

Abstract
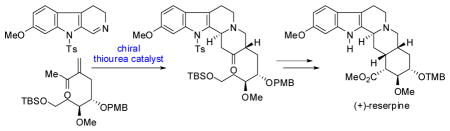
A catalytic, enantioselective synthesis of (+)-reserpine is reported. The route features a highly diastereoselective, chiral catalyst-controlled formal aza-Diels–Alder reaction between a 6-methoxytryptamine-derived dihydro-β-carboline and an enantioenriched α-substituted enone to form a key tetracyclic intermediate. This approach addresses the challenge of setting the C3 stereogenic center by using catalyst control. Elaboration of the tetracycle to (+)-reserpine includes an intramolecular aldol cyclization and a highly diastereoselective hydrogenation of a sterically hindered enoate.
Reserpine (1) has represented an iconic target for organic synthesis since its isolation six decades ago.1 The stereochemically complex pentacyclic structure of this indole alkaloid continues to serve as a forum for exploring the frontiers of stereoselective synthesis, and has inspired some of the most important work in the history of the field.2 Despite the scope of this effort, each of the successful approaches to this molecule has relied on the same fundamental strategy, namely, a late-stage generation of the C-ring and its embedded C3 stereocenter from a 2,3-seco-derivative (2, Scheme 1).3 This approach is generally complicated by preferential formation of the C3 center with the undesired, thermodynamically favored relative stereochemistry.4 This limitation was overcome in a notable and most elegant manner in the Stork synthesis, wherein the desired C3 configuration was installed through a kinetically-controlled cyclization of an amino-nitrile 2,3-seco-derivative. 3i,q,5
Scheme 1.
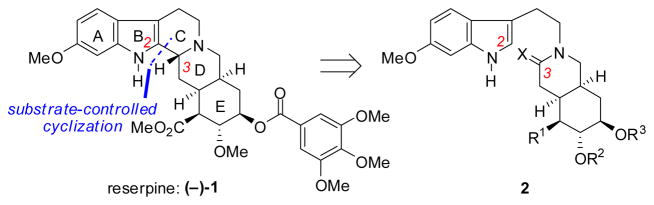
Synthetic strategy common to all previous successful approaches to reserpine
We envisioned an alternative approach to the reserpine framework focused on specifically targeting control over the C3 stereogenic center by means of a stereoselective formal aza-Diels–Alder (FADA) reaction between two fragments of comparable size (3 and 4, Scheme 2).6 Although high levels of substrate-induced stereocontrol were deemed unlikely in such a coupling reaction, we were encouraged by the prospect of selectively introducing the key C3 stereocenter through the use of the chiral catalyst-controlled formal aza-Diels–Alder (FADA) reaction discovered recently in our laboratories.7 Herein, we describe the successful application of the asymmetric FADA methodology to a catalytic enantioselective total synthesis of (+)-reserpine.8
Scheme 2.
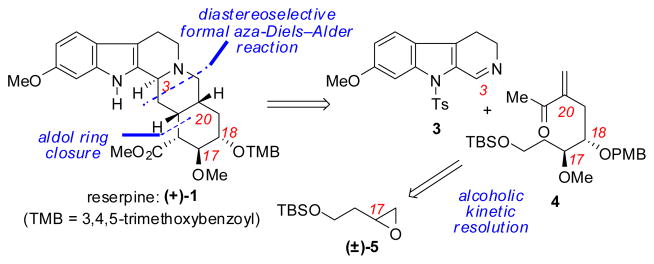
Retrosynthetic analysis of (+)-reserpine
The synthetic efforts toward the requisite enone component 4 began with a highly selective alcoholic kinetic resolution of racemic terminal epoxide 5. The differentially protected 4-carbon triol 6 was obtained in 96% ee through the use of oligomeric cobalt salen catalyst 8,9 employing benzyl alcohol as the nucleophile (Scheme 3). This procedure proved more efficient and reliable in our hands than routes originating from malic acid,10 particularly when applied on multi-gram scale. Elaboration of protected alcohol 6 to aldehyde 7 was accomplished in a 3-step sequence consisting of methylation of the secondary alcohol, subsequent hydrogenolysis of the benzyl ether, and Swern oxidation of the resulting primary alcohol. The α-methoxy group of aldehyde 7 then served to direct a chelation-controlled diastereoselective allylation, thereby installing the adjacent C18 stereogenic center and providing vinyl bromide 9 as a single diastereomer.11 Finally, elaboration to enone 4 was accomplished via protection of the C18 alcohol, followed by lithium-halogen exchange and subsequent addition into N-methoxy-N-methylacetamide.
Scheme 3.

Stereoselective synthesis of enone 4
The key coupling of enone 4 and 6-methoxytryptamine-derived dihydro-β-carboline 37 to generate tetracyclic ketone 11 was then examined under a series of conditions (Scheme 4). The FADA reaction could be carried out with a small excess of enone 4 (1.2 equivalents) relative to imine 3 only with primary amine catalysts, consistent with previous observations employing simple, hindered enone derivatives.12 The degree of intrinsic substrate-induced diastereocontrol was evaluated using achiral amine promoters. With stoichiometric n-hexylamine,13 ketones 11 and 12, which contain a trans-relationship between the newly formed C3 and C20 stereocenters, were generated in a 1:1 diastereomeric ratio (dr) (entry 1). In contrast, high levels of chiral catalyst-controlled diastereoselectivity were observed in the presence of 20 mol % aminothiourea 10,7 providing the desired diastereomer 11 in 76% isolated yield. Notably, the enantiomeric primary aminothiourea ent-10 induced a reversal of diastereoselectivity in the FADA reaction to afford ketone 12 selectively (entry 3).
Scheme 4.
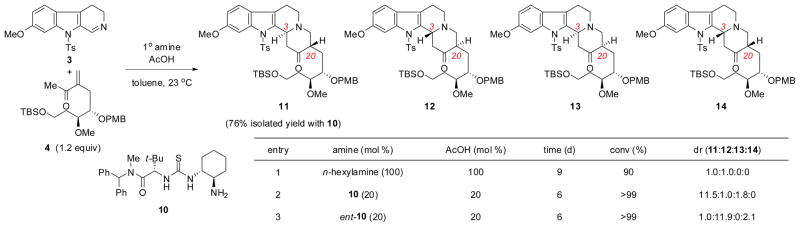
Diastereoselective thiourea-catalyzed formal aza-Diels–Alder reactions
The closure of the E-ring to complete the carbon skeleton of reserpine was effected by an intramolecular aldol reaction of keto-aldehyde 15 (Scheme 5). Intermediate 15 was obtained in two steps from FADA adduct 11 through cleavage of the primary TBS ether with pyridine-buffered HF and oxidation of the resulting primary alcohol with the Dess–Martin periodinane. Treatment of crude aldehyde 15 with piperidine and catalytic TsOH resulted in an intramolecular enamine aldol reaction to afford C15 tertiary alcohol 16 as a single diastereomer. Pinnick oxidation of aldehyde 16 to the corresponding carboxylic acid followed by esterification with diazomethane provided methyl ester 17.
Scheme 5.

Completion of the synthesis of (+)-reserpine
Having efficiently accessed the pentacyclic framework of reserpine by means of the thiourea-catalyzed FADA reaction and a subsequent aldol cyclization, the key remaining challenge involved reduction of the hindered C15 alcohol of 17 with introduction of the desired cis-fusion at the D- and E-ring junction. While attempts to obtain the desired reduction product directly through radical deoxygenation were not fruitful, an elimination/reduction strategy proved successful. Regioselective elimination to α,β-unsaturated ester 18 was induced via trifluoroacetylation of alcohol 17 and subsequent elimination with DBU. After extensive evaluation of both homogeneous and heterogeneous catalytic systems under a range of conditions, cationic iridium complex 20,14 bearing the non-coordinating BArF counteranion, was identified as uniquely effective in the reduction of enoate 18.15 In addition, hydrogenation proceeded with a significant degree of facial selectivity (6:1 dr), ultimately affording the desired saturated ester 19 in 44% isolated yield (81% based on recovered olefin 18). The stereochemical assignment of compound 19 was confirmed by X-ray crystallographic analysis.
With the fully elaborated pentacycle in hand, completion of the synthesis required only a global deprotection and installation of the trimethoxybenzoyl ester on the E-ring. Thus, treatment of 19 sequentially with TfOH and sodium-mercury amalgam resulted in cleavage of the PMB ether and tosyl protective groups, respectively. The resulting C18 secondary alcohol was esterified using previously reported conditions to deliver reserpine ((+)-1).3i
The enantioselective total synthesis of reserpine was accomplished in 19 steps in the longest linear sequence from racemic epoxide 5. The convergent approach relied on chiral catalysts to provide access to coupling component 4 and to address the historically problematic installation of the C3 stereogenic center. Further application of the aminothiourea-catalyzed formal aza-Diels–Alder reaction in the synthesis of complex alkaloids of both natural and synthetic origin is anticipated.
Supplementary Material
Acknowledgments
This work was supported by the NIGMS (GM-43214, GM-59316, and GM-69721), and by predoctoral fellowship support to N.S.R from Boehringer Ingelheim and to M.A.M. from Eli Lilly. We thank Dr. Shao-Liang Zheng at the Center for Crystallographic Studies at Harvard University for X-ray data collection and structure determination. We thank Dr. Yu-Sheng Chen at ChemMatCARS, APS, for his assistance with single-crystal data. ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant number NSF/CHE 0822838. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information Available Complete experimental procedures, characterization data, and 1H and 13C NMR spectra of all isolated intermediates, and crystallographic data for compound 19. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Müller J, Schlittler E, Bein H. Experientia. 1952;8:338. doi: 10.1007/BF02174406. [DOI] [PubMed] [Google Scholar]
- 2.For a review of synthetic efforts directed toward reserpine, see: Chen FE, Huang J. Chem Rev. 2005;105:4671. doi: 10.1021/cr050521a.For more recent approaches, see: Barcan AG, Patel A, Houk KN, Kwon O. Org Lett. 2012;14:5388. doi: 10.1021/ol302265z.Yar M, Arshad M, Akhtar MN, Shahzad SA, Khan IU, Khan ZA, Ullah N, Ninomiya I. Eur J Org Chem. 2012;3:26.Huang J, Chen F-E. Helv Chim Acta. 2007;90:1366.
- 3.(a) Woodward RB, Bader FE, Bickel H, Kierstead RW. Tetrahedron. 1958;2:1. [Google Scholar]; (b) Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. J Am Chem Soc. 1956;78:2023. [Google Scholar]; (c) Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. J Am Chem Soc. 1956;78:2657. [Google Scholar]; (d) Pearlman BA. J Am Chem Soc. 1979;101:6398. [Google Scholar]; (e) Wender PA, Schaus JM, White AW. J Am Chem Soc. 1980;102:6157. [Google Scholar]; (f) Wender PA, Schaus JM, White AW. Heterocycles. 1987;25:263. [Google Scholar]; (g) Martin SF, Grzejszczak S, Rueger H, Williamson SA. J Am Chem Soc. 1985;107:4072. [Google Scholar]; (h) Martin SF, Rueger H, Williamson SA, Grzejszczak S. J Am Chem Soc. 1987;109:6124. [Google Scholar]; (i) Stork G. Pure & Appl Chem. 1989;61:439. [Google Scholar]; (j) Gomez AM, Lopez JC, Fraser-Reid B. J Org Chem. 1994;59:4048. [Google Scholar]; (k) Gomez AM, Lopez JC, Fraser-Reid B. J Org Chem. 1995;60:3859. [Google Scholar]; (l) Chu C-S, Liao C-C, Rao PD. Chem Commun. 1996:1537. [Google Scholar]; (m) Hanessian S, Pan JW, Carnell A, Bouchard H, Lesage L. J Org Chem. 1997;62:465. doi: 10.1021/jo961713w. [DOI] [PubMed] [Google Scholar]; (n) Mehta G, Reddy DS. J Chem Soc, Perkin Trans 1. 2000:1399. [Google Scholar]; (o) Sparks SM, Shea KJ. Org Lett. 2001;3:2265. doi: 10.1021/ol015988w. [DOI] [PubMed] [Google Scholar]; (p) Sparks SM, Gutierrez AJ, Shea KJ. J Org Chem. 2003;68:5274. doi: 10.1021/jo0341362. [DOI] [PubMed] [Google Scholar]; (q) Stork G, Tang PC, Casey M, Goodman B, Toyota M. J Am Chem Soc. 2005;127:16255. doi: 10.1021/ja055744x. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhang LH, Gupta AK, Cook JM. J Org Chem. 1989;54:4708. [Google Scholar]; (b) Lounasmaa M, Berner M, Tolvanen A. Heterocycles. 1998;48:1275. [Google Scholar]
- 5.A related cyclization of an amino-nitrile intermediate was recently employed in the syntheses of C3-epimeric natural products venenatine and alstovenine: Lebold TP, Wood JL, Deitch J, Lodewyk MW, Tantillo DJ, Sarpong R. Nat Chem. 2012 doi: 10.1038/nchem.1528.
- 6.Analogous fragment couplings have been applied in the synthesis of related alkaloids: (±)-deserpidine: Szántay C, Blaskó G, Honty K, Baitz-Gács E, Tamás J, Töke L. Leibigs Ann Chem. 1983;8:1292.(±)-yohimbine congeners: Danishefsky S, Langer ME, Vogel C. Tetrahedron Lett. 1985;26:5983.Itoh T, Yokoya M, Miyauchi K, Nagata K, Ohsawa A. Org Lett. 2006;8:1533. doi: 10.1021/ol0530575.Nagata K, Ishikawa H, Tanaka A, Miyazaki M, Kanemitsu T, Itoh T. Heterocycles. 2010;81:1791.
- 7.Lalonde MP, McGowan MA, Rajapaksa NS, Jacobsen EN. J Am Chem Soc. doi: 10.1021/ja310718f. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The three previous reported syntheses of (−)-reserpine have relied on either classical resolution or chiral pool approaches (refs 3i, 3j, 3m).
- 9.White DE, Jacobsen EN. Tetrahedron: Asymmetry. 2003;14:3633. [Google Scholar]
- 10.Pattenden G, González MA, Little PB, Millan DS, Plowright AT, Tornos JA, Ye T. Org Biomol Chem. 2003;1:4173. doi: 10.1039/b308305e. [DOI] [PubMed] [Google Scholar]
- 11.The sequence of allylsilane addition and subsequent PMB protection was adapted from: Evans DA, Rajapakse HA, Stenkemp D. Angew Chem, Int Ed. 2002;41:4569. doi: 10.1002/1521-3773(20021202)41:23<4569::AID-ANIE4569>3.0.CO;2-V.
- 12.No catalysis was observed with proline or related secondary amine catalysts. The proline-catalyzed formal aza-Diels–Alder reaction between dihydro-β-carboline and enones has been shown to require a large excess of enone (30 equivalents) relative to imine in those cases where catalysis is observed: See Refs. 6c and 6d.
- 13.Very low conversions (<10% after 6 d) were obtained using 20 mol % n-hexylamine and 20 mol % acetic acid.
- 14.(a) Vazquez-Serrano LD, Owens BT, Buriak JM. Inorg Chim Acta. 2006;359:2786. [Google Scholar]; (b) Wustenberg B, Pfaltz A. Adv Synth Catal. 2008;350:174. [Google Scholar]
- 15.Attempts to effect this transformation by means of conjugate reductions resulted instead in elimination of the C17 methoxy group.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.