′ is tethered to its helices αB and αC through hydrophobic interactions between residue I370 on helix αB′ and a hydrophobic platform comprised of residues P375, W378 on helix αB and L419 on helix αC (Fig. 1C). In addition, residue W378 on helix αB of IL-17RB is hydrogen bonded to Q366 on αB′ (distance of atom Nε1 of W378 and atom Oε1 of Q366 is 2.8 Å), which further stabilizes and locks αB′ and αB helices into a rigid conformation. Residue W378 is strictly conserved among SEFIR containing IL-17Rs and Act1 (Supplemental Fig. 2). Residues equivalent to IL-17RB Q366 are either a glutamate or serine in other SEFIR proteins. The helix αB′ seems to carry a unique role in SEFIR mediated IL-17 signaling (see below).
IL-17RB SEFIR domain does not contain a true TRAF6 binding motif
The availability of the high-resolution structure of IL-17RB-SEFIR allows us to re-assess some previous functional data. The recruitment of TRAF6 to IL-17RB was shown through direct binding to a putative TRAF6 binding site Pro-X-Glu-XX-(aromatic/acidic residue) in IL-17RB SEFIR domain. The putative TRAF6 binding site contains residues 336–341 (VYPSEICF) and an alanine substitution of residue E338 of IL-17RB attenuated IL-17RB-mediated NF-κB activation but not MAPK (ERK, JNK and p38) activation (12). Residues located at P-2, P0 and P+3 positions in canonical TRAF6 binding motif were shown as critical for binding (23). The corresponding residues in IL-17RB are P336, E338 and F341. In our structure, the putative TRAF6 binding sequence in IL-17RB is located at AA′ loop. Both P336 and F341 are completely buried inside the SEFIR domain, involving in extensive hydrophobic interactions with other internal residues of the protein and play important structural roles, excluding any possibilities of interacting with other proteins such as TRAF6 on the surface (Fig. 1D). We then carried out co-immunoprecipation assay with MEFs stably expression IL-17RB, and found that mutations (S337A, E338A, I339A) within putative TRAF6 binding sequence did not have any effect on IL-17RB and TRAF6 interaction (Fig. 2A). In addition, our in vitro size exclusion chromatography analysis, purified IL-17RB SEFIR domain did not form complex with TRAF6 (Supplemental Fig. 3). Therefore, our structure-function studies suggest that the SEFIR domain of IL-17RB does not contain a true TRAF6 binding motif. However our data does not exclude the possibility that other regions of IL-17RB interact with TRAF6 directly. On the other hand, it is also possible that IL-17RB recruits TRAF6 indirectly, perhaps through either IL-17RA or the adaptor protein Act1 for NF-κB activation.
Fig. 2. Mutagenesis studies on IL-17RB-SEFIR.

(A) IL-17RB-deficient MEFs were reconstituted with empty vector, Myc-tagged mouse RB mutants as indicated, by retroviral infection, after which cells were treated with IL-25 for 10 min, followed by immunoprecipation with antibody against Myc and Western analysis with indicated antibodies. (B) Same cells (as indicated in panel A) were treated by IL-25 for 3 hours. The abundances of il-13 mRNAs were measured by real-time RT-PCR, and the fold-induction in mRNA amounts was calculated as a ratio of the amount of mRNA in the treated sample compared to that in the untreated sample. Data are the means ± SEMs from three experiments.
Key IL-17RB residues for Act1 interaction are located on helix αC
The adaptor protein Act1 mediates IL-17RB signaling in a SEFIR-dependent manner (7, 9), via hetero-dimerization of Act1 and IL-17RB. The deletion of SEFIR domain from either protein abolished their interaction. In the current study, we indeed detected direct interaction between the SEFIR domains from IL-17RB and Act1 by using Surface Plasmon Resonance (SPR, Fig. 3A). The crystallized mutant of IL-17RB harboring alanine substitutions at four lysine residues (K364, K367, K368 and K369, see Materials and Methods) on helix αB′ had no obvious impact on the ability of IL-17RB-SEFIR to interact with Act1-SEFIR (Fig. 3A).
Fig. 3. SEFIR-mediated Act1:IL-17RB interaction.
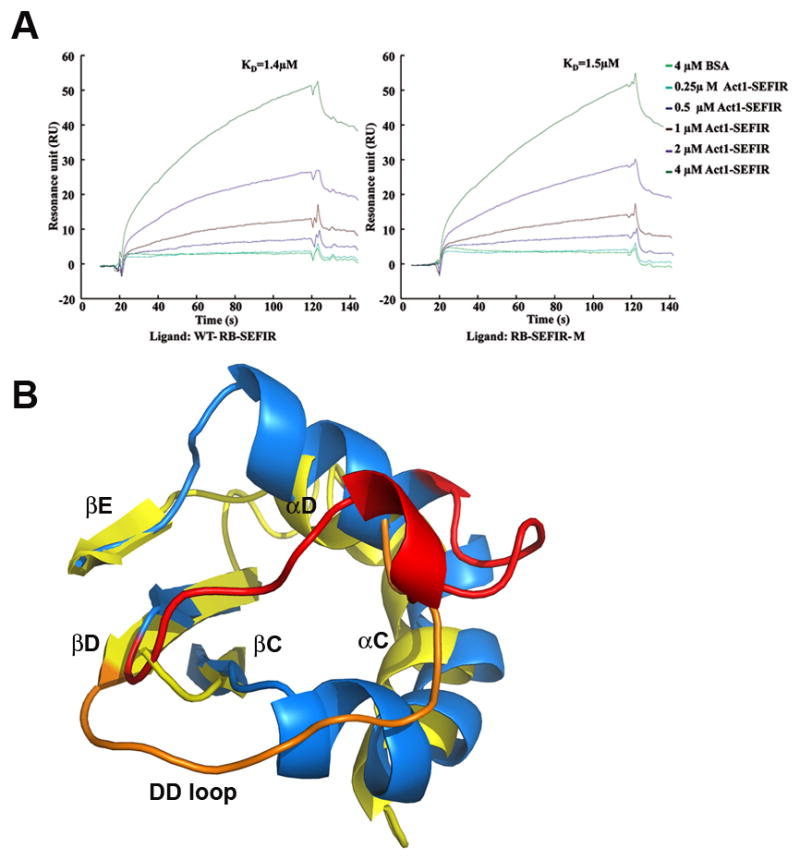
(A) SPR analysis of the binding of the Act1-SEFIR to the IL-17RB-SEFIR domain. Purified Act1-SEFIR (0.25 to 4μM) and bovine serum albumin (BSA, 4μM) were injected over the surface to which WT IL-17RB-SEFIR or IL-17RB-SEFIR-M (K364A, K367A, K368A and K369A quadruple mutant) was immobilized. Binding signal is recorded as resonance units (RU). (B) Significant conformational changes between the SEFIR and TIR domains. The SEFIR structure is shown in yellow and its DD′-loop in orange. The structure of the TIR domain from TLR10 is shown in blue and its DD′ loop in red. The DD′-loop of SEFIR shifted by more than 10Å, with respect to that in the TIR.
To identify residues on IL-17RB-SEFIR that are key to its function, we generated 16 mutants of IL-17RB-SEFIR and performed reconstitution and CO-IP experiments in MEFs that lack IL-17RB expression. The mutation sites are grouped into the following four regions: 1) putative TRAF6 motif (S337A, E338A and I339A); 2) CC′ loop (E409R, G410D, S411R, E414R, N415D, S416R and Q417R); 3) αC (D418V, L419D, P421R and L422D); 4) αB′ (Q366D and W378R). We found mutations of Q366D and W378R on IL-17RB-SEFIR only partly decreased its interaction with Act1, and attenuated the gene expression stimulated by IL-25 (Fig. 2A and B). In contrast, single mutation of L419D or L422D of IL-17RB completely abolished the interactions with Act1 and gene expression. Both L419 and L422 are located on the surface of αC helix and solvent accessible, implying their potential roles in protein interactions. Interestingly, all other mutations did not show significant effects, including the point mutations of CC′ loop. It is intriguing that we previously showed that the CC′-deletion mutant of IL-17RB displayed decreased interaction with Act1. The structure of IL-17RB-SEFIR now suggests that the loss of function by the CC′-deletion mutant of IL-17RB in our previous studies might be due to perturbation of the structural folding. Taken together, the results here implicate that αC helix of IL-17RB is the most critical region for its heterotypic interaction with Act1.
Key Act1 residues for IL-17R interactions are also located on helix αC
The structural information of IL-17RB-SEFIR allows us again to re-assess the functional data on Act1-SEFIR that we reported recently. In our previous study (11), we have identified four residues on Act1-SEFIR, where single A.A. substitutions (L474D, H475D, K477D and Y478A within the sequence of LDEDEHGLHTKY, with L474, H475, K477 and Y478 highlighted) were shown to abolish the interactions with IL-17RA and could not restore IL-17A- and IL-17E (IL-25)-mediated gene expression in Act1−/− MEFs (11), suggesting these residues as critical for heterotypic SEFIR-SEFIR interactions. In addition, we showed that a cell-permeable peptide derived from mAct1-SEFIR (containing LDEDEHGLHTKY) disrupts the Act1-IL-17RA interaction and inhibits IL-17- and IL-25-dependent signaling. To be noted, these residues were modeled to situate on the C-terminal end of the CC′ loop in the previously homology-modeled structure, which was based on a distantly related TIR template (11). Therefore, we previously termed this peptide as CC′ loop decoy. To obtain better structural insights, we decided to remodel the structure of mAct1 since we have now the high-resolution crystal structure of IL-17RB-SEFIR, one of the SEFIR family members.
Based on the crystal structure of IL-17RB-SEFIR, the structure of mouse Act1 was re-modeled by homology modeling using SWISS-Model interface (http://swissmodel.expasy.org) (24). All four key residues (L474, H475, K477 and Y478) are now present on the N-terminal segment of αC helix. Residues L474, K477 and Y478 are exposed to solvent, while H475 is partially exposed, suggesting their potential roles in interactions with other SEFIR domains from IL-17 receptors. Collectively, the structural and functional data from IL-17RB and Act1 SEFIR domains suggest that αC helices in both proteins serve as the ‘hot spot’ for heterotypic SEFIR-SEFIR interactions. In addition, structure based family sequence alignment shows that αC helix is also conserved in IL-17RA and RC, suggesting this helix is a common structural signature for IL-17 receptor signaling (Supplemental Fig. 2).
SEFIR vs TIR
The structure of IL-17RB-SEFIR was submitted for structure similarity search using ssmserver (http://www.ebi.ac.uk/msd-srv/ssm) (25). The result showed that the overall fold of the current structure resembles closest the TIR domains of human TLR10 (PDB 2J67) and human TLR2 (PDB 1o77), though the shared amino acid sequence identity is less than 10% in each pair-wise comparison. The r.m.s. deviation of 104 equivalent Cα atoms between the current structure and the top scoring human TLR10 TIR domain (7% identity) is about 2.6 Å. The central 5-stranded β sheet as well as three helices αA, αB and αE from both SEFIR and TIR structures superimpose well (Supplemental Fig. 1C). The canonical BB′ loop in a TIR domain structure adopts a short helix (αB′) in the current structure (residue 363–373), which is nearly perpendicular to helix αB. The largest difference between TIR and SEFIR domains lies in the CC′ region, helices αC, αD and DD′ loop (Fig. 3B). In the TIR domain structures, the layer of secondary structures containing strand βC, helix αC and CC′ loop are abutting the layer comprised of strand βD, helixαD and DD′ loop, tightly packed without any knot features. However, in SEFIR structure, the DD′ loop shifted by more than 10 Å, occupying the space where the CC′ loop of TIR is located. The structure based sequence alignment shows the 20 A.A. CC′ segment between helix αC and strand βC is a unique insertion in all SEFIR- containing proteins, which is absent from TLR10-TIR (Supplemental Fig. 2).
Discussion
In the present study, we report the structure of the SEFIR domain from IL-17RB at 1.8Å resolution, providing the first atomic view at the unique signaling module that is signature for IL-17 intracellular signaling. The structure of IL-17RB-SEFIR revealed that SEFIR domains adopt similar overall architecture as TIR domains despite overall low sequence homology. However, the SEFIR domain displays large conformational differences compared to its closest resembled structure of TL10-TIR, in helices αC, αD and DD′ loop. The structure of IL-17RB-SEFIR revealed key elements involved in IL-17RB-mediated signaling. Our structural and functional analysis on IL-17RB-SEFIR identified the helix αC as a critical structural motif for heterotypic SEFIR-SEFIR interactions between Act1 and IL-17RB. Specifically, we show single mutations at residue L419 or L422 that locates on the surface of helix αC of IL-17RB-SEFIR abolished the interaction with Act1 and IL-25-induced gene expression.
The availability of the high-resolution structure of IL-17RB-SEFIR allows us to re-assess some previous functional data. We have previously identified a unique region (residues 467–478) in Act1-SEFIR critical for heterotypic association with IL-17RA-SEFIR. In the absence of structural information on SEFIR, we used a distantly related TIR domain as the template for homology modeling, which predicted the secondary structure of this region as a loop, termed CC′ loop. Deletion or mutations in this region of Act1-SEFIR disrupted its hetero-dimerization with IL-17RA and suppressed IL-17A- and IL-17E (IL-25)-dependent signaling (11). In addition, a cell-permeable decoy peptide derived from the A.A. sequence of this CC′ loop disrupts the Act1-IL-17RA interaction and inhibits IL-17A- and IL-17E (IL-25)-induced pulmonary inflammation. The high-resolution crystal structure of IL-17RB-SEFIR provides a better structural template for the new attempt of the homology modeling of Act1-SEFIR. The functionally essential residues (L474, H475, K477 and Y478), located on the previously defined CC′ loop in Act1-SEFIR domain, are now all mapped on the N-terminal section of the helix αC. Future studies will be to test the importance of the C-terminal section of the helix αC, which may be helpful to improve the design of peptidomimetics as inhibitors for IL-17- and IL-25-dependennt autoimmune inflammatory diseases. Nevertheless, our structure based sequence alignment shows that the helix αC is a conserved structural feature for SEFIR domains among IL-17RA, RB, RC and Act1. Collectively, our data suggest that the helix αC might be a conserved structural motif that is key to heterotypic SEFIR-SEFIR interactions.
The structure of IL-17RB-SEFIR provides insights into the revolutionary pathway of the SEFIR and TIR domains as well as the structural basis for the specificity of SEFIR versus TIR domain in their respective signaling pathways. Although various structural motifs were identified as important elements in mediating TIR-TIR complex assembly, the BB′ loop was found as one of the most critical structural elements in mediating TIR mediated interactions. The BB′ loops in TIR domains were observed in different conformations suggesting its flexibility in nature (10, 26). In contrast, the counterpart in IL-17RB-SEFIR is helix αB′, which is tethered to αB and αC through hydrophobic interactions and a hydrogen bond between W378 and Q366, suggesting a stable and relatively rigid conformation. We previously showed that the helix αB′ of Act1 was not essential to heterotypic SEFIR-SEFIR interactions, since the deletion of this segment from Act1 did not have significant impact on its interaction with IL-17RA (11). We now show that the helix αB′ in SEFIR is instead essential for homotypic SEFIR-SEFIR interactions, since the deletion of the helix αB′ (D-BB′, residues 425–432 deleted) in Act1-SEFIR domain greatly reduced its self-association and IL-25 (IL-17E)-mediated gene expression (Supplemental Fig. 4).
To provide a better picture on SEFIR-mediated protein-protein interactions that are important for IL-17 receptor signaling, we constructed a model of Act1-SEFIR:IL-17RB-SEFIR hetero-dimer complex. We used HADDOCK server (http://haddock.science.uu.nl) (27) to dock the crystal structure of IL-17RB-SEFIR with the homology modeled Act1-SEFIR, applying the identified key functional residues as constraints. In this model, the interactions between Act1 and IL-17RB-SEFIR domains are predominantly clustered at helix αC and the tip of helix αB (Fig. 4A). The αB and αC helices from IL-17RB-SEFIR domain are almost perpendicular to their counterparts in Act1, forming a four-helix bundle at the interface, involving hydrophobic and charge-charge interactions. Specifically, residue I434 (αB), the aliphatic side chain of residue R481 and residues M482 (αC) of Act1 are stacked on a hydrophobic groove on the surface of IL-17RB-SEFIR, comprised of residues P375, V376 from αB and L422 and L426 from αC, involving extensive hydrophobic interactions (Fig. 4B). Y478 of Act1 is also abutting this hydrophobic platform with its edges. The basic residues K477 and R481 are forming favorable interactions with a negatively charged patch on the surface of IL-17RB-SEFIR, contributed mainly by two acidic residues, E414 and D418. The model of Act1-IL-17RB heterodimer suggests K477 and Y478 of Act1 as important residues for mediating heterotypic SEFIR-SEFIR interactions, which agrees with our previously reported data on the interaction of Act1 with IL-17RA (11). L474 and H475 of Act1 however do not appear from this model to contribute much to the binding of Act1 to IL-17RB, suggesting their possible roles in receptor binding specificity since these residues were found as critical for IL-17RA binding and signaling. Clearly, more mutagenesis analysis will be needed in the future to fully characterize the interactions between Act1 and IL-17 receptors.
Fig. 4. A model of heterodimer of Act1-SEFIR and IL-17RB-SEFIR domains.

(A) Act1-SEFIR structure was modeled by SWISS-Model interface (http://swissmodel.expasy.org), using the crystal structure of IL-17RB-SEFIR as the template. Subsequent docking of Act1-SEFIR: IL-17RB-SEFIR was carried out with HADDOCK server (http://haddock.science.uu.nl), using identified key functional residues as constraints. Notice the helices αB′ (red) from both Act1-SEFIR and IL-17RB-SEFIR are exposed and not involved in the hetero-dimerization. (B) IL-17RB-SEFIR crystal structure is shown as electropotential surface, while residues on Act1-SEFIR are shown as sticks (yellow ones on αC and magenta ones onαB). The contact residues from Act1-SEFIR and IL-17RB-SEFIR are labeled in black and blue, respectively.
Interestingly, the modeled complex structure of IL-17RB-SEFIR and Act1-SEFIR implies a dual ligand-binding mode for SEFIR domain with differential structural motifs (helix αC versus helix αB′) engaging in either hetero- or homo- SEFIR-SEFIR interactions. Specifically, in this model, the αB′ helices from both Act1 and IL-17RB SEFIR domains are located distal to the four-helix bundle core that is comprised of αB/αC helices from both proteins, in a back-to-back fashion, not participating in any direct interactions at the hetero-dimer interface. However the αB′ helices from both proteins are largely exposed, suggesting the hypothesis that they could be potentially involved in homo-dimerization of SEFIR domains. The model is in consistent with our observation that the deletion of the αB′ helix from Act1 did not affect its heterotypic interaction with IL-17RA (11), but instead reduced the homotypic association (Supplemental Fig. 4). Nevertheless, the homotypic association of SEFIR domains is rather complex. The exact stoichiometry and detailed mechanism by which SEFIR domain containing proteins associate with each other are not known. The purified IL-17RB-SEFIR displays as predominantly monomeric in solution, based on size exclusion chromatography (Supplemental Fig. 3) and dynamic light scattering. It is also a monomer in the crystal structure as considered by PISA server (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (28). However, Act1 was shown to form homo-dimer or homo-oligomers through its SEFIR domain (29). The cumulative data from the past and current work suggest that Act1 self-association is required for IL-17E (IL-25) signaling and this homotypic interaction could be mediated through helix αB′ in the SEFIR domain.
Supplementary Material
Table I.
Data and Statistics.
| Se Peak | Native | |
|---|---|---|
| Beamline | 19-ID, APS | 19-ID, APS |
| Wavelength (Å) | 0.97918 | 0.97918 |
| Space Group | I222 | I222 |
| Cell Parameters: a, b, c (Å) | 54.8,65.8,89.9 | 55.0,65.8,89.6 |
| Resolution (Å) | 50 - 2.0 | 50 -1.8 |
| Total Reflections | 75,371 | 104,869 |
| Unique Reflections | 11,167 (973) | 15,902 (1,478) |
| Redundancy | 6.7 (5.1) | 6.6 (4.9) |
| Completeness (%) | 98.2 (87.0) | 99.4 (94.9) |
| I/σ | 19.8 (3.0) | 19.2 (2.3) |
| Rsym (%) | 9.6 (40.6) | 10.0 (55.5) |
|
| ||
| Refinement Statistics | ||
|
| ||
| Resolution range used, (Å) | 32.9-1.8 | |
| No. reflections used | 15,387 | |
| Rwork/Rfree | 18.0/22.3 | |
| R.m.s. deviations | ||
| bond lengths (Å) | 0.007 | |
| bond angles (°) | 1.003 | |
| Ramachandran Values | ||
| Preferred regions (%) | 97.0 | |
| Allowed regions (%) | 3.0 | |
Values in parentheses are for the highest resolution shell, 2.07–2.0 Å (Se Peak) and 1.86–1.80 Å (native).
Rsym = Σ |Iobs − Iavg|/ Σ Iavg; Rwork = Σ|| Fobs | - |Fcalc||/Σ Fobs
Rfree was calculated using 5% of data.
Acknowledgments
We thank the staff of beamline 19ID at the Advanced Photon Source for their support. We also thank Xiaolan Zhao (Genomics Core, Lerner Research Institute, Cleveland Clinic, OH, USA) for plasmid sequencing.
J.D. is supported by NIH grants AI081928, and by the Oklahoma Agricultural Experiment Station at Oklahoma State University under project OKL02618. X.L is supported by a Senior Investigator Award from the American Asthma Foundation, NIH Grants R01HL098935-01A1 and R01NS071996.
Footnotes
Data deposition: Atomic coordinates and structure factors have been deposited with the Protein Data Bank, www.rcsb.org (PDB accession code 3VBC).
All authors declare no conflict of interest.
References
- 1.Angkasekwinai P, Park H, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, Sturton G, Wong SH, McKenzie AN. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120:1324–1331. doi: 10.1016/j.jaci.2007.07.051. [DOI] [PubMed] [Google Scholar]
- 3.Cheung PF, Wong CK, Ip WK, Lam CW. IL-25 regulates the expression of adhesion molecules on eosinophils: mechanism of eosinophilia in allergic inflammation. Allergy. 2006;61:878–885. doi: 10.1111/j.1398-9995.2006.01102.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 6.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 7.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, Chen A, Ren NY, Wang H, Siebenlist U. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. Journal of immunology (Baltimore, Md : 1950) 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nature immunology. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 9.Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, Aronica M, Li X. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. Journal of immunology (Baltimore, Md : 1950) 2009;182:1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 11.Liu C, Swaidani S, Qian W, Kang Z, Sun P, Han Y, Wang C, Gulen MF, Yin W, Zhang C, Fox PL, Aronica M, Hamilton TA, Misra S, Deng J, Li X. A CC′ loop decoy peptide blocks the interaction between Act1 and IL-17RA to attenuate IL-17- and IL-25-induced inflammation. Sci Signal. 2011;4:ra72. doi: 10.1126/scisignal.2001843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maezawa Y, Nakajima H, Suzuki K, Tamachi T, Ikeda K, Inoue J, Saito Y, Iwamoto I. Involvement of TNF receptor-associated factor 6 in IL-25 receptor signaling. Journal of immunology (Baltimore, Md : 1950) 2006;176:1013–1018. doi: 10.4049/jimmunol.176.2.1013. [DOI] [PubMed] [Google Scholar]
- 13.Krumm B, Meng X, Li Y, Xiang Y, Deng J. Structural basis for antagonism of human interleukin 18 by poxvirus interleukin 18-binding protein. Proc Natl Acad Sci U S A. 2008;105:20711–20715. doi: 10.1073/pnas.0809086106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr D Biol Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 15.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 16.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley PaCK. Coot: model building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 20.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
- 22.Lee J, Lee D, Park H, Coutsias EA, Seok C. Protein loop modeling by using fragment assembly and analytical loop closure. Proteins. 2011;78:3428–3436. doi: 10.1002/prot.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike JW, Darnay BG, Choi Y, Wu H. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418:443–447. doi: 10.1038/nature00888. [DOI] [PubMed] [Google Scholar]
- 24.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics (Oxford, England) 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 26.Tao X, Xu Y, Zheng Y, Beg AA, Tong L. An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochemical and biophysical research communications. 2002;299:216–221. doi: 10.1016/s0006-291x(02)02581-0. [DOI] [PubMed] [Google Scholar]
- 27.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nature protocols. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 28.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 29.Mauro C, Vito P, Mellone S, Pacifico F, Chariot A, Formisano S, Leonardi A. Role of the adaptor protein CIKS in the activation of the IKK complex. Biochemical and biophysical research communications. 2003;309:84–90. doi: 10.1016/s0006-291x(03)01532-8. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.