

Summary
CD4+ T-helper (Th) cells are a major cell population that plays an important role in governing acquired immune responses to a variety of foreign antigens as well as inducing some types of autoimmune diseases. There are at least four distinct Th cell subsets (Th1, Th2, Th17, and inducible T-regulatory cells), each of which has specialized functions to control immune responses. Each of these cell types emerge from naive CD4+ T cells after encounter with foreign antigens presented by dendritic cells (DCs). Each Th cell subset expresses a unique set of transcription factors and produce hallmark cytokines. Both T-cell receptor (TCR)-mediated stimulation and the cytokine environment created by activated CD4+ T cells themselves, by `partner' DCs and/or other cell types during the course of differentiation, play an important role in the fate decisions towards distinct Th subsets. Here, we review how TCR-mediated signals in collaboration with the cytokine environment influence the fate decisions of naive CD4+ T cells towards distinct Th subsets at the early stages of activation. We also discuss the roles of TCR-proximal signaling intermediates and of the Notch pathway in regulating the differentiation to distinct Th phenotypes.
Keywords: T-helper cells, TCR signals, cytokines, transcription factors, Notch pathway
Introduction
When naive CD4+ T cells recognize a foreign antigen-derived peptide presented in the context of major histocompatibility complex (MHC) class II on dendritic cells (DCs), they undergo massive proliferation and differentiation into distinct T-helper (Th) cell subsets. There is still considerable uncertainty as to the number of these subsets, the precursor-product relationships among them, and their ability to convert from one to another. At least four different Th cell subsets [Th1, Th2, Th17, and inducible T-regulatory (iTreg) cells] have been studied in great detail. Each cell subset expresses a unique set of transcription factors as well as hallmark cytokines. Indeed, the cytokine environment created by activated CD4+ T cells themselves, by `partner' DCs, and/or other cell types during the course of differentiation is one of the key determinants for the fate decision towards distinct Th subsets (1).
The analysis of how various signaling pathways impinge on Th differentiation is best understood in the context of a two-phase process, consisting of a T-cell receptor (TCR)-driven induction phase, in which key transcription factors are induced or activated, and a cytokine-driven polarization phase, in which the expression of the key factors is amplified and their differentiation completed. Indeed, each subset utilizes a master regulatory transcription factor and a particular signal transducer and activator of transcription (STAT). The relationships are as follows: Th2, GATA-3/STAT5; Th1, T-bet/STAT4; Th17, retinoid orphan receptor γt (RORγt)/STAT3; iTreg, forkhead box protein 3 (Foxp3)/STAT5. Recent studies suggest that Tfh cells may also fit the paradigm with the factors being Bcl-6/STAT3. Interestingly, in many instances, the STAT involved also plays a role in the induction of the master transcriptional regulator (Fig. 1). In this review, we discuss how the signals mediated by the TCR in collaboration with the cytokine environment influence the fate decisions of naive CD4+ T cells towards distinct Th subsets at the early stages of activation as well as the roles of TCR-proximal signaling intermediates and of the Notch pathway in regulating Th subset differentiation.
Fig. 1. T-helper cell subsets.
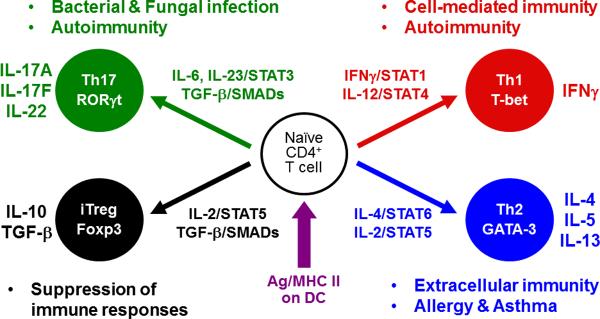
Upon TCR recognition of a foreign antigen-derived peptide presented in the context of MHC class II on dendritic cells, naive CD4+ T cells undergo massive proliferation and differentiation into distinct T-helper (Th) cell subsets. There are at least four different Th cell subsets (Th1, Th2, Th17 and iTreg) that have distinct functions to regulate immune responses. Each cell subset expresses a unique set of transcription factors as well as hallmark cytokines. The cytokine environment created by activated CD4+ T cells themselves, by dendritic cells, and/or other cell types during the course of differentiation plays a critical role in the Th fate decisions. It still remains controversial whether Tfh cells (not depicted in the figure) originate directly from naive CD4+ T cells as a distinct subset similar to Th1, Th2, Th17, and iTreg cells or whether Tfh cells emerge from CD4+ T cells that have already determined their fates toward either Th1, Th2, or Th17 cell subset.
TCR signal strength
Early decision between Th1 and Th2 subsets
Strength of TCR signaling during in vitro differentiation regulates Th1/Th2 polarization. In general, weak signaling favors Th2 differentiation, and stronger signaling leads to Th1 differentiation (2). Priming AND TCR-transgenic CD4+ T cells, specific for moth cytochrome C (MCC), with the altered peptide ligand (APL) K99R, which has a weaker affinity for the AND TCR than does the MCC peptide, preferentially induces Th2 differentiation (3). Jorritsma et al. (4) showed that K99R stimulates weak and transient extracellular signal-regulated kinase (ERK) activation compared to the MCC peptide. The reduced activation of ERK by stimulation with K99R is associated with early IL-4 production by naive AND CD4+ T cells and with a distinct pattern of DNA-binding activity of AP-1 to the Il4 promoter, dominated by a JunB homodimer (4). This is consistent with a previous report showing that JunB directly binds to the Il4 promoter and synergizes with c-Maf to activate an Il4 luciferase reporter gene (5).
We observed that when naive CD4+ T cells of the 5C.C7 TCR-transgenic mouse (also specific for MCC) are stimulated with low concentrations of a peptide from the related protein pigeon cytochrome C (PCC), ERK activation is weak and transient. These `low concentration-stimulated' T cells rapidly induce the expression of the Th2 master regulatory transcription factor GATA-3 and produce IL-2 that in turn activates STAT5. GATA-3 and STAT5 synergize to result in TCR-dependent, IL-4-independent early IL-4 transcription (the induction phase). These low concentration-stimulated T cells undergo the completion of their differentiation into Th2 cells by responding to the endogenously produced IL-4 and continued STAT5 activation (the polarization phase) (6) (Fig. 2). By contrast, stimulating naive 5C.C7 CD4+ T cells with high concentrations of cognate peptide results in failure to undergo Th2 differentiation. TCR-dependent IL-4-independent early GATA-3 expression is suppressed and IL-2R-mediated STAT5 activation is transiently blocked, resulting in failure to produce early IL-4. Under these stimulation conditions, strong and prolonged ERK activation is observed. Blockade of the ERK pathway with a MEK inhibitor allows T cells stimulated with high concentrations of PCC peptide to express early GATA-3 and to respond to endogenously produced IL-2, leading to early IL-4 production, completion of the induction phase and subsequent Th2 polarization process. Thus, strong ERK activation prevents early GATA-3 production and `desensitizes' the IL-2 receptor, thus blocking the Th2 induction phase (Fig. 3). Although these `high concentration-stimulated' T cells express basal levels of GATA-3 and become able to strongly activate STAT5 in response to endogenously produced IL-2 after 40 h of stimulation, they still fail to produce IL-4, implying some mechanism(s), possibly epigenetic gene regulation, through which high concentration stimulation `permanently' silences the Th2 differentiation program.
Fig. 2. Two-step regulation of Th2 differentiation initiated by weak TCR signals.
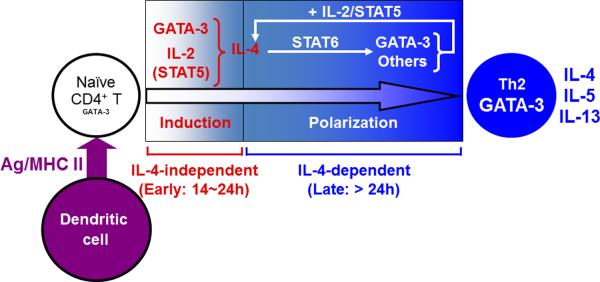
When naive CD4+ T cells receive weak TCR signals, these `weakly stimulated' T cells rapidly induce the expression of the Th2 master regulatory transcription factor GATA-3 in an IL-4/STAT6-independent manner and produce IL-2 that in turn activates STAT5. GATA-3 cooperates with STAT5 to induce TCR-dependent, IL-4/STAT6-independent early IL-4 production that occurs 14-24 h after activation (the induction phase). The endogenously produced IL-4 acts on the weakly stimulated T cells through IL-4R to further upregulate GATA-3 expression in a STAT6-dependent manner. The upregulated GATA-3, in collaboration with continued STAT5 activation, amplifies the production of IL-4. This positive feedback loop leads to the completion of differentiation into Th2 cells (the polarization phase).
Fig. 3. Early Th2 fate decision regulated by strength of TCR signals.
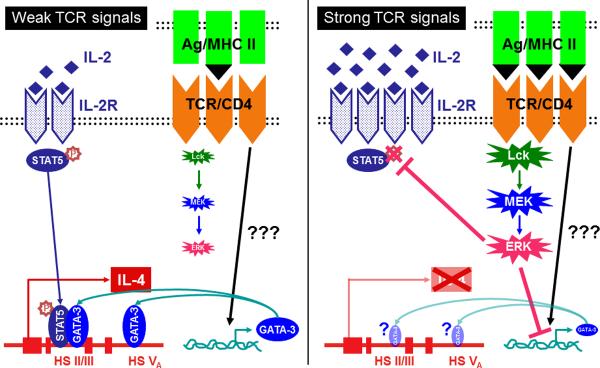
When naive 5C.C7 CD4+ T cells receive strong TCR signals, strong and prolonged ERK activation is induced. This intense ERK activation results in failure to produce early IL-4 by suppressing IL-4-independent TCR-driven early GATA-3 expression and by transiently blocking IL-2R-mediated STAT5 activation (Right). By contrast, when naive 5C.C7 CD4+ T cells receive weak TCR signals, the level of ERK activation is not so strong as to suppress early GATA-3 expression or to inhibit the IL-2/STAT5 pathway. GATA-3 and STAT5 subsequently translocate into the nucleus and bind to the DNase hypersensitivity sites II/III and V in the Il4 gene locus, leading to the induction of early IL-4 production, completion of the induction phase and subsequent Th2 polarization process (Left).
IL-2 also upregulates IL-4Rα expression on activated CD4+ T cells in a STAT5-dependent manner, enhancing the capacity of IL-4 to induce signals in developing Th2 cells. Leonard and colleagues (7) showed that IL-2 activation of STAT5 results in the binding of STAT5 to the IFNγ-activated sequence (GAS) 3 motif in the Il4ra locus during the early stage of Th2 differentiation. IL-4 itself also upregulates IL-4Rα expression, so that the relative importance of IL-2 and IL-4 in enhancing the developing CD4+ T cell's sensitivity to IL-4 will almost certainly depend upon the timing of the availability of these cytokines during the Th2 differentiation process.
The physiological relevance of TCR signal strength-mediated regulation of in vivo Th1/Th2 differentiation has been an open question. Two recent studies revealed that Omega-1, a T2 ribonuclease derived from Schistosoma mansoni egg antigen (SEA), acts on DCs to suppress IL-12 production and to diminish intensity of TCR-mediated signals that naive CD4+ T cells receive, implying that manipulating the function of DCs may result in weak TCR signals even if antigen amount is not limiting, thus favoring in vivo Th2 differentiation (8, 9).
What is the biological significance of strong TCR signal-mediated suppression of Th2 differentiation? We propose that this could be a mechanism by which naive CD4+ T cells are prevented from undergoing excessive Th2 differentiation when they encounter large amounts of foreign antigen that often lead to massive T-cell proliferation and could induce a dangerously high level of allergic sensitization.
Early decision between Th17 and iTreg subset differentiation
Th17 cells play an important role in protection from bacterial and fungal infection and in the development of autoimmunity. It has been proposed that there are three steps that control the differentiation of naive CD4+ T cells into Th17 cells: (i) TGF-β/IL-6-induced differentiation, (ii) IL-21-driven amplification, followed by (iii) IL-23-mediated stabilization (10, 11). We would map the differentiation step as equivalent to the induction phase described above and the amplification and stabilization steps as making up the polarization phase. During the differentiation step, TGF-β and IL-6 in combination with TCR stimulation cause naive CD4+ T cells to express the IL-23R and to induce the major Th17 master regulatory transcription factor RORγt, as well as to produce IL-17A, IL-17F, and IL-21 (12–15). Induction of IL-21 production depends on IL-6-driven STAT3 activation and inducible T-cell costimulator (ICOS) stimulation (16, 17). During the amplification step, IL-21 cooperates with TGF-γ to further upregulate IL-17 production and IL-23R expression in a STAT3-dependent manner. Deletion of either IL-21 or the IL-21R results in diminished Th17 differentiation both in vitro and in vivo (16, 18, 19). If T-regulatory (Treg) cells are depleted by anti-CD25 treatment prior to immunization, endogenous IL-21 is capable of inducing Th17 responses in vivo even in the absence of IL-6, although to a lesser degree than if IL-6 is available (18). Thus, IL-21 appears to play an important role in the positive feedback regulation of Th17 differentiation through its activation of STAT3. However, this conclusion has been challenged by a study reporting that mice deficient in either IL-21 or the IL-21R are still capable of mounting Th17 immune responses in vivo, implying a dispensable role for IL-21 in Th17 differentiation when pro-inflammatory cytokines such as IL-6, IL-1, and TNF-α are abundantly available (20).
The IL-2/STAT5 pathway has been demonstrated to block differentiation to the Th17 fate. When IL-2 is exogenously provided to Th17-polarizing cultures that contain IL-6 and TGF-β, the generation of IL-17-producing cells is impaired, while there is an increase in the frequency of cells that express Foxp3, the master regulatory transcription factor for Treg cells (21). Indeed, TGF-β is essential for the conversion of peripheral naive CD4+ T cells into Foxp3+ cells with regulatory capacity, often referred to as induced Treg (iTreg) cells (22); IL-2 is generally required for such TGF-β-mediated iTreg differentiation (23). Naive CD4+ T cells from Rag1−/− OT-II TCR transgenic mice on a scurfy background, which lack functional Foxp3, still fail to undergo Th17 differentiation under Th17-polarizing conditions if IL-2 is added to the culture (24). STAT5 has been shown to compete with STAT3 for binding to multiple sites within the Il17a-Il17f locus; STAT5 binding to these sites is associated with repressive epigenetic marks across the Il17a promoter region and enhancer elements (24), suggesting that STAT5-binding blocks STAT3 function, thus providing a mechanism through which IL-2 directly represses Th17 differentiation.
Similar to the regulation of the Th1/Th2 fate decision, TCR signal strength has been shown to be a key factor in determining the choice between iTreg and Th17 differentiation. Indeed, if naive CD4+ T cells receive weak TCR signals, they fail to differentiate into Th17 cells but instead express Foxp3, even though they were exposed to Th17-inducing cytokines (25–27). As discussed earlier, strong TCR signals have been demonstrated to transiently inhibit IL-2-driven STAT5 activation despite abundant production of IL-2 by activated CD4+ T cells, thereby blocking Th2 development (6, 28). Thus, it is conceivable that the early fate decision toward the Th17 subset may require strong TCR signals to block the IL-2/STAT5 pathway so that the inhibitory effects of IL-2 can be abrogated. It will be of great importance to clarify the molecular basis underlying TCR signal strength-mediated control of IL-2/STAT5 pathway to better understand the delicate fate balance between Th17 and iTreg subsets that are reciprocally regulated.
TGF-βR-mediated phosphorylation of the receptor-regulated SMAD proteins (R-SMADs) SMAD2 and SMAD3 consists of two aspects, TGF-β-induced phosphorylation of C-terminal serines, leading to association with SMAD4, inducing transcription of TGFβ target genes, and mitogen-activated protein kinase (MAPK)-mediated phosphorylation of a short region that links N-terminal and C-terminal MAD homology domains of R-SMAD suppressing the TGF-β signaling pathway (29–32). MAPK kinase kinases MEKK2 and MEKK3 are the upstream regulators of ERK, p38, and c-Jun N-terminal protein kinases. Su and colleagues (33) reported that in naive CD4+ T cells from mice in which both MEKK2 and MEKK3 have been conditionally deleted, Foxp3 is induced more efficiently in response to limiting amounts of TGF-β than in such cells from wildtype mice. Loss of MEKK2 and MEKK3 in CD4+ T cells results in the impaired phosphorylation of the R-SMADs at their short linker regions in response to TCR/CD28 stimulation, resulting in increased serine phosphorylation at their C-terminus domain in response to TGF-β (33). Moreover, Su and colleagues (33) showed that ERK is responsible for the MEKK2/3-mediated phosphorylation of the short linker region of the R-SMADs, suggesting a negative regulation of the TGF-β signaling pathway by strong TCR-mediated stimulation. This implies that during the differentiation into iTreg cells, weak TCR stimulation more efficiently allows naive CD4+ T cells to receive signals through both the IL-2R and TGF-βR for induction of Foxp3.
Early decision toward Tfh cells
T follicular helper (Tfh) cells `help' T-cell-dependent humoral immune responses by promoting germinal center formation, immunoglobulin class switching, affinity maturation of antibody-secreting B cells and long-lived antibody responses (34). Tfh cells express the transcriptional repressor B-cell lymphoma 6 (Bcl-6) as their master regulator (35–37), the CXC-chemokine receptor 5 (CXCR5) (38, 39), large amounts of programmed cell death 1 (PD-1) (40), B and T-lymphocyte attenuator (BTLA) (41), SLAM-associated protein (SAP) (42), and ICOS (38, 39), but downregulate B lymphocyte-induced maturation protein 1 (Blimp-1) (36). It still remains controversial whether Tfh cells originate directly from naive CD4+ T cells as a distinct subset similar to Th1, Th2, Th17, and iTreg cells or whether Tfh cells emerge from CD4+ T cells that have already determined their fates toward either Th1, Th2, or Th17 cell subset. One key issue regarding the acquisition of Tfh identity is to clarify the timing and the mechanism by which responding CD4+ T cells acquire expression of Bcl-6 and CXCR5. CXCR5 expression is essential for activated CD4+ T cells to migrate to the follicles. In the follicles, the CD4+ T cells interact with B cells that express the same cognate peptide that had been presented by DCs in the priming process that led to their becoming functional Tfh cells.
Mice with a T cell-specific deletion of Stat3 (Stat3fl/fl × CD4Cre-transgenic) exhibit a substantial diminution in the frequency of CD4+CXCR5+ cells in vivo in response to immunization with KLH in complete Freund's adjuvant (CFA), a phenotype resembling that seen in IL-6 or IL-21 deficiency (43). By contrast, mice deficient in either IL-6 or IL-21 have recently been reported to generate Tfh cells normally upon infection with lymphocytic choriomeningitis virus (LCMV), indicating a redundant role of these cytokines in Tfh cell development (44). Although the combined absence of IL-6 and IL-21 results in the failure to secrete antigen-specific IgG upon LCMV infection, such mice display only a modest reduction in Tfh cell generation (45). Since STAT3 dependence of LCMV-generated Tfh cells has not been determined, it is uncertain as to whether IL-6 and IL-21 may be replaced by another STAT3 activator in this response or whether a STAT3-independent pathway for Tfh generation exists.
Two days after LCMV infection of mice that have received CD4+ T cells expressing a SMARTA TCR specific for an LCMV glycoprotein, the donor CD4+ T cells fall into two subpopulations: CD25hi cells, expressing high levels of Blimp1, undergoing a program of T-effector (Teff) cell differentiation, and CD25int cells, expressing Bcl-6 and CXCR5, undergoing differentiation to Tfh cells (46). The failure of CD25hi cells to become Tfh cells suggests an inhibitory role for IL-2-generated signals in the development of Tfh cells in vivo. Subsequently, four groups (47–50) independently reported that the IL-2/STAT5 pathway blocks the generation of Tfh cells by inducing Blimp1, resulting in suppression of Bcl-6 expression. Interestingly, this crucial role for the IL-2/STAT5 pathway in the fate decision toward either Teff or Tfh cells through regulation of the expression of the two mutually exclusive transcriptional regulators, Blimp1 and Bcl-6, resembles that observed for the differentiation into either iTreg or Th17 subset, in which Foxp3 and RORγt reciprocally regulate one another. Given the evidence shown by McHeyzer-Williams and colleagues (51) that the generation and function of Tfh cells depends on the strength of TCR binding to a foreign peptide/MHC class II complex, TCR signal strength appears to be one of the key determinants for the early fate decision toward Tfh cells rather than Teff cells as well through the transient blockade of IL-2-driven STAT5 activation, thereby abrogating the inhibitory effects of IL-2 on Bcl-6 expression and preventing Blimp-1 from being induced.
TCR-proximal signaling intermediates
Src-family tyrosine kinases Lck and Fyn
Lck and Fyn are Src-family tyrosine kinases involved in optimal T-cell activation. Lck associates with the cytoplasmic tail of CD4 and becomes activated upon TCR ligation, leading to phosphorylation of tyrosine residues in the CD3ζ chain to which ζ-chain-associated protein kinase 70-kDa (ZAP-70) is recruited via its Src homology region 2 (SH2) domain. Lck−/− mice exhibit a prominent but incomplete developmental arrest at the transition from the double negative (DN) to the double positive (DP) stage of thymocyte development. The few peripheral T cells that do develop have markedly impaired responses to TCR stimulation (52). The importance of Lck in T-cell development makes difficult the assessment of the role of Lck in peripheral T-cell activation in such mice. To inactivate the function of Lck in peripheral T cells without affecting thymocyte development, a transgenic mouse line was generated that expressed a dominant negative enzymatically inactive form of Lck under the control of the distal Lck promoter (DLGKR); DLGKR is expressed in peripheral T cells but not in thymocytes. Nakayama and colleagues (53) reported that CD4+ T cells from DLKGR transgenic mice exhibit impaired Th2 differentiation in vitro. Moreover, CD4+ T cells from mice expressing a transgene encoding a dominant-negative H-Ras under the control of the proximal Lck promoter show a phenotype similar to that of DLGKR-expressing cells, leading the authors to conclude that TCR-mediated Lck/Ras/ERK activation is required for Th2 differentiation through enhancing STAT6 activation in response to IL-4 (54) and/or preventing ubiquitin/proteasome-mediated degradation of GATA-3 by inhibiting the activity of Mdm2, an E3 ubiquitin ligase that acts on GATA-3 (55). These results provide a conflicting view of the role of ERK function in Th2 differentiation to that described in the previous section of this review and need to be considered in terms of our two-phase model of Th2 polarization. Indeed, studied in detail, naive CD4+ T cells from the DLKGR transgenic mice actually produce significantly more IL-4 than do those from the littermate control mice during the Th2 induction phase (53), indicating an inhibitory role of the Lck/Ras/ERK cascade in TCR-driven IL-4 production. However, IL-2 production by naive CD4+ T cells from the DLKGR transgenic mice is significantly diminished (53). Reduced IL-2 production could account for the poorer Th2 differentiation observed in such cells, since STAT5 activation is essential for both the induction and polarization phases of Th2 differentiation; the degree of STAT5 activation may have fallen below the threshold level during the polarization phase in cells in which Lck or Ras is not functional. Indeed, this result is consistent with data we have reported showing that blockade of the ERK pathway leads to a substantial diminution in IL-2 production by 5C.C7 CD4+ T cells stimulated with low concentrations of PCC peptide (6) and that in the presence of a MEK inhibitor, Th2 differentiation initiated by weak TCR signals requires addition of IL-2. Interestingly, the expression of Mdm2 is aberrantly induced by loss of the Stat5 gene in CD4+ T cells, implying that the IL-2/STAT5 pathway suppresses Mdm2 expression, protecting GATA-3 from degradation during Th2 differentiation (7), and that Mdm2-mediated degradation of GATA-3 could be secondary to the diminished IL-2 production in cells treated with a MEK inhibitor.
Fyn also plays a critical role in signal transduction through the TCR. Fyn−/− mice show virtually no abnormality in thymocyte development and in the size of the compartment of peripheral T cells (56, 57). Nariuchi and colleagues reported that naive CD4+ T cells from Fyn−/− C57BL/6 mice have enhanced Th2 polarization upon TCR/CD28 stimulation under neutral conditions, where no exogenous polarizing cytokines are added, although the molecular basis for Th2 skewing by loss of Fyn was not identified in their study (58). DeFranco and colleagues (59) found that CD4+ T cells from Fyn−/− DO11.10 TCR transgenic BALB/c mice have greater skewing toward IL-4-producing cells than cells from wildtype DO11.10 mice upon stimulation with cognate peptide in vitro. These reports imply that Fyn plays an inhibitory role in Th2 differentiation.
By contrast, Schwartzberg and Veillette and their colleagues (60, 61) reported that Fyn−/− naive CD4+ T cells are poorer at undergoing Th2 differentiation compared to wildtype counterparts. A possible explanation of their results is based on the recruitment of Fyn to the SH2 domain of SAP when it is bound to the SLAM family receptors that are homotypically engaged during a cognate interaction between a CD4+ T cell and a DC. SAP then phosphorylates the cytoplasmic tail of the SLAM family receptor, recruiting protein kinase Cθ (PKCθ) and leading to the translocation of the NF-κB1 (NF-κB p50) subunit to the nucleus. CD4+ T cells from Nfkb1−/− or Prkcq−/− mice have been reported to be substantially defective in Th2 differentiation in vitro and in vivo due to impaired GATA-3 expression (62, 63).
Since NF-κB1 has no transactivation domain, it must form a complex with other protein(s) to activate NF-κB1-dependent gene expression. Boothby and colleagues (64) identified Bcl-3, which belongs to the IκB family proteins and possesses a transactivation domain, as the partner of NF-κB1 for binding to the κB-like DNA consensus sequence located at 310 to 301 bp upstream of the Gata3 transcription initiation site, indicating that NF-κB1 and Bcl-3 may be important in regulating cell/cell interaction-driven GATA-3 expression (65) and thus indirectly TCR-driven GATA-3 expression. However, given the critical role for the NF-κB pathway in IL-2 production and CD25 expression (66), entities also essential for Th2 differentiation, a careful study is needed to determine the relative importance of NF-κB-mediated GATA-3 upregulation versus enhanced STAT5 phosphorylation during Th2 cell differentiation induced by this pathway. Similarly, the disagreements regarding the effect of deleting Fyn on Th2 differentiation require resolution; we are actively studying this problem.
LAT
Linker for activation of T cells (LAT) is a transmembrane adapter protein that assembles many substrates of ZAP-70 including SH2 domain-containing leukocyte protein of 76-kDa (SLP-76) and phospholipase Cγ1 (PLC-γ1), leading to T-cell development, activation, survival, and homeostasis. Love's and Malissen's groups (67, 68) independently reported that mice with a point mutation converting tyrosine 136 of LAT to phenylalanine (LatY136F mice), which abrogates its interaction with PLC-γ1, exhibit abnormal thymocyte development with impaired positive and negative selection but later develop lymphoproliferative disorders with massive Th2 immune responses in which T cells produce large amounts of Th2 cytokines and B cells secrete high levels of IgG1 and IgE and of autoantibodies. T cells from the LatY136F mice have severe defect in TCR-induced activation of PLC-γ1, nuclear factor of activated T cells (NFAT), Ca2+ influx, IL-2 production, and cell death, whereas TCR-induced ERK activation is intact. These data have been interpreted to indicate that LAT plays a critical role in many signaling intermediates downstream of TCR and that LAT contributes to the suppression of Th2 differentiation.
Given that the phenotype of the LatY136F mice resembles that of Scurfy mice, it was possible that the LatY136F mice might not have Treg cells. Indeed, Zhang and colleagues (69) reported that the LatY136F mice lack Foxp3+ Treg cells in the thymus as well as in the peripheral lymphoid organs. LatY136F bone marrow (BM) cells that were co-transferred with wildtype BM cells into Lat−/− mice fail to develop into Foxp3+ Treg cells. Moreover, adoptive transfer of wildtype CD4+CD25+ T cells into neonatal LatY136F mice protects the recipients from lymphoproliferative disorders. These results indicated that the LAT/PLC-γ1 interaction plays a critical role in the development of Foxp3+ Treg cells (69). However, Malissen and colleagues (70) challenged this conclusion by showing that LatY136F mice on a Foxp3-eGFP reporter background still have Foxp3+ Treg cells in the thymus although threefold diminished in absolute cell number compared to wildtype Foxp3-eGFP reporter mice. Moreover, they showed that adoptive transfer of wildtype Foxp3-eGFP+ Treg cells into neonatal LatY136F mice failed to rescue the recipient mutant mice from lymphoproliferative disorders, implying that LatY136F CD4+ T cells are capable of escaping from the suppression by wildtype Foxp3+ Treg cells (70).
Although we cannot provide a reasonable explanation for the discrepancy between these two studies, we would like to propose a different interpretation for the massive Th2 lymphoproliferative responses in LatY136F mice. IL-2 production by T cells from LatY136F mice is abrogated given the critical role of LAT/PLC-γ1 interaction in the activation of signaling intermediates essential for the induction of IL-2 production (67, 68). Although we discussed in the previous section that the IL-2/STAT5 pathway play a critical role in the differentiation of naïve CD4+ T cells into Th2 subset, this IL-2 function can be substituted by other STAT5-activating cytokines such as IL-7 and thymic stromal lymphopoietin (TSLP). However, IL-2 is indispensable for Foxp3+ Treg cells to exert their suppressor function to responder T cells (71); neither IL-7 nor TSLP can replace IL-2's role in activating the suppressor function of Foxp3+ Treg cells. Indeed, mice lacking either the Il2 or Il2ra gene, in which Foxp3+ Treg cells are present but not functional, develop abnormalities resembling those observed in the LatY136F mice (72–74). Thus, the abrogated IL-2 production by responder T cells carrying the LATY136F mutation could lead to the failure of adoptively transferred wildtype Foxp3+ Treg cells to control lymphoproliferative disorders of the neonatal LatY136F recipient mice.
Itk
T cells express three Tec-family kinases Itk, Rlk, and Tec. The role of Itk (IL-2-inducible T-cell kinase) during T-cell activation and Th1/Th2 differentiation has been extensively studied. Following TCR ligation, Itk is recruited to LAT/SLP-76/PLC-γ1 multi-molecular complex where it activates PLC-γ1, inducing hydrolysis of phosphatidylinositol 4,5-bisphosphate to generate inositol 1,4,5-trisphosphate and diacylglycerol leading to Ca2+ release from the endoplasmic reticulum and to PKCθ activation, respectively (75). Itk−/− mice have diminished in vivo Th2 responses to Leishmania major, and CD4+ T cells from these mice show impaired Th2 differentiation in vitro due to a deficit in the Ca2+/NFATc1 pathway (76). Indeed, T cells from mice lacking NFATc1 in the lymphoid system generated by blastocyst complementation exhibit impaired Th2 cytokine production. Sera from these mice display reduced IgG1 and IgE levels, indicating a critical requirement of NFATc1 for Th2 immune responses (77, 78).
Berg and colleagues reported that CD4+ T cells from Itk−/− 5C.C7 TCR transgenic Rag2−/− mice show aberrant expression of T-bet and polarize toward the Th1 fate in response to weak TCR signals that would normally induce a Th2 response, further arguing for a critical role for Itk in Th2 differentiation (79). Recently, Schwartzberg and colleagues (80) found that when naive Itk−/− CD4+ T cells are stimulated under Th17-polarizing conditions, a defect in the Ca2+/NFATc1 pathway leads to substantial diminution in IL-17A production without affecting IL-17F production or RORγt expression.
A caveat to all of these studies is that a null mutation in the Itk gene leads to abnormal thymocyte development (81). As a consequence, the number of peripheral CD4+ T cells is very low in Itk−/− mice, and the frequency of cells of activated/memory phenotype, as judged by the surface expression of CD44 and CD62L, is greatly increased. Moreover, even if naive CD4+ T cells are carefully prepared by cell sorting, these cells express fewer TCRs than do their wildtype counterparts. The impaired expression of TCR resulting from loss of Itk is corrected by crossing Itk−/− mice onto 5C.C7 TCR transgenic mice on a Rag2−/− background. We carefully purified naive CD4+ T cells from Itk−/− 5C.C7 transgenic Rag2−/− mice and stimulated them with low and high concentrations of PCC peptide presented by CD11c+CD49b− DCs under neutral conditions, where no exogenous polarizing cytokines are added. Itk−/− cells polarized toward the Th2 phenotype comparably to wildtype cells at low peptide concentration but those cells underwent Th2 differentiation at high peptide concentration that induced robust Th1 differentiation in wildtype cells (manuscript in preparation by Pamela Schwartzberg and colleagues). Our results conflict with those reported by Berg and colleagues, but ours appear to make more sense given the critical role of Itk in activating PLC-γ1, which subsequently activates many signaling intermediates including the ERK pathway that is responsible for strong TCR signal-mediated suppression of Th2 differentiation. Generation of conditional Itk-deficient mice will provide an improved picture of the role of Itk in Th subset differentiation.
Notch pathway
The Notch pathway has been demonstrated to play a crucial role in a number of biological events including development of the central nervous system and the vascular system. The mammalian Notch family has four members: Notch1, 2, 3, and 4. They are expressed on the cell surface following various post-translational modifications, including fucosylation by Pofut, glucosylation by Fringe, and S1-cleavage by a Furin-like protease. In mammals, there are five Notch ligands: Jagged (Jag) 1 and 2, and Delta-like (Dll) 1, 3, and 4. When a Notch interacts with a Notch ligand, the γ-secretase complex proteolytically releases the Notch intracellular domain (NICD). NICD translocates into the nucleus, where it displaces a co-repressor complex from CSL (CBF1/RBP-Jκ in vertebrates, Suppressor of Hairless in Drosophila, Lag1 in C. elegans) and recruits a co-activator complex leading to Notch-dependent gene transcription (82).
In the immune system, Notch has been extensively studied in thymic T-cell differentiation and in the development of marginal zone B cells (83). There is accumulating evidence showing that Notch also plays an important role in regulating differentiation of naive CD4+ T cells into distinct Th subsets (Fig. 4). Yasutomo and colleagues (84) first reported that the interaction of Notch3 on CD4+ T cells with the Dll1 ligand plays a critical role in Th1 differentiation. Nussenzweig and colleagues (85) found that lipopolysaccharide (LPS) induces myeloid differentiation factor 88 (MyD88)-dependent Dll4 expression on the CD8α− DC subset and that these Dll4-expressing DCs direct Th1 differentiation in an IL-12-independent but a Notch-dependent manner. Pearce and colleagues (86) reported that enforced expression of either Dll1 or Dll4 on IL-12 p40−/− BM-derived DCs promotes Th1 differentiation in a T-bet-dependent manner and suppresses Th2 development. Osborne and colleagues (87, 88) found that Notch1 ICD (N1ICD) can form a complex with NF-κB1 and c-Rel, allowing these NF-κB isoforms to be retained in the nucleus and to activate IFNγ expression as a result of the binding of the N1ICD/NF-κB complex to the IFNγ promoter. They also showed that the N1ICD binds to the T-bet promoter and claimed that T-bet gene expression is directly regulated by the Notch1 pathway (89).
Fig. 4. Canonical and noncanonical Notch pathways.
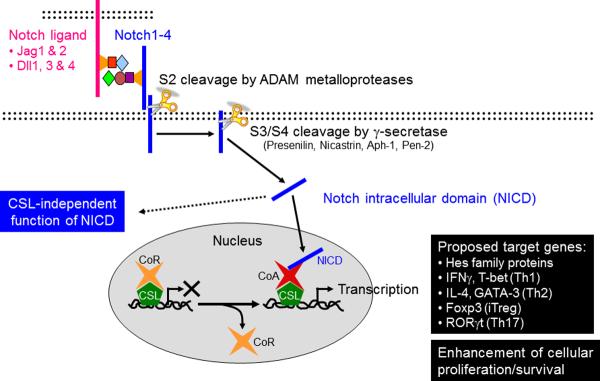
In mammals, there are four Notch receptors, Notch 1–4, and five Notch ligands, Jagged (Jag) 1 and 2, and Delta-like (Dll) 1, 3, and 4. When a Notch interacts with a Notch ligand, the γ-secretase complex proteolytically releases the Notch intracellular domain (NICD). NICD translocates into the nucleus, where it displaces a co-repressor complex from CSL and recruits a co-activator complex leading to Notch-dependent gene transcription. CSL-independent functions of NICD have been also reported. The Notch pathway has been proposed to govern differentiation of naive CD4+ T cells into each of the Th fates by directly controlling the expression of unique master regulatory transcription factors and hallmark cytokines. An alternative view is that the Notch pathway does not directly instruct Th fate decisions but regulates the basal T-cell activation, T-cell expansion and survival, and cytokine production of differentiated Th cells upon secondary stimulation.
Several reports have indicated that Notch also has a role in directing Th2 lineage commitment. CD4+ T cells from mice with a conditional deletion of Rbpj, encoding CSL, by Cre-recombinase driven by the CD4-promoter, fail to undergo Th2 differentiation under non-polarizing conditions (90, 91). Flavell and colleagues (91) proposed a model in which Notch-mediated signals instruct naive CD4+ T cells to undergo differentiation to distinct Th fates by recognizing different Notch ligands with the Dll- and Jag- ligands favoring Th1 and Th2 differentiation, respectively. They identified a CSL binding site in the HS V site of the Il4 locus and found that the binding of a retrovirally overexpressed N1ICD to this site upregulates Il4 gene expression (91). Moreover, Flavell's and Pear's groups (92, 93) independently found that the N1ICD binds to an alternative Gata3 promoter, ~10 kb upstream of a translational start site of the Gata3 gene, and proposed that such binding, in combination with TCR stimulation, can directly induce Gata3 gene transcription in a STAT6-independent manner.
Notch activation has also been reported to regulate differentiation towards other Th phenotypes. Pre-treatment of CD4+ T cells with a γ-secretase inhibitor diminishes the frequency of Foxp3+ cells induced by TGF-β1 due to a reduction in the N1ICD binding to the Foxp3 promoter (94). γ-secretase inhibitor treatment also blocks the recruitment of SMAD proteins to the Foxp3 promoter (94), consistent with a previous finding that N1ICD associates with SMAD3 to integrate Notch and TGF-β signals in myogenic cells (95). The interaction of Notch with Dll4 has been reported to cause a substantial increase in the expression of Th17-related genes in CD4+ T cells stimulated under Th17-polarizing conditions (96). Binding of CSL to the Rorc(t) and Il17a promoter regions is greatly enhanced by Notch/Dll4 interaction but is abrogated by treatment with γ-secretase inhibitor.
The Notch pathway appears to govern differentiation of naive CD4+ T cells into each of the Th fates by directly controlling the expression of unique master regulatory transcription factors and hallmark cytokines. However, given the common machinery activated by Notch interaction with any Notch ligand, it is hard to provide a reasonable explanation for how different Notch ligands instruct naive CD4+ T cells to undergo such a diverse set of Th differentiation outcomes. An alternative view is that the Notch pathway does not directly instruct Th fate decisions but regulates T-cell expansion during the priming period and cytokine production of differentiated Th cells upon secondary stimulation. Kopan, Murphy, and colleagues (97) reported that neither Dll1- nor Jag1-expressing artificial APCs instruct naive DO11.10 CD4+ T cells to differentiate into Th1 or Th2 cells under non-polarizing conditions. Conditional deletion of CSL or of the presenilins, components of the γ-secretase complex, in T cells did not affect T-bet or GATA-3 expression at day 6 of priming under Th1- and Th2-polarizing conditions, respectively. However, the loss of CSL or the presenilins reduces the capacity of differentiated Th cells to secrete effector cytokines upon recall challenge. This reduced cytokine-secretion is associated with and may be due to decreased T-cell proliferation during the priming period (97). These results are consistent with those in earlier reports suggesting that Notch signaling controls global T-cell activation. Osborne and colleagues (88) showed that inhibition of Notch activation dramatically decreases TCR-driven cell division by both CD4+ and CD8+ T cells. Moreover, the inhibition of γ-secretase decreases IL-2 production and CD25 expression, resulting in diminished proliferation of activated CD4+ T cells (98). These data imply an important costimulatory role for the Notch pathway in controlling optimal T-cell activation, although it remains unclear how this pathway controls the basal activation of T cells.
Our unpublished observations indicate that the Notch pathway is required for the Th2 differentiation induced by weak TCR signal under non-polarizing conditions. However, in contrast to previous reports by Flavell's and Pear's groups (91–93), we found that early induction of both GATA-3 expression and IL-4 production by weak TCR signals during the induction phase was intact in naive CD4+ T cells from the mice deficient in either CSL or the presenilins, but IL-2 production was greatly impaired. Consistent with our previous reports demonstrating the central role of IL-2 in Th2 differentiation (6, 99), CD4+ T cells lacking CSL or presenilins failed to complete the polarization phase of Th2 differentiation since IL-4-dependent amplification of IL-4 production and GATA-3 expression during the polarization phase was diminished due to the reduced IL-2 production resulting in limited STAT5 activation. Exogenous IL-2 added to the culture after the induction phase restored IL-4 production and subsequent GATA-3 expression and thus corrected the impaired Th2 differentiation in CD4+ T cells deficient in CSL or presenilins. These results imply that during Th2 differentiation, TCR-dependent GATA-3 expression and IL-4 production do not require direct binding of the Notch/CSL complex to these gene loci. In the absence of Notch signaling, the failure to produce sufficient amounts of IL-2 to sustain IL-4 production during the polarization phase is responsible for defective Th2 differentiation. We also found that while Dll4 and Jag1 are expressed on splenic myeloid DCs (CD11c+CD49b−DCIR2+CD11b+CD8α−) used to stimulate naive CD4+ T cells with a cognate peptide, only Dll4 was responsible for inducing Th2 differentiation initiated by weak TCR signals, arguing against the proposed model where Dll and Jag ligands play distinct roles in instructing naive CD4+ T cells towards Th1 and Th2 phenotypes, respectively (91).
Concluding remarks
For the last decade or two, the advance of technology has allowed us to address the questions of how the differentiation of naive CD4+ T cells towards distinct Th subsets is regulated. It has become clear that the interplay of the critical master regulators and of the stimuli that control their activation is at the center of these differentiation decisions. Here, we have emphasized the role of TCR signal strength in controlling these processes and in helping to shape the cytokine environment that is often deterministic in the polarization process. Such information will be of great value both in efforts to understand the basis of polarization in response to pathogen infection and to develop interventions that can control the `direction' of polarization or alter the polarized state after it has been established.
Acknowledgments
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA.
Footnotes
The authors declare no competing financial interests.
References
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 3.Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J Immunol. 1997;158:4237–4244. [PubMed] [Google Scholar]
- 4.Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naive CD4+ T cells. J Immunol. 2003;170:2427–2434. doi: 10.4049/jimmunol.170.5.2427. [DOI] [PubMed] [Google Scholar]
- 5.Li B, Tournier C, Davis RJ, Flavell RA. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 1999;18:420–432. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. 2005;202:793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao W, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor α-chain expression. Nat Immunol. 2008;11:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everts B, et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. 2009;206:1673–1680. doi: 10.1084/jem.20082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinfelder S, et al. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1) J Exp Med. 2009;206:1681–1690. doi: 10.1084/jem.20082462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of TH17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 12.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Mangan PR, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 14.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 15.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 16.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 17.Bauquet AT, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou L, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2008;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 20.Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–1838. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]
- 21.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 24.Yang XP, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iezzi G, et al. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci USA. 2009;106:876–881. doi: 10.1073/pnas.0810769106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-κ-dependent manner. J Immunol. 2011;186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee IH, Li WP, Hisert KB, Ivashkiv LB. Inhibition of interleukin 2 signaling and signal transducer and activator of transcription (STAT)5 activation during T cell receptor-mediated feedback inhibition of T cell expansion. J Exp Med. 1999;190:1263–1274. doi: 10.1084/jem.190.9.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 30.Kretzschmer M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 32.Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005;44:12546–12553. doi: 10.1021/bi050560g. [DOI] [PubMed] [Google Scholar]
- 33.Chang X, Liu F, Wang X, Lin A, Zhao H, Su B. The kinases MEKK2 and MEKK3 regulate transforming growth factor-β-mediated helper T cell differentiation. Immunity. 2011;34:201–212. doi: 10.1016/j.immuni.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 35.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cells. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Breitfeld D, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaerli P, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haynes NM, et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 41.M'Hidi H, et al. High expression of the inhibitory receptor BTLA in T-follicular helper cells and in B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Am J Clin Pathol. 2009;132:589–596. doi: 10.1309/AJCPPHKGYYGGL39C. [DOI] [PubMed] [Google Scholar]
- 42.Yusuf I, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150) J Immunol. 2010;185:190–202. doi: 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poholek AC, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010;185:313–326. doi: 10.4049/jimmunol.0904023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eto D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi YS, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. 2012;13:405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ballesteros-Tato A, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. doi: 10.1084/jem.20111174. (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nurieva RI, et al. STAT5 protein negatively regulates T follicular hlper (Tfh) cell generation and function. J Biol Chem. 2012;287:11234–11239. doi: 10.1074/jbc.M111.324046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molina TJ, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 53.Yamashita M, Hashimoto K, Kimura M, Kubo M, Tada T, Nakayama T. Requirement for p56lck tyrosine kinase activation in Th subset differentiation. Int Immunol. 1998;10:577–591. doi: 10.1093/intimm/10.5.577. [DOI] [PubMed] [Google Scholar]
- 54.Yamashita M, et al. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc Natl Acad Sci USA. 1999;96:1024–1029. doi: 10.1073/pnas.96.3.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamashita M, et al. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem. 2005;280:29409–29419. doi: 10.1074/jbc.M502333200. [DOI] [PubMed] [Google Scholar]
- 56.Stein PL, Lee HM, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y. [DOI] [PubMed] [Google Scholar]
- 57.Appleby MW, Gross JA, Cooke MP, Levin SD, Qian X, Perlmutter RM. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell. 1992;70:751–763. doi: 10.1016/0092-8674(92)90309-z. [DOI] [PubMed] [Google Scholar]
- 58.Tamura T, et al. Impairment in the expression and activity of Fyn during differentiation of naive CD4+ T cells into the Th2 subset. J Immunol. 2001;167:1962–1969. doi: 10.4049/jimmunol.167.4.1962. [DOI] [PubMed] [Google Scholar]
- 59.Mamchak AA, et al. Normal development and activation but altered cytokine production of Fyn-deficient CD4+ T cells. J Immunol. 2008;181:5374–5385. doi: 10.4049/jimmunol.181.8.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cannons JL, et al. SAP regulates TH2 differentiation and PKC-θ-mediated activation of NF-κB1. Immunity. 2004;21:693–706. doi: 10.1016/j.immuni.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 61.Davidson D, et al. Genetic evidence linking SAP, the X-linked lymphoproliferative gene product, to Src-related kinase FynT in TH2 cytokine regulation. Immunity. 2004;21:707–717. doi: 10.1016/j.immuni.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 62.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-κB in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 63.Marsland BJ, Soos TJ, Späth G, Littman DR, Kopf M. Protein kinase C θ is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J Exp Med. 2004;200:181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nolan GP, et al. The bcl-3 proto-oncogene encodes a nuclear Iκ-like molecule that preferentially interacts with NF-κB p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 67.Aguado E, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- 68.Sommers CL, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 69.Koonpaew S, Shen S, Flowers L, Zhang W. LAT-mediated signaling in CD4+CD25+ regulatory T cell development. J Exp Med. 2006;203:119–129. doi: 10.1084/jem.20050903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, et al. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J Immunol. 2008;180:1565–1575. doi: 10.4049/jimmunol.180.3.1565. [DOI] [PubMed] [Google Scholar]
- 71.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 73.Sadlack B, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- 74.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 75.Schwartzberg PL, Finkelstein LD, Readinger JA. TEC-family kinases: regulators of T-helper-cell differentiation. Nat Rev Immunol. 2005;5:284–295. doi: 10.1038/nri1591. [DOI] [PubMed] [Google Scholar]
- 76.Fowell DJ, et al. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 1999;11:399–409. doi: 10.1016/s1074-7613(00)80115-6. [DOI] [PubMed] [Google Scholar]
- 77.Yoshida H, et al. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity. 1998;8:115–124. doi: 10.1016/s1074-7613(00)80464-1. [DOI] [PubMed] [Google Scholar]
- 78.Ranger AM, et al. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity. 1998;8:125–134. doi: 10.1016/s1074-7613(00)80465-3. [DOI] [PubMed] [Google Scholar]
- 79.Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity. 2004;21:67–80. doi: 10.1016/j.immuni.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 80.Gomez-Rodriguez J, et al. Differential expression of IL-17A and IL-17F is coupled to TCR signaling via Itk-mediated regulation of NFATc1. Immunity. 2009;16:587–597. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity. 1995;3:757–769. doi: 10.1016/1074-7613(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 82.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stanley P, Guidos CJ. Regulation of Notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. 2009;230:201–215. doi: 10.1111/j.1600-065X.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- 84.Maekawa Y, et al. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–559. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 85.Skokos D, Nussenzweig MC. CD8− DCs induce IL-12-independent Th1 differentiation through Delta 4 Notch-like ligand in response to bacterial LPS. J Exp Med. 2007;204:1525–1531. doi: 10.1084/jem.20062305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun J, Krawczyk CJ, Pearce EJ. Suppression of Th2 cell development by Notch ligands Delta1 and Delta4. J Immunol. 2008;180:1655–1661. doi: 10.4049/jimmunol.180.3.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shin HM, et al. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Palaga T, Miele L, Golde TE, Osborne BA. TCR-mediated Notch signaling regulates proliferation and IFN-γ production in peripheral T cells. J Immunol. 2003;171:3019–3024. doi: 10.4049/jimmunol.171.6.3019. [DOI] [PubMed] [Google Scholar]
- 89.Minter LM, et al. Inhibitors of γ-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol. 2005;6:680–688. [PubMed] [Google Scholar]
- 90.Tanigaki K, et al. Regulation of αβ/γδ T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity. 2004;20:611–622. doi: 10.1016/s1074-7613(04)00109-8. [DOI] [PubMed] [Google Scholar]
- 91.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 92.Amsen D, et al. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–110. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samon JB, et al. Notch1 and TGFβ1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112:1813–1821. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blokzijl A, et al. Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163:723–728. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol. 2009;182:7381–7388. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ong CT, Sedy JR, Murphy KM, Kopan R. Notch and presenilin regulate cellular expansion and cytokine secretion but cannot instruct Th1/Th2 fate acquisition. PLoS One. 2008;3:e2823. doi: 10.1371/journal.pone.0002823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Adler SH, et al. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J Immunol. 2003;171:2896–2903. doi: 10.4049/jimmunol.171.6.2896. [DOI] [PubMed] [Google Scholar]
- 99.Cote-Sierra J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]