

Abstract
Accurate ante mortem diagnosis in frontotemporal lobar degeneration (FTLD) is crucial to the development and implementation of etiology-based therapies. Several neurodegenerative disease-associated proteins, including the major protein constituents of inclusions in Alzheimer's disease (AD) associated with amyloid-beta (Aβ1−42) plaque and tau neurofibrillary tangle pathology, can be measured in cerebrospinal fluid (CSF) for diagnostic applications. Comparative studies using autopsy-confirmed samples suggest that CSF total-tau (t-tau) and Aβ1−42 levels can accurately distinguish FTLD from AD, with a high t-tau to Aβ1−42 ratio diagnostic of AD; however, there is also an urgent need for FTLD-specific biomarkers. These analytes will require validation in large autopsy-confirmed cohorts and face challenges of standardization of within- and between-laboratory sources of error. In addition, CSF biomarkers with prognostic utility and longitudinal study of CSF biomarker levels over the course of disease are also needed. Current goals in the field include identification of analytes that are easily and reliably measured and can be used alone or in a multi-modal approach to provide an accurate prediction of underlying neuropathology for use in clinical trials of disease modifying treatments in FTLD. To achieve these goals it will be of the utmost importance to view neurodegenerative disease, including FTLD, as a clinicopathological entity, rather than exclusively a clinical syndrome.
Keywords: cerebrospinal fluid, biomarker, tau, Aβ1−42, frontotemporal dementia, primary progressive aphasia, Alzheimer's disease
Introduction
Most neurodegenerative diseases are characterized by specific abnormally-modified protein aggregates, with resulting neuronal cell loss and gliosis. The gold standard for diagnosis is microscopic examination at autopsy; however, there is considerable variability of clinical manifestations associated with underlying neuropathological diagnoses, as clinical symptoms most often reflect the regional burden of pathology within the central nervous system (CNS) rather than the specific underlying proteinopathy. This is especially true in the heterogeneous family of frontotemporal lobar degeneration (FTLD) clinical syndromes.
Two main pathologic FTLD subtypes exist (Figures 1A, 2): cases with inclusions formed from the microtubule-binding protein tau (FTLD-tau) and those with TAR DNA binding protein-43 (TDP-43) pathology (FTLD-TDP) (Mackenzie et al., 2010). FTLD-tau includes the following tauopathies (Figures 2A–D): Pick's disease (PiD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), FTD and parkinsonism linked to chromosome 17 (pathogenic MAPT mutations; FTDP-17), and unclassifiable tauopathies (Mackenzie et al., 2010). FTLD-TDP (Figures 2E–G) can be subdivided into four subtypes (A–D) based on the morphology and distribution of lesions (Mackenzie et al., 2011) and can also be associated with TDP-43 inclusions in the anterior horn of the spinal cord and gliosis of the corticospinal tracts, suggesting a continuum of FTLD with amyotrophic lateral sclerosis (ALS; FTLD-ALS) (Geser et al., 2008, 2009). A smaller number of FTLD cases are associated with inclusions of another DNA-binding protein, fused-in-sarcoma protein (FUS; FTLD-FUS), or other rare, less-defined pathologies (FTLD-UPS, FTLD-ni) (Mackenzie et al., 2010). The major genetic etiologies resulting in FTLD are exclusively associated with specific underlying neuropathologies (Figure 1B), despite heterogeneous expression of FTLD clinical syndromes, and include pathogenic mutations in the gene for progranulin (GRN) (Baker et al., 2006; Cruts et al., 2006), tau (MAPT) (Hutton et al., 1998), and C9orf72 (C9orf72) (Dejesus-Hernandez et al., 2011; Renton et al., 2011). Less common genetic etiologies of FTLD include: valosin-containing protein (VCP) resulting in inclusion body myopathy with Paget's disease of bone and frontotemporal dementia with FTLD-TDP subtype D neuropathology, TARDBP coding for TDP-43 protein and causing ALS or ALS-FTLD (rarely FTLD-TDP alone), CHMP2B coding for charged mutlivesciular body protein 2B and resulting in FTLD-UPS, and mutations in FUS causing FTLD-FUS (Figure 1B) (Mackenzie et al., 2010).
Figure 1.

Clinicopathological and genetic associations in FTLD. (A) Neuropathological classification of FTLD-tau and FTLD-TDP subtypes (PSP, progressive supranuclear palsy; CBD, corticobasal degeneration; PiD, Pick's disease; FTDP17, frontotemporal dementia with Parkinsonism linked to chromosome 17; Tauopathy NOS, unclassifiable tauopathy; Subtypes A–D, morphological subtypes of FTLD-TDP; ALS-FTLD, amyotrophic lateral sclerosis with FTLD-TDP; FTLD-FUS, FTLD with fused in sarcoma protein inclusions; FTLD-UPS, FTLD with tau- and TDP-43-negative ubiquitinated inclusions; FTLD-ni, FTLD in the absence of significant neuropathological inclusions), (B) pathogenic mutation associations with underlying neuropathology (dashed-line separates less common molecular etiologies of FTLD; MAPT, tau resulting in FTDP-17; C90rf72, pathogenic hexanucleotide expansion resulting in FTLD and/or ALS associated with FTLD-TDP B; GRN, progranulin resulting in FTLD-TDP type A; TARDP, TDP-43 resulting in ALS ± FTLD and less commonly FTLD; VCP, valosin-containing protein resulting in inclusion body myopathy with Paget's disease of bone and frontotemporal dementia with FTLD-TDP subtype D; FUS, fused-in sarcoma protein resulting in FTLD-FUS; and CHMP2B, charged mutlivesciular body protein 2B resulting in FTLD-UPS), (C) clinicopathological correlations of FTLD (colored regions of clinical syndromes represent relative percentages of neuropathological subtypes found in autopsy studies; AD, Alzheimer's disease; bvFTD, behavioral variant of FTLD; PPA, primary progressive aphasia; svPPA, semantic variant PPA; naPPA, nonfluent agrammatic variant PPA; lvPPA, logopenic variant PPA; +ALS, co-morbid amyotrophic lateral sclerosis; +EPS, co-morbid extra-pyramidal Parkinsonian symptoms: i.e., features of akinetic-rigid syndromes of PSP or corticobasal syndrome).
Figure 2.
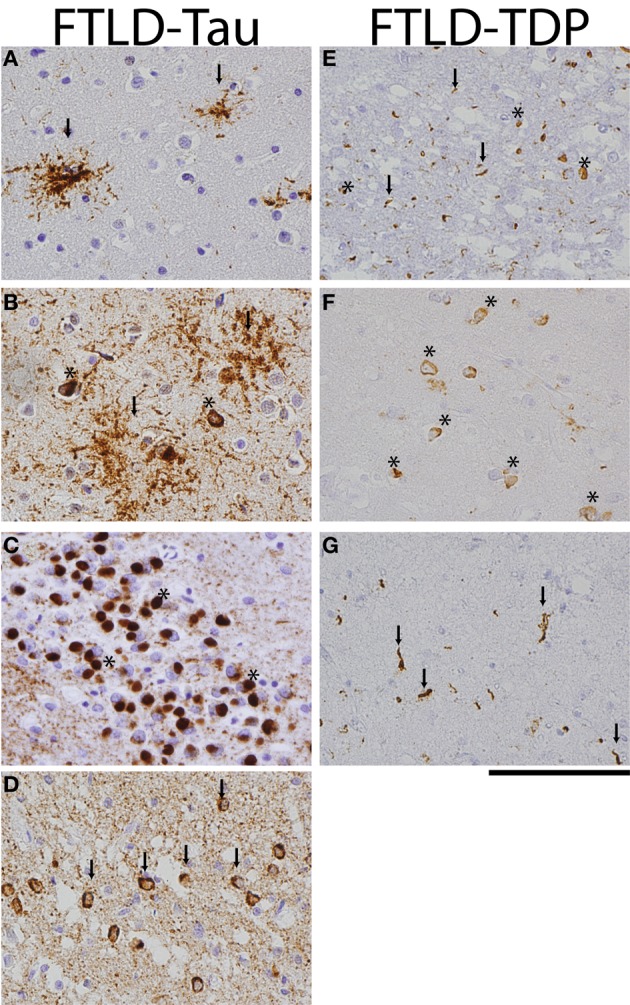
FTLD-Tau and FTLD-TDP histology. Photomicrographs of FTLD-tau (A–D) and FTLD-TDP (E–G) visualized with immunohistochemistry (PHF-1 and pTDP 409/410 for tau and TDP, respectively). (A) PSP frontal cortex with tau-positive tufted astrocytes (arrows), (B) CBD temporal cortex with diffuse astrocytic plaques (arrows) and neuronal tangles (asterisks), (C) Pick's disease with round tau-positive Pick bodies (asterisks) in the dentate nucleus of the hippocampus, (D) FTDP-17 case with p.P301L pathogenic mutation with tau-positive neuronal tangles (arrows) and diffuse neuropil threads in temporal cortex, (E) FTLD-TDP subtype A with cytoplasmic neuronal inclusions (asterisks) and short dystrophic neurites (arrows) in superficial layers of frontal cortex, (F) FTLD-TDP subtype B with prominent cytoplasmic inclusions (asterisks) in deep temporal cortical layer, and (G) long dystrophic neurites (arrows) in superficial layers of mid-frontal cortex of a patient with FTLD-TDP subtype C. Scale bar = 100 μm.
Clinically, FTLD can be broadly divided into two main subtypes, those with predominant behavioral and social comportment disorder (behavioral-variant frontotemporal dementia, bvFTD) (Rascovsky et al., 2011) and those with primary language disturbances (primary progressive aphasia, PPA) (Mesulam, 1982, 2001). Among PPA patients, three subgroups have been recently divided (Gorno-Tempini et al., 2011) into the logopenic (lvPPA) (Gorno-Tempini et al., 2004, 2008), semantic (svPPA) (Hodges and Patterson, 2007), and non-fluent aggramatic variant (naPPA) (Turner et al., 1996). Clinicopathological correlations of these syndromes are complex (Josephs, 2008; Grossman, 2010). For example, large studies of autopsy-confirmed FTLD (behavioral and aphasic variants) find roughly equal numbers of FTLD-tau and FTLD-TDP (Hodges et al., 2004; Kertesz et al., 2005; Knopman et al., 2005; Shi et al., 2005; Forman et al., 2006). Furthermore, a primary neuropathological diagnosis of Alzheimer's disease (AD) has been found in up to 30% of autopsy-confirmed clinically defined FTLD cohorts (Kertesz et al., 2005; Knopman et al., 2005; Forman et al., 2006; Knibb et al., 2006). Examination of focal presentations of AD found it to be the primary diagnosis in 7% of bvFTD, 44% of naPPA, 10% of svPPA, and 50% of the extrapyramidal and cognitive disorder, corticobasal syndrome (CBS) patients (Alladi et al., 2007). Others have also found a substantial proportion of AD in PPA cases (Forman et al., 2006; Knibb et al., 2006) especially in lvPPA (Grossman et al., 2008; Mesulam et al., 2008; Grossman, 2010) and also CBS (Lee et al., 2011). Thus, differentiation of AD and FTLD spectrum disorders poses a serious diagnostic challenge for clinicians.
Within the FTLD neuropathological spectrum, examination of the specific clinical subtypes finds varying degrees of association with FTLD-tau and FTLD-TDP (Figure 1C). FTLD-tau has been overrepresented in some naPPA cohorts (Hodges et al., 2004; Josephs et al., 2006a,b; Knibb et al., 2006; Snowden et al., 2007; Mesulam et al., 2008; Grossman et al., 2012), especially when associated with apraxia of speech (Josephs et al., 2006a; Snowden et al., 2007) and svPPA has been predominantly associated with TDP-43 pathology (Hodges et al., 2004; Josephs et al., 2006a; Snowden et al., 2007; Grossman et al., 2008); while bvFTD contains similar proportions of FTLD-tau and FTLD-TDP (Forman et al., 2006; Josephs et al., 2006b; Snowden et al., 2007). Extrapyramidal symptoms may predict a tauopathy (Forman et al., 2006; Josephs et al., 2006b) while co-morbid ALS is almost certainly due to TDP-43 aggregation (Shi et al., 2005; Forman et al., 2006; Josephs et al., 2006b). Clinicopathological associations from these large autopsy studies are summarized in Figure 1C.
A major challenge in the development and implementation of disease-modifying therapy in FTLD is the accurate identification of the neuropathological diagnosis during life, including differentiation from AD, so that patients may be triaged to the appropriate protein-targeted therapy (i.e., tau or TDP-43 targeted agents).
Biofluid biomarkers have the potential to optimize diagnostic accuracy and detect disease earlier in the course of an illness and possibly pre-symptomatically, such as prior to structural changes of neurodegeneration seen on neuroimaging (Hu et al., 2010a; Jack et al., 2010), making further exploration in this area promising for the development of disease modifying treatments. In addition, some clinical measures of disease progression in FTLD, including functional scales, may be limited by floor- and ceiling-effects (Knopman et al., 2008), so biofluid biomarkers are potentially attractive surrogate end points for use in clinical trials (Boxer et al., 2012b). The cerebrospinal fluid (CSF) is relatively easy to obtain and contains a direct connection to the pathological milieu in central nervous system, making it a desirable biofluid for study. In this review we will discuss the current state of CSF biomarker research in FTLD in terms of differentiation from AD and future directions and challenges for the field in development of FTLD-specific biomarkers.
Alzheimer's disease related csf biomarkers: Aβ1−42 and tau
Studies in alzheimer's disease
As a first step in biofluid-based biomarker assessment of neurodegenerative disease, it is valuable to distinguish broadly between AD and FTLD. CSF values of the major constituents of AD pathology, tau and β-amyloid, (Aβ1−42) have been widely studied using immune-based analytical platforms in AD and amnestic mild cognitive impairment (MCI) patients, with lower Aβ1−42 values and higher levels of total- and phosphorylated-tau (t-tau, p-tau) compared with controls across multiple large studies (Shaw et al., 2009, 2011; De Meyer et al., 2010; Trojanowski et al., 2010; Weiner et al., 2010). Furthermore, our group has shown prognostic utility of these markers by accurately predicting MCI conversion to AD (Shaw et al., 2009; De Meyer et al., 2010).
The majority of atypical clinical presentations of AD in early-onset patients consisting of predominantly visuo-spatial difficulties (i.e., consistent with poster cortical atrophy) or asymmetric apraxia/rigidity (i.e., consistent with CBS) may have a similar CSF biomarker profile to that of typical amnestic-AD (De Souza et al., 2011; Seguin et al., 2011), with a further elevated t-tau level in one study (Koric et al., 2010). Elevated CSF t-tau and low Aβ1−42 levels have also been described in some PPA patients (i.e., lvPPA) (Bibl et al., 2011; De Souza et al., 2011) most likely due to underlying AD neuropathology in these individuals; however, to our knowledge no autopsy-confirmed studies of atypical clinical AD presentations have been performed.
The exact relationship between AD neuropathologic change (i.e., tau neurofibrillary pathology and Aβ1−42 extracellular plaques) and observed measurement of these analytes in CSF is unclear; however, the total tau level is thought to reflect underlying neurodegeneration and neuron loss, as elevations are also seen in other CNS insults (Otto et al., 1997; Hesse et al., 2000; Jin et al., 2006; Ost et al., 2006; Krut et al., 2013). Lower Aβ1−42 CSF levels may be the result of sequestration of soluble interstitial brain Aβ1−42 into extracellular plaques as there is an inverse correlation of CSF Aβ1−42 levels and the degree of cortical plaque pathology (Tapiola et al., 2009; Patel et al., 2012; Seppala et al., 2012) and in vivo neuroimaging evidence of amyloidosis (Fagan et al., 2006). Phosphorylated epitopes of tau (p-tau) can be measured in CSF as well; while most phospho-epitopes of tau are also found in healthy non-diseased brains and are not AD-specific, pathological tau species overall are highly phosphorylated in AD (Matsuo et al., 1994) and this altered state reflects the elevated levels of p-tau seen in AD. The most commonly studied p-tau epitopes are serine 181 (p-tau181) (Vanmechelen et al., 2000), and threonine 231 (p-tau231) (Buerger et al., 2002a,b).
Studies in frontotemporal lobar degeneration
FTLD is not characterized pathologically by cerebral Aβ1−42 amyloidosis, and only FTLD-tau is characterized by significant tau inclusions. From this perspective, measures of CSF t-tau and Aβ1−42 may have helpful diagnostic utility in excluding AD neuropathology. Indeed, in clinically-defined cohorts AD cases have higher levels of t-tau, p-tau181 and lower levels of Aβ1−42 compared to FTLD and controls in group-wise comparisons (Blennow et al., 1995; Arai et al., 1997; Green et al., 1999; Sjogren et al., 2000a, 2001; Vanmechelen et al., 2000; Riemenschneider et al., 2002; Clark et al., 2003; Pijnenburg et al., 2004, 2007; Schoonenboom et al., 2004, 2012; Engelborghs et al., 2006; Bibl et al., 2007, 2011; Kapaki et al., 2008; Verwey et al., 2010; De Souza et al., 2011; Gabelle et al., 2011; Van Harten et al., 2011).
A major challenge in FTLD CSF biomarker studies is the heterogeneity of the condition (Figure 1), making autopsy-confirmation of diagnostic classification a crucial issue. As mentioned previously, up to 30% of clinically-defined FTLD cohorts may have underlying AD neuropathologic change as the etiology of their symptoms (Kertesz et al., 2005; Knopman et al., 2005; Forman et al., 2006; Knibb et al., 2006) and contamination with these atypical AD cases could influence results significantly. Indeed, examination of diagnostic accuracy of CSF t-tau and Aβ1−42 in a large autopsy-confirmed dementia cohort found that use of the clinical diagnosis, rather than neuropathological diagnosis as the gold standard for biomarker performance resulted in a 10–20% underestimation of biomarker accuracy (Toledo et al., 2012). Furthermore, since 1995 there has been over a 10-fold increase in the number of FTLD manuscripts published (NLM/NIH, 2012) and due to this exponential increase in research in the field and our expanding knowledge of FTLD, clinical criteria (Gorno-Tempini et al., 2011; Rascovsky et al., 2011) have evolved resulting in refinement of our clinical definitions. Indeed, the emergence of the new clinical variant of PPA, lvPPA (Gorno-Tempini et al., 2008, 2011), which is most often associated with AD neuropathology (Mesulam et al., 2008; Rabinovici et al., 2008; Grossman, 2010) (Figure 1C), and therefore suggested to be excluded from FTLD clinical trials (Knopman et al., 2008), could influence group-wise CSF tau and Aβ1−42 results. Thus, the makeup of clinical cohorts used in earlier studies may not be entirely translatable to newer studies, limiting the meaningful interpretation of the literature of clinically-derived cohorts.
As such, study of autopsy/genetic-confirmed cases has been a focus for our center. In an early study of autopsy-confirmed cases by our group, AD was differentiated from a mixed dementia cohort (including 13 FTLD cases) with reasonable sensitivity (72%) and specificity (69%) using CSF t-tau levels (Clark et al., 2003). Focused analysis of FTLD (with autopsy confirmation in 9 cases) in a later study found lower levels of t-tau and higher levels of Aβ1−42 than AD, and roughly 30% of FTLD cases had significantly decreased t-tau from controls (Grossman et al., 2005). In a follow-up large autopsy/genetically confirmed FTLD series (n = 30) t-tau levels were significantly lower in FTLD than AD, while similar to controls on group-wise comparison; individual-case analysis revealed that a considerable subset of FTLD patients had markedly low t-tau values (Bian et al., 2008). Interestingly, FTLD cases with substantially lower t-tau levels included both FTLD-tau and FTLD-TDP (Bian et al., 2008), although a non-significant trend was found for lower t-tau in FTLD-tau (Hu et al., 2011). Furthermore, FTLD was differentiated from AD with high accuracy using the t-tau/Aβ1−42 ratio; that is, FTLD cases had a lower ratio (lower t-tau and higher Aβ1−42) (Bian et al., 2008).
Measurement of these analytes in the CSF in most studies utilizes one of two immune-based platforms: enzyme-linked immunosorbent assay (ELISA; Innotest, Innogenetics), and a multiplex assay based on flow-cytometry of antibody-coated fluorescent beads (INNO-BIA AlzBio3 xMAP; Luminex, Innogenetics). Absolute values obtained from these platforms differ because the coefficient of variance (%CV) with the xMAP Luminex platform is much narrower than with ELISA, but they are highly correlated (Olsson et al., 2005; Lewczuk et al., 2009; Fagan et al., 2011; Wang et al., 2012) and have similar levels of diagnostic accuracy for AD (Fagan et al., 2011; Wang et al., 2012) and differentiating AD from FTLD (Toledo et al., 2012). Thus, values from one platform can be effectively transformed into equivalent units of the other using a conversion factor (Fagan et al., 2011) Indeed, we were able to transform values obtained from ELISA to equivalent xMAP units using linear regression to create a larger autopsy/genetic-confirmed FTLD dataset and help confirm our pervious observations of the diagnostic utility of the t-tau/Aβ1−42 ratio to differentiate FTLD from AD (Irwin et al., 2012b). Maximizing available data is crucial for these extremely valuable and well-annotated research samples. In summary, in multiple large-scale autopsy-confirmed studies we have demonstrated the diagnostic utility of CSF t-tau, p-tau, and Aβ1−42 in differentiation of AD and FTLD (Bian et al., 2008; Irwin et al., 2012b; Toledo et al., 2012).
Few other CSF studies have used autopsy-confirmed cohorts of FTLD patients (Table 1). One study included 10 autopsy-confirmed FTLD patients and found similar results of lower t-tau and p-tau181 levels in FTLD compared with AD, with high diagnostic accuracy of p-tau181 (Koopman et al., 2009). Another study including 12 confirmed FTLD patients described “slightly elevated tau levels” in several patients compared to an age-dependent reference range and low compared to the majority of AD cases (Brunnstrom et al., 2010). Neuropathological subgroups of FTLD (FTLD-TDP, n = 5 and FTLD-tau, n = 7) had similar mean values, with 4/12 patients below the reference limit by >70 pg/ml (Brunnstrom et al., 2010). Thus, this study also found a subset of individual FTLD patients with lower than normal t-tau levels. The diagnostic utility of t-tau/Aβ1−42 in differentiating FTLD was not systematically explored in this small group of AD cases (n = 8). Finally, to our knowledge the only additional studies utilizing autopsy-confirmed FTLD cohorts included a small number of FTLD cases (<10) in a non-AD category, with no direct comparison of FTLD and AD (Engelborghs et al., 2008; Tapiola et al., 2009; Schoonenboom et al., 2012). Thus, further study is required in large prospective, autopsy-confirmed samples to confirm our observations.
Table 1.
Comparative studies of CSF biomarkers in autopsy/genetic-confirmed FTLD and AD cohorts.
| Study | Patients | Aβ1−42 | t-tau | p-tau181 | Diagnostic accuracy (AD vs. FTLD) |
|---|---|---|---|---|---|
| Clark et al., 2003 | (10) FTLD(74) AD*73(4) CN | AD < FTLD, CN | CN < FTLD < AD | NA | No statistical analysis of FTLD diagnostic accuracy performed |
| Grossman et al., 2005 | 73 (11) FTLD(17) AD13 CN | AD < FTLD, CN | CN, FTLD < AD | CN, FTLD < AD | t-tau |
| AUC = 0.86, sens = 74%, spec = 82.4% | |||||
| Bian et al., 2008 | (30) FTLD(19) AD13 CN | AD < FTLD, CN | CN, FTLD < AD | NA | t-tau/Aβ1−42 |
| AUC = 0.93, sens = 78.9%, spec = 96.6% | |||||
| Engelborghs et al., 2008 | (2) FTLD(73) AD*100 CN | NA | NA | NA | No statistical analysis of FTLD diagnostic accuracy performed |
| Koopman et al., 2009 | (10) FTLD(95) AD | AD < FTLD | FTLD < AD | FTLD < AD | p-tau181 |
| AUC = 0.85, sens = 91%, spec = 80% | |||||
| Tapiola et al., 2009 | (9) FTLD(83) AD | NA | NA | NA | No statistical analysis of FTLD diagnostic accuracy performed |
| Brunnstrom et al., 2010 | (12) FTLD(8) AD* | NA | NA | NA | No statistical analysis of FTLD diagnostic accuracy performed |
| Irwin et al., 2012b | (20) FTLD(41) AD* | NA | NA | NA | t-tau/Aβ1−42 |
| AUC = 0.99, sens = 90–100%, spec = 90–96% | |||||
| Toledo et al., 2012 | (71) AD(29) FTLD66 CN | AD < FTLD < CN | CN, FTLD < AD | CN, FTLD < AD | t-tau/Aβ1−42 (ELISA) |
| AUC = 0.96, sens = 90, spec = 82% | |||||
| p-tau181/Aβ1−42 (xMAP) | |||||
| AUC = 0.98, sens = 100%, spec = 88% |
Other diagnostic groups that may be present in some studies are omitted and only direct comparisons of FTLD group to AD or CN are reported. “<” or “>” denotes significant difference between groups and “,” denotes non-significant difference between groups, () denotes autopsy/genetic confirmed cohort.
CN, non-demented controls;
, AD group contains cases with co-morbid Lewy Body or Vascular Disease; NA, Not assessed; AUC, Area under the curve for receiver operating curve analysis; ELISA, enzyme-linked immunosorbent assay; xMAP, luminex multiplex assay.
The higher Aβ1−42 in FTLD compared to AD most likely reflects the absence of significant cerebral amyloidosis while the biological basis for observed low CSF t-tau in some FTLD patients is uncertain. One possibility is related to cortical tau depletion (Zhukareva et al., 2001, 2003; Grossman et al., 2005) through sequestration into the neuronal and glial inclusions in the absence of significant extracellular tau pathology (FTLD-tau) Dickson, 2004, such as extracellular “ghost tangles” as seen in AD (Schmidt et al., 1988), or altered post-translational stability of tau in FTLD-TDP (Zhukareva et al., 2001, 2003); furthermore, CSF t-tau does appear related to underlying FTLD pathophysiology as t-tau levels in FTLD patients correlated to areas of frontal and temporal cortical atrophy on magnetic resonance imaging (MRI) (Grossman et al., 2005; McMillan et al., 2013). Further study of CSF protein dynamics in animal models of disease may help clarify these seemingly discordant associations of low tau levels with underlying neuropathology in FTLD-tau and FTLD-TDP.
Despite the clear distinction of t-tau and Aβ1−42 levels between AD and FTLD, there is more variability in the literature for the relationship of these markers in FTLD compared with non-demented controls (Table 1). There are several reasons for these discrepancies; first, even in most autopsy-based studies, autopsy data on controls is lacking (Table 1) and a significant proportion of non-demented elderly can have underlying AD neuropathology (Davis et al., 1999), and thus influence CSF analyte measures. Next, even with pathologic confirmation, patient classification in FTLD is challenging, as another potential confounding issue is the presence of mixed pathologies in dementia patients. Indeed, our group has shown in a large autopsy-confirmed sample that mixed pathology is present in roughly 30% of cases, and that FTLD patients with significant AD neuropathologic change can influence the CSF t-tau and Aβ1−42 levels, causing higher t-tau and lower Aβ1−42 in cases with mixed FTLD and AD pathology compared to “pure” FTLD (Toledo et al., 2012). Additionally, a recent largely clinically-defined cohort study found an AD CSF biomarker profile in 30% of FTLD (Schoonenboom et al., 2012) which may be due, in part, to mixed pathology or inclusion of atypical AD cases mimicking the FTLD clinical syndrome (Toledo et al., 2012). Thus, the use of autopsy-confirmed samples is essential for in-depth study and validation of the diagnostic accuracy of potential biomarkers in FTLD.
Finally, variability in measurement between studies is another potential issue as significant variation between centers in absolute values measured in “spiked” pooled CSF control samples with known concentrations of analyte has been described (Shaw et al., 2011). These discrepancies are most likely due to sources of variation in CSF collection, handling and storage (pre-analytical), equipment, reagents and methods of analysis (analytical), and data management and interpretation (post-analytical) (Mattsson et al., 2011). For these reasons, large scale studies of measurement precision of these analytes and coordinated multi-center quality control programs with standard operating procedures to minimize these sources of variation have been conducted (Mattsson et al., 2011; Shaw et al., 2011).
Despite these issues, we have demonstrated (Bian et al., 2008; Irwin et al., 2012b; Toledo et al., 2012) that these AD-specific analytes (t-tau to Aβ1−42 ratio) may perform within the range of sensitivity and specificity (>80%) for use in clinical trials (Trojanowski and Growdon, 1998) to differentiate FTLD from AD; however, these analytes are not as effective for differentiation of FTLD from normal controls (Bian and Grossman, 2007; Toledo et al., 2012). Although patients may present with decompensated psychiatric issues or other non-progressive non-degenerative etiologies resembling FTLD (phenocopy syndrome) (Kipps et al., 2010), these patients may be identified with serial clinical exams and neuroimaging (Kipps et al., 2010). The more urgent need is for FTLD-specific biomarkers and those that can differentiate between the two major neuropathologic subtypes (FTLD-tau and FTLD-TDP) (Hu et al., 2011).
Future directions
Further study of CSF tau and Aβ1−42
Previous work in large cross-sectional studies in AD suggests a temporal progression of dynamic biomarker change in AD (Jack et al., 2010, 2012), as Aβ1−42 amyloidosis, and resultant lower CSF Aβ1−42, is thought to occur decades before clinical symptoms emerge in AD, while increased CSF t-tau is thought to be a later event in disease progression and correlates more closely with cognitive decline. It is likely that t-tau, p-tau and potential novel CSF biomarkers could display similar changes throughout the course of disease in FTLD and could correlate with clinical symptoms. Few studies have examined the change in CSF biomarkers over time or their relation to clinical symptoms. One study included a follow up CSF analysis in one FTLD-tau patient, with similar t-tau and Aβ1−42, roughly 18 months between CSF collections (Brunnstrom et al., 2010). Interestingly, a recent study of bvFTD patients found a significant correlation with Aβ1−42 levels and cognitive performance, even after removal of patients with CSF profile suggestive of AD neuropathology (Koedam et al., 2012). These results could suggest an influence of co-morbid AD neuropathology; however autopsy information in these cases was lacking. Other studies in clinical series without autopsy confirmation found no association of these markers and clinical measures or disease severity (Riemenschneider et al., 2002; Engelborghs et al., 2006; De Souza et al., 2011). Further study of clinical correlates of CSF biomarkers and longitudinal profiles of CSF analyte change throughout the course of disease will be helpful.
Similar to the dominantly-inherited AD network (DIAN) initiative to study patients with known pathogenic mutations to cause AD (Bateman et al., 2012), study of prodromal FTLD patients with pathogenic mutations may provide additional insights into the temporal sequence of biomarkers in FTLD (Boxer et al., 2012a). Furthermore, CSF analyte levels in symptomatic patients with genetic forms of FTLD have not been explored in detail and could potentially differ from sporadic cases. Indeed, we found a more rapid rate of progression in cognitive measures corresponding to more severe neurodegeneration in C9orf72-associated FTLD (Irwin et al., 2013) and others have described unique neuroimaging patterns of atrophy across different genetic forms of FTLD (Whitwell et al., 2012). This evidence of biologic differences in genetic and sporadic FTLD suggest alterations in CSF biomarker profiles are also a possibility, although one study found similar levels of CSF tau and Aβ1−42 in genetically-confirmed FTDP-17 (n = 9) compared to sporadic FTLD (n = 17) (Rosso et al., 2003).
Development of FTLD-specific biomarkers
In the context of disease-modifying therapies targeting a specific histopathologic abnormality, an important goal is to distinguish between FTLD due to TDP-43 and FTLD due to tau. Exploratory analyses for novel biomarkers that have diagnostic utility in FTLD are ongoing and include several basic approaches. First, measurement of biologically relevant molecules is the most straightforward approach, as tau and Aβ1−42 have been successful biomarker candidates in AD. Using this rationale, the two most obvious candidates for FTLD-specific biomarkers are TDP-43 progranulin. Indeed, TDP-43 has been detected in human CSF (Steinacker et al., 2008; Kasai et al., 2009) and serum (Foulds et al., 2008), suggesting elevated levels may occur in some patients with TDP-43 proteinopathies, but initial studies show limited diagnostic accuracy. Low serum progranulin may identify FTLD patients with a pathogenic GRN mutation resulting in progranulin haploinsufficiency (Ghidoni et al., 2008), which could be useful in monitoring potential progranulin-replacing therapies in development for FTLD (Boxer et al., 2012b).
Other biologically relevant potential biomarkers for FTLD include specific isoforms or neoepitopes of tau. Tau undergoes multiple post-translational modifications thought to contribute to tangle formation. Indeed, we found acetylation of tau at a specific residue in the microtubule-binding domain (MTBD) to be exclusively found in tauopathies, providing promise for this epitope as a useful marker of AD and FTLD-tau (Cohen et al., 2011; Irwin et al., 2012a). Translating these immunohistochemical observations to clinical assays may prove difficult, as levels of tau in CSF are near the lower limits of biologic detection (Hampel et al., 2010) limiting the further identification of a specific subset of tau in the form of a neoepitope; although one group has found promising evidence for diagnostic utility of specific C-truncated isoforms of tau in PSP through immunoprecipitation and western blotting techniques (Borroni et al., 2008, 2009) and others have developed assays to measure 3- and 4R tau in CSF (Luk et al., 2012). Alternatively-truncated forms of Aβ1−42 may also have diagnostic importance in FTLD (Pijnenburg et al., 2007; Bibl et al., 2011, 2012; Gabelle et al., 2011) and cytoskeletal proteins, such as neurofilament have also been explored (Sjogren et al., 2000b; De Jong et al., 2007). These potential biomarkers warrant further study and validation.
Another, possible approach is to screen a large number of potential analytes without an a priori biologic rationale in a proteomic analysis of CSF in FTLD. Indeed, using an immune-based multiplex approach our group found promising CSF biomarker candidates to differentiate FTLD-TDP and FTLD-tau with high sensitivity and specificity, but these candidate analytes need further study to confirm their utility as FTLD biomarkers (Hu et al., 2010b). Finally, other non-immune based methods, such as mass-spectrometry are also being explored to identify novel biofluid biomarkers in FTLD (Mattsson et al., 2008).
Potential FTLD-specific biofluid biomarkers will be faced with the same challenges of testing reliability and sources of variation (i.e., analytical, pre-/post-analytical) currently experienced by CSF t-tau and Aβ1−42 measurements. As such, coordinated and cooperative efforts between multiple centers will undoubtedly be necessary to help validate potential FTLD-specific CSF biomarkers prior to clinical use.
Most likely, a multimodal assessment incorporating potential novel biofluid biomarker values with clinical, neuroimaging and genetic markers may be the most effective approach to accurately identify FTLD subtypes. Neuropsychological testing can help differentiate AD from FTLD (Rascovsky et al., 2008; Libon et al., 2011) as routine cognitive measures may not be sensitive enough to detect the behavioral and language deficits in FTLD. Indeed, our group has explored quantitative approaches to language (Ash et al., 2006, 2009; Gunawardena et al., 2010) and social cognition (Massimo et al., 2009, 2013; Grossman et al., 2010; Eslinger et al., 2012; McMillan et al., 2012b) to examine brain-behavior relationships and improve diagnostic accuracy in FTLD. Neuroimaging is another potential method with diagnostic utility alone, or as an adjunct to clinical and biofluid biomarkers in FTLD; we have found combining neuropsychological testing and MRI can improve diagnostic accuracy in PPA (Hu et al., 2010c); and others find combination of CSF tau isoform levels and midbrain atrophy improve identification of PSP (Borroni et al., 2010). Multiple modalities of MRI methods, including diffusion-tensor imaging (DTI) of white matter may help identify FTLD patients in dementia cohorts. We have demonstrated increased diagnostic sensitivity to differentiate AD from FTLD cases using a combination of gray matter (GM) density and DTI measures (McMillan et al., 2012a). In addition, we have also discovered promising diagnostic utility for differentiating FTLD-tau and FTLD-TDP using DTI (unpublished data). Cortical atrophy and CSF biomarker levels appear to be highly correlated as we have recently demonstrated that GM density could predict CSF t-tau and Aβ1−42 levels, and these predicted values could accurately distinguish AD and FTLD (McMillan et al., 2013). These results indicate that MRI could potentially serve as a surrogate for CSF, which would have significant utility for patients where lumbar puncture would be difficult or for clinical trial endpoints where repeated lumbar puncture may be needed. Finally, recent genome-wide association studies (GWAS) have found risk alleles associated with FTLD-TDP (Van Deerlin et al., 2010) and FTLD-tau (Hoglinger et al., 2011). Further knowledge of clinical, neuroimaging, and biofluid correlates of these risk alleles in FTLD could provide further useful diagnostic and prognostic information. Thus, comparative studies of clinical, genetic, biofluid, and neuroimaging biomarkers in longitudinally followed, well-annotated, autopsy-confirmed subjects will be a powerful method for improving our understanding of the pathophysiology of FTLD and further directing diagnostic and treatment efforts.
Summary
CSF measurements of Aβ1−42, t-tau, and p-tau in FTLD differ significantly from the abnormal levels seen in AD, and in a subset of both FTLD-tau and FTLD-TDP there are extremely low levels of t-tau of unclear etiology. These properties allow for accurate distinction of FTLD from AD in autopsy-confirmed cohorts, while FTLD-specific markers are still lacking.
As we move toward therapies that impact the progression of the disease and target the underlying pathophysiology in FTLD and other neurodegenerative disorders it will be essential for clinicians to view these disorders as clinicopathological entities with the underlying neuropathological substrate in mind. Indeed, new clinical criteria for AD incorporate this ideology with the designation of “pre-symptomatic AD” (Sperling et al., 2011). In the study of the complex clinicopathological spectrum of FTLD disorders, where heterogeneity is the rule, useful markers to develop homogenous clinical, genetic, and neuropathologic subgroups will be crucial to further our goals toward meaningful treatments that could potential slow disease progression and limit patient disability.
Conflict of interest statement
Dr. John Q. Trojanowski reports single consulting services to Pfizer, J&J, MetLife, and BMS; Royalty payments through Penn licenses; and research support from AstraZeneca and BMS. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients studied here and their families who made the research reviewed here possible. Funding for this study was provided by the National Institutes of Health grants P30 AG10124, AG17586, and T32-AG000255 as well as the Wyncote Foundation.
Glossary
Abbreviations
- FTLD
frontotemporal lobar degeneration
- AD
Alzheimer's disease
- Aβ
amyloid-beta
- CSF
cerebrospinal fluid
- CNS
central nervous system
- FTLD-tau
FTLD with tau pathology
- TDP-43
TAR DNA binding protein-43
- FTLD-TDP
FTLD with TDP pathology
- PiD
Pick's disease
- CBD
corticobasal degeneration
- PSP
progressive supranuclear palsy
- pathogenic MAPT mutations-FTDP-17
FTD and parkinsonism linked to chromosome 17
- ALS
amyotrophic lateral sclerosis
- FUS
fused-in-sarcoma protein
- FTLD-FUS
FTLD with FUS pathology
- FTLD-UPS
FTLD with tau- and TDP-43-negative ubiquitinated inclusions
- FTLD-ni
FTLD in the absence of significant neuropathological inclusions
- GRN
progranulin gene
- MAPT
tau gene
- C9orf72
C9orf72 gene
- VCP
valosin-containing protein gene
- TARDBP
TDP-43 gene
- FTLD-ALS
clinical FTLD with ALS
- CHMP2B
charged mutlivesciular body protein 2B gene
- bvFTD
behavioral-variant frontotemporal dementia
- PPA
primary progressive aphasia
- lvPPA
logopenic-variant PPA
- svPPA
semantic-variant PPA
- naPPA
non-fluent aggramatic variant PPA
- CBS
corticobasal syndrome
- Aβ1−42
β-amyloid
- MCI
mild cognitive impairment
- t-tau
total-tau
- p-tau
phosphorylated-tau
- p-tau181
phosphorylated tau at serine 181
- p-tau231
phosphorylated tau at threonine 231
- ELISA
enzyme-linked immunosorbent assay
- xMAP
luminex flow immunoassay
- MRI
magnetic resonance imaging
- DIAN
dominantly-inherited AD network
- MTBD
microtubule-binding domain
- DTI
diffusion-tensor imaging
- GM
gray matter
- GWAS
genome-wide association studies.
References
- Alladi S., Xuereb J., Bak T., Nestor P., Knibb J., Patterson K., et al. (2007). Focal cortical presentations of Alzheimer's disease. Brain 130, 2636–2645 10.1093/brain/awm213 [DOI] [PubMed] [Google Scholar]
- Arai H., Morikawa Y., Higuchi M., Matsui T., Clark C. M., Miura M., et al. (1997). Cerebrospinal fluid tau levels in neurodegenerative diseases with distinct tau-related pathology. Biochem. Biophys. Res. Commun. 236, 262–264 10.1006/bbrc.1997.6908 [DOI] [PubMed] [Google Scholar]
- Ash S., Moore P., Antani S., McCawley G., Work M., Grossman M. (2006). Trying to tell a tale: discourse impairments in progressive aphasia and frontotemporal dementia. Neurology 66, 1405–1413 10.1212/01.wnl.0000210435.72614.38 [DOI] [PubMed] [Google Scholar]
- Ash S., Moore P., Vesely L., Gunawardena D., McMillan C., Anderson C., et al. (2009). Non-Fluent Speech in Frontotemporal Lobar Degeneration. J. Neurolinguistics 22, 370–383 10.1016/j.jneuroling.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M., Mackenzie I. R., Pickering-Brown S. M., Gass J., Rademakers R., Lindholm C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Bateman R. J., Xiong C., Benzinger T. L., Fagan A. M., Goate A., Fox N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian H., Grossman M. (2007). Frontotemporal lobar degeneration: recent progress in antemortem diagnosis. Acta Neuropathol. 114, 23–29 10.1007/s00401-007-0235-4 [DOI] [PubMed] [Google Scholar]
- Bian H., Van Swieten J. C., Leight S., Massimo L., Wood E., Forman M., et al. (2008). CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology 70, 1827–1835 10.1212/01.wnl.0000311445.21321.fc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibl M., Gallus M., Welge V., Esselmann H., Wolf S., Ruther E., et al. (2012). Cerebrospinal fluid amyloid-beta 2-42 is decreased in Alzheimer's, but not in frontotemporal dementia. J. Neural Transm. 119, 805–813 10.1007/s00702-012-0801-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibl M., Mollenhauer B., Lewczuk P., Esselmann H., Wolf S., Otto M., et al. (2011). Cerebrospinal fluid tau, p-tau 181 and amyloid-beta38/40/42 in frontotemporal dementias and primary progressive aphasias. Dement. Geriatr. Cogn. Disord. 31, 37–44 10.1159/000322370 [DOI] [PubMed] [Google Scholar]
- Bibl M., Mollenhauer B., Wolf S., Esselmann H., Lewczuk P., Kornhuber J., et al. (2007). Reduced CSF carboxyterminally truncated Abeta peptides in frontotemporal lobe degenerations. J. Neural Transm. 114, 621–628 10.1007/s00702-006-0618-z [DOI] [PubMed] [Google Scholar]
- Blennow K., Wallin A., Agren H., Spenger C., Siegfried J., Vanmechelen E. (1995). Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol. Chem. Neuropathol. 26, 231–245 10.1007/BF02815140 [DOI] [PubMed] [Google Scholar]
- Borroni B., Gardoni F., Parnetti L., Magno L., Malinverno M., Saggese E., et al. (2009). Pattern of Tau forms in CSF is altered in progressive supranuclear palsy. Neurobiol. Aging 30, 34–40 10.1016/j.neurobiolaging.2007.05.009 [DOI] [PubMed] [Google Scholar]
- Borroni B., Malinverno M., Gardoni F., Alberici A., Parnetti L., Premi E., et al. (2008). Tau forms in CSF as a reliable biomarker for progressive supranuclear palsy. Neurology 71, 1796–1803 10.1212/01.wnl.0000335941.68602.39 [DOI] [PubMed] [Google Scholar]
- Borroni B., Malinverno M., Gardoni F., Grassi M., Parnetti L., Agosti C., et al. (2010). A combination of CSF tau ratio and midsaggital midbrain-to-pons atrophy for the early diagnosis of progressive supranuclear palsy. J. Alzheimers Dis. 22, 195–203 10.3233/JAD-2010-100333 [DOI] [PubMed] [Google Scholar]
- Boxer A. L., Gold M., Huey E., Gao F. B., Burton E. A., Chow T., et al. (2012a). Frontotemporal degeneration, the next therapeutic frontier: Molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. [Epub ahead of print]. 10.1016/j.jalz.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer A. L., Gold M., Huey E., Hu W. T., Rosen H., Kramer J., et al. (2012b). The advantages of frontotemporal degeneration drug development (part 2 of frontotemporal degeneration: the next therapeutic frontier). Alzheimers Dement. [Epub ahead of print]. 10.1016/j.jalz.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnstrom H., Rawshani N., Zetterberg H., Blennow K., Minthon L., Passant U., et al. (2010). Cerebrospinal fluid biomarker results in relation to neuropathological dementia diagnoses. Alzheimers Dement. 6, 104–109 10.1016/j.jalz.2009.12.005 [DOI] [PubMed] [Google Scholar]
- Buerger K., Teipel S. J., Zinkowski R., Blennow K., Arai H., Engel R., et al. (2002a). CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology 59, 627–629 [DOI] [PubMed] [Google Scholar]
- Buerger K., Zinkowski R., Teipel S. J., Tapiola T., Arai H., Blennow K., et al. (2002b). Differential diagnosis of Alzheimer disease with cerebrospinal fluid levels of tau protein phosphorylated at threonine 231. Arch. Neurol. 59, 1267–1272 10.1001/archneur.59.8.1267 [DOI] [PubMed] [Google Scholar]
- Clark C. M., Xie S., Chittams J., Ewbank D., Peskind E., Galasko D., et al. (2003). Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch. Neurol. 60, 1696–1702 10.1001/archneur.60.12.1696 [DOI] [PubMed] [Google Scholar]
- Cohen T. J., Guo J. L., Hurtado D. E., Kwong L. K., Mills I. P., Trojanowski J. Q., et al. (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2:252 10.1038/ncomms1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., Van Der Zee J., Engelborghs S., Wils H., Pirici D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- Davis D. G., Schmitt F. A., Wekstein D. R., Markesbery W. R. (1999). Alzheimer neuropathologic alterations in aged cognitively normal subjects. J. Neuropathol. Exp. Neurol. 58, 376–388 [DOI] [PubMed] [Google Scholar]
- De Jong D., Jansen R. W., Pijnenburg Y. A., Van Geel W. J., Borm G. F., Kremer H. P., et al. (2007). CSF neurofilament proteins in the differential diagnosis of dementia. J. Neurol. Neurosurg. Psychiatry 78, 936–938 10.1136/jnnp.2006.107326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer G., Shapiro F., Vanderstichele H., Vanmechelen E., Engelborghs S., De Deyn P. P., et al. (2010). Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch. Neurol. 67, 949–956 10.1001/archneurol.2010.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza L. C., Lamari F., Belliard S., Jardel C., Houillier C., De Paz R., et al. (2011). Cerebrospinal fluid biomarkers in the differential diagnosis of Alzheimer's disease from other cortical dementias. J. Neurol. Neurosurg. Psychiatry 82, 240–246 10.1136/jnnp.2010.207183 [DOI] [PubMed] [Google Scholar]
- Dejesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D. (2004). Sporadic tauopaties: Pick's disease, corticobasal degeneration, progressive supranuclear palsy and argyrophilic grain disease, in The Neuropathology of Dementia 2nd Edn, eds Esiri M., Lee V. M-Y., Trojanowski J. Q. (New York, NY: Cambridge University Press; ), 227–256 [Google Scholar]
- Engelborghs S., De Vreese K., Van De Casteele T., Vanderstichele H., Van Everbroeck B., Cras P., et al. (2008). Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol. Aging 29, 1143–1159 10.1016/j.neurobiolaging.2007.02.016 [DOI] [PubMed] [Google Scholar]
- Engelborghs S., Maertens K., Vloeberghs E., Aerts T., Somers N., Marien P., et al. (2006). Neuropsychological and behavioural correlates of CSF biomarkers in dementia. Neurochem. Int. 48, 286–295 10.1016/j.neuint.2005.11.002 [DOI] [PubMed] [Google Scholar]
- Eslinger P. J., Moore P., Antani S., Anderson C., Grossman M. (2012). Apathy in frontotemporal dementia: behavioral and neuroimaging correlates. Behav. Neurol. 25, 127–136 10.3233/BEN-2011-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan A. M., Mintun M. A., Mach R. H., Lee S. Y., Dence C. S., Shah A. R., et al. (2006). Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 59, 512–519 10.1002/ana.20730 [DOI] [PubMed] [Google Scholar]
- Fagan A. M., Shaw L. M., Xiong C., Vanderstichele H., Mintun M. A., Trojanowski J. Q., et al. (2011). Comparison of analytical platforms for cerebrospinal fluid measures of {beta}-amyloid 1-42, total tau, and P-tau181 for identifying alzheimer disease amyloid plaque pathology. Arch. Neurol. 68, 1137–1144 10.1001/archneurol.2011.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M. S., Farmer J., Johnson J. K., Clark C. M., Arnold S. E., Coslett H. B., et al. (2006). Frontotemporal dementia: clinicopathological correlations. Ann. Neurol. 59, 952–962 10.1002/ana.20873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds P., McAuley E., Gibbons L., Davidson Y., Pickering-Brown S. M., Neary D., et al. (2008). TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer's disease and frontotemporal lobar degeneration. Acta Neuropathol. 116, 141–146 10.1007/s00401-008-0389-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelle A., Roche S., Geny C., Bennys K., Labauge P., Tholance Y., et al. (2011). Decreased sAbetaPPbeta, Abeta38, and Abeta40 cerebrospinal fluid levels in frontotemporal dementia. J. Alzheimers Dis. 26, 553–563 10.3233/JAD-2011-110515 [DOI] [PubMed] [Google Scholar]
- Geser F., Brandmeir N. J., Kwong L. K., Martinez-Lage M., Elman L., McCluskey L., et al. (2008). Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. 65, 636–641 10.1001/archneur.65.5.636 [DOI] [PubMed] [Google Scholar]
- Geser F., Martinez-Lage M., Robinson J., Uryu K., Neumann M., Brandmeir N. J., et al. (2009). Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch. Neurol. 66, 180–189 10.1001/archneurol.2008.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R., Benussi L., Glionna M., Franzoni M., Binetti G. (2008). Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239 10.1212/01.wnl.0000325058.10218.fc [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini M. L., Brambati S. M., Ginex V., Ogar J., Dronkers N. F., Marcone A., et al. (2008). The logopenic/phonological variant of primary progressive aphasia. Neurology 71, 1227–1234 10.1212/01.wnl.0000320506.79811.da [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini M. L., Dronkers N. F., Rankin K. P., Ogar J. M., Phengrasamy L., Rosen H. J., et al. (2004). Cognition and anatomy in three variants of primary progressive aphasia. Ann. Neurol. 55, 335–346 10.1002/ana.10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini M. L., Hillis A. E., Weintraub S., Kertesz A., Mendez M., Cappa S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A. J., Harvey R. J., Thompson E. J., Rossor M. N. (1999). Increased tau in the cerebrospinal fluid of patients with frontotemporal dementia and Alzheimer's disease. Neurosci. Lett. 259, 133–135 10.1016/S0304-3940(98)00904-5 [DOI] [PubMed] [Google Scholar]
- Grossman M. (2010). Primary progressive aphasia: clinicopathological correlations. Nat. Rev. Neurol. 6, 88–97 10.1038/nrneurol.2009.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M., Eslinger P. J., Troiani V., Anderson C., Avants B., Gee J. C., et al. (2010). The role of ventral medial prefrontal cortex in social decisions: converging evidence from fMRI and frontotemporal lobar degeneration. Neuropsychologia 48, 3505–3512 10.1016/j.neuropsychologia.2010.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M., Farmer J., Leight S., Work M., Moore P., Van Deerlin V., et al. (2005). Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer's disease. Ann. Neurol. 57, 721–729 10.1002/ana.20477 [DOI] [PubMed] [Google Scholar]
- Grossman M., Powers J., Ash S., McMillan C., Burkholder L., Irwin D., et al. (2012). Disruption of large-scale neural networks in non-fluent/agrammatic variant primary progressive aphasia associated with frontotemporal degeneration pathology. Brain Lang. [Epub ahead of print]. 10.1016/j.bandl.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M., Xie S. X., Libon D. J., Wang X., Massimo L., Moore P., et al. (2008). Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology 70, 2036–2045 10.1212/01.wnl.0000303816.25065.bc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena D., Ash S., McMillan C., Avants B., Gee J., Grossman M. (2010). Why are patients with progressive nonfluent aphasia nonfluent? Neurology 75, 588–594 10.1212/WNL.0b013e3181ed9c7d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H., Blennow K., Shaw L. M., Hoessler Y. C., Zetterberg H., Trojanowski J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp. Gerontol. 45, 30–40 10.1016/j.exger.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse C., Rosengren L., Vanmechelen E., Vanderstichele H., Jensen C., Davidsson P., et al. (2000). Cerebrospinal fluid markers for Alzheimer's disease evaluated after acute ischemic stroke. J. Alzheimers Dis. 2, 199–206 [DOI] [PubMed] [Google Scholar]
- Hodges J. R., Davies R. R., Xuereb J. H., Casey B., Broe M., Bak T. H., et al. (2004). Clinicopathological correlates in frontotemporal dementia. Ann. Neurol. 56, 399–406 10.1002/ana.20203 [DOI] [PubMed] [Google Scholar]
- Hodges J. R., Patterson K. (2007). Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol. 6, 1004–1014 10.1016/S1474-4422(07)70266-1 [DOI] [PubMed] [Google Scholar]
- Hoglinger G. U., Melhem N. M., Dickson D. W., Sleiman P. M., Wang L. S., Klei L., et al. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705 10.1038/ng.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W. T., Chen-Plotkin A., Arnold S. E., Grossman M., Clark C. M., Shaw L. M., et al. (2010a). Biomarker discovery for Alzheimer's disease, frontotemporal lobar degeneration, and Parkinson's disease. Acta Neuropathol. 120, 385–399 10.1007/s00401-010-0723-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W. T., Chen-Plotkin A., Grossman M., Arnold S. E., Clark C. M., Shaw L. M., et al. (2010b). Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 75, 2079–2086 10.1212/WNL.0b013e318200d78d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W. T., McMillan C., Libon D., Leight S., Forman M., Lee V. M., et al. (2010c). Multimodal predictors for Alzheimer disease in nonfluent primary progressive aphasia. Neurology 75, 595–602 10.1212/WNL.0b013e3181ed9c52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W. T., Trojanowski J. Q., Shaw L. M. (2011). Biomarkers in frontotemporal lobar degenerations–progress and challenges. Prog. Neurobiol. 95, 636–648 10.1016/j.pneurobio.2011.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., et al. (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- Irwin D. J., Cohen T. J., Grossman M., Arnold S. E., Xie S. X., Lee V. M., et al. (2012a). Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain 135, 807–818 10.1093/brain/aws013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. J., McMillan C. T., Toledo J. B., Arnold S. E., Shaw L. M., Wang L. S., et al. (2012b). Comparison of cerebrospinal fluid levels of tau and Abeta 1-42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch. Neurol. 69, 1018–1025 10.1001/archneurol.2012.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. J., McMillan C. T., Brettschneider J., Libon D. J., Powers J., Rascovsky K., et al. (2013). Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 84, 163–169 10.1136/jnnp-2012-303507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Knopman D. S., Jagust W. J., Shaw L. M., Aisen P. S., Weiner M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Vemuri P., Wiste H. J., Weigand S. D., Lesnick T. G., Lowe V., et al. (2012). Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch. Neurol. 69, 856–867 10.1001/archneurol.2011.3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K., Takeda A., Shiga Y., Sato S., Ohnuma A., Nomura H., et al. (2006). CSF tau protein: a new prognostic marker for Guillain-Barre syndrome. Neurology 67, 1470–1472 10.1212/01.wnl.0000240119.29939.c7 [DOI] [PubMed] [Google Scholar]
- Josephs K. A. (2008). Frontotemporal dementia and related disorders: deciphering the enigma. Ann. Neurol. 64, 4–14 10.1002/ana.21426 [DOI] [PubMed] [Google Scholar]
- Josephs K. A., Duffy J. R., Strand E. A., Whitwell J. L., Layton K. F., Parisi J. E., et al. (2006a). Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 129, 1385–1398 10.1093/brain/awl078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs K. A., Whitwell J. L., Jack C. R., Parisi J. E., Dickson D. W. (2006b). Frontotemporal lobar degeneration without lobar atrophy. Arch. Neurol. 63, 1632–1638 10.1001/archneur.63.11.1632 [DOI] [PubMed] [Google Scholar]
- Kapaki E., Paraskevas G. P., Papageorgiou S. G., Bonakis A., Kalfakis N., Zalonis I., et al. (2008). Diagnostic value of CSF biomarker profile in frontotemporal lobar degeneration. Alzheimer Dis. Assoc. Disord. 22, 47–53 10.1097/WAD.0b013e3181610fea [DOI] [PubMed] [Google Scholar]
- Kasai T., Tokuda T., Ishigami N., Sasayama H., Foulds P., Mitchell D. J., et al. (2009). Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 117, 55–62 10.1007/s00401-008-0456-1 [DOI] [PubMed] [Google Scholar]
- Kertesz A., McMonagle P., Blair M., Davidson W., Munoz D. G. (2005). The evolution and pathology of frontotemporal dementia. Brain 128, 1996–2005 10.1093/brain/awh598 [DOI] [PubMed] [Google Scholar]
- Kipps C. M., Hodges J. R., Hornberger M. (2010). Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ‘bvFTD phenocopy syndrome’. Curr. Opin. Neurol. 23, 628–632 10.1097/WCO.0b013e3283404309 [DOI] [PubMed] [Google Scholar]
- Knibb J. A., Xuereb J. H., Patterson K., Hodges J. R. (2006). Clinical and pathological characterization of progressive aphasia. Ann. Neurol. 59, 156–165 10.1002/ana.20700 [DOI] [PubMed] [Google Scholar]
- Knopman D. S., Boeve B. F., Parisi J. E., Dickson D. W., Smith G. E., Ivnik R. J., et al. (2005). Antemortem diagnosis of frontotemporal lobar degeneration. Ann. Neurol. 57, 480–488 10.1002/ana.20425 [DOI] [PubMed] [Google Scholar]
- Knopman D. S., Kramer J. H., Boeve B. F., Caselli R. J., Graff-Radford N. R., Mendez M. F., et al. (2008). Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 131, 2957–2968 10.1093/brain/awn234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koedam E. L., Van Der Vlies A. E., Van Der Flier W. M., Verwey N. A., Koene T., Scheltens P., et al. (2012). Cognitive correlates of cerebrospinal fluid biomarkers in frontotemporal dementia. Alzheimers Dement. [Epub ahead of print]. 10.1016/j.jalz.2011.12.007 [DOI] [PubMed] [Google Scholar]
- Koopman K., Le Bastard N., Martin J. J., Nagels G., De Deyn P. P., Engelborghs S. (2009). Improved discrimination of autopsy-confirmed Alzheimer's disease (AD) from non-AD dementias using CSF P-tau(181P). Neurochem. Int. 55, 214–218 10.1016/j.neuint.2009.02.017 [DOI] [PubMed] [Google Scholar]
- Koric L., Felician O., Guedj E., Hubert A. M., Mancini J., Boucraut J., et al. (2010). Could clinical profile influence CSF biomarkers in early-onset Alzheimer disease? Alzheimer Dis. Assoc. Disord. 24, 278–283 10.1097/WAD.0b013e3181d712d9 [DOI] [PubMed] [Google Scholar]
- Krut J. J., Zetterberg H., Blennow K., Cinque P., Hagberg L., Price R. W., et al. (2013). Cerebrospinal fluid Alzheimer's biomarker profiles in CNS infections. J. Neurol. 260, 620–626 10.1007/s00415-012-6688-y [DOI] [PubMed] [Google Scholar]
- Lee S. E., Rabinovici G. D., Mayo M. C., Wilson S. M., Seeley W. W., Dearmond S. J., et al. (2011). Clinicopathological correlations in corticobasal degeneration. Ann. Neurol. 70, 327–340 10.1002/ana.22424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewczuk P., Zimmermann R., Wiltfang J., Kornhuber J. (2009). Neurochemical dementia diagnostics: a simple algorithm for interpretation of the CSF biomarkers. J Neural Transm 116, 1163–1167 10.1007/s00702-009-0277-y [DOI] [PubMed] [Google Scholar]
- Libon D. J., Rascovsky K., Gross R. G., White M. T., Xie S. X., Dreyfuss M., et al. (2011). The Philadelphia Brief Assessment of Cognition (PBAC): a validated screening measure for dementia. Clin. Neuropsychol. 25, 1314–1330 10.1080/13854046.2011.631585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk C., Compta Y., Magdalinou N., Marti M. J., Hondhamuni G., Zetterberg H., et al. (2012). Development and assessment of sensitive immuno-PCR assays for the quantification of cerebrospinal fluid three- and four-repeat tau isoforms in tauopathies. J. Neurochem. 123, 396–405 10.1111/j.1471-4159.2012.07911.x [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Neumann M., Baborie A., Sampathu D. M., Du Plessis D., Jaros E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R., Neumann M., Bigio E. H., Cairns N. J., Alafuzoff I., Kril J., et al. (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4 10.1007/s00401-009-0612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimo L., Libon D. J., Chandrasekaran K., Dreyfuss M., McMillan C. T., Rascovsky K., et al. (2013). Self-appraisal in behavioural variant frontotemporal degeneration. J. Neurol. Neurosurg. Psychiatry, 84, 148–153 10.1136/jnnp-2012-303153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimo L., Powers C., Moore P., Vesely L., Avants B., Gee J., et al. (2009). Neuroanatomy of apathy and disinhibition in frontotemporal lobar degeneration. Dement. Geriatr. Cogn. Disord. 27, 96–104 10.1159/000194658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo E. S., Shin R. W., Billingsley M. L., Van Devoorde A., O'Connor M., Trojanowski J. Q., et al. (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron 13, 989–1002 10.1016/0896-6273(94)90264-X [DOI] [PubMed] [Google Scholar]
- Mattsson N., Andreasson U., Persson S., Arai H., Batish S. D., Bernardini S., et al. (2011). The Alzheimer's association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 7, 386–395 e386. 10.1016/j.jalz.2011.05.2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N., Ruetschi U., Pijnenburg Y. A., Blankenstein M. A., Podust V. N., Li S., et al. (2008). Novel cerebrospinal fluid biomarkers of axonal degeneration in frontotemporal dementia. Mol. Med. Report. 1, 757–761 10.3892/mmr_00000025 [DOI] [PubMed] [Google Scholar]
- McMillan C. T., Avants B., Irwin D. J., Toledo J. B., Wolk D. A., Van Deerlin V. M., et al. (2013). Can MRI screen for CSF biomarkers in neurodegenerative disease? Neurology, 80, 132–138 10.1212/WNL.0b013e31827b9147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan C. T., Brun C., Siddiqui S., Churgin M., Libon D., Yushkevich P., et al. (2012a). White matter imaging contributes to the multimodal diagnosis of frontotemporal lobar degeneration. Neurology 78, 1761–1768 10.1212/WNL.0b013e31825830bd [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan C. T., Rascovsky K., Khella M. C., Clark R., Grossman M. (2012b). The neural basis for establishing a focal point in pure coordination games. Soc. Cogn. Affect. Neurosci. 7, 881–887 10.1093/scan/nsr070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M., Wicklund A., Johnson N., Rogalski E., Leger G. C., Rademaker A., et al. (2008). Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann. Neurol. 63, 709–719 10.1002/ana.21388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M. M. (1982). Slowly progressive aphasia without generalized dementia. Ann. Neurol. 11, 592–598 10.1002/ana.410110607 [DOI] [PubMed] [Google Scholar]
- Mesulam M. M. (2001). Primary progressive aphasia. Ann. Neurol. 49, 425–432 [PubMed] [Google Scholar]
- NLM/NIH. (2012). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/ [Accessed 10/27/12 2012].
- Olsson A., Vanderstichele H., Andreasen N., De Meyer G., Wallin A., Holmberg B., et al. (2005). Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 51, 336–345 10.1373/clinchem.2004.039347 [DOI] [PubMed] [Google Scholar]
- Ost M., Nylen K., Csajbok L., Ohrfelt A. O., Tullberg M., Wikkelso C., et al. (2006). Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67, 1600–1604 10.1212/01.wnl.0000242732.06714.0f [DOI] [PubMed] [Google Scholar]
- Otto M., Wiltfang J., Tumani H., Zerr I., Lantsch M., Kornhuber J., et al. (1997). Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci. Lett. 225, 210–212 10.1016/S0304-3940(97)00215-2 [DOI] [PubMed] [Google Scholar]
- Patel S., Lee E. B., Xie S. X., Law A., Jackson E. M., Arnold S. E., et al. (2012). Phosphorylated tau/amyloid beta 1-42 ratio in ventricular cerebrospinal fluid reflects outcome in idiopathic normal pressure hydrocephalus. Fluids Barriers CNS 9:7 10.1186/2045-8118-9-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnenburg Y. A., Schoonenboom N. S., Rosso S. M., Mulder C., Van Kamp G. J., Van Swieten J. C., et al. (2004). CSF tau and Abeta42 are not useful in the diagnosis of frontotemporal lobar degeneration. Neurology 62, 1649 [DOI] [PubMed] [Google Scholar]
- Pijnenburg Y. A., Schoonenboom S. N., Mehta P. D., Mehta S. P., Mulder C., Veerhuis R., et al. (2007). Decreased cerebrospinal fluid amyloid beta (1-40) levels in frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 78, 735–737 10.1136/jnnp.2006.105064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici G. D., Jagust W. J., Furst A. J., Ogar J. M., Racine C. A., Mormino E. C., et al. (2008). Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann. Neurol. 64, 388–401 10.1002/ana.21451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K., Hodges J. R., Knopman D., Mendez M. F., Kramer J. H., Neuhaus J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K., Salmon D. P., Hansen L. A., Galasko D. (2008). Distinct cognitive profiles and rates of decline on the Mattis Dementia Rating Scale in autopsy-confirmed frontotemporal dementia and Alzheimer's disease. J. Int. Neuropsychol. Soc. 14, 373–383 10.1017/S135561770808051X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider M., Wagenpfeil S., Diehl J., Lautenschlager N., Theml T., Heldmann B., et al. (2002). Tau and Abeta42 protein in CSF of patients with frontotemporal degeneration. Neurology 58, 1622–1628 [DOI] [PubMed] [Google Scholar]
- Rosso S. M., Van Herpen E., Pijnenburg Y. A., Schoonenboom N. S., Scheltens P., Heutink P., et al. (2003). Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch. Neurol. 60, 1209–1213 10.1001/archneur.60.9.1209 [DOI] [PubMed] [Google Scholar]
- Schmidt M. L., Gur R. E., Gur R. C., Trojanowski J. Q. (1988). Intraneuronal and extracellular neurofibrillary tangles exhibit mutually exclusive cytoskeletal antigens. Ann. Neurol. 23, 184–189 10.1002/ana.410230212 [DOI] [PubMed] [Google Scholar]
- Schoonenboom N. S., Pijnenburg Y. A., Mulder C., Rosso S. M., Van Elk E. J., Van Kamp G. J., et al. (2004). Amyloid beta(1-42) and phosphorylated tau in CSF as markers for early-onset Alzheimer disease. Neurology 62, 1580–1584 [DOI] [PubMed] [Google Scholar]
- Schoonenboom N. S., Reesink F. E., Verwey N. A., Kester M. I., Teunissen C. E., Van De Ven P. M., et al. (2012). Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology 78, 47–54 10.1212/WNL.0b013e31823ed0f0 [DOI] [PubMed] [Google Scholar]
- Seguin J., Formaglio M., Perret-Liaudet A., Quadrio I., Tholance Y., Rouaud O., et al. (2011). CSF biomarkers in posterior cortical atrophy. Neurology 76, 1782–1788 10.1212/WNL.0b013e31821ccc98 [DOI] [PubMed] [Google Scholar]
- Seppala T. T., Nerg O., Koivisto A. M., Rummukainen J., Puli L., Zetterberg H., et al. (2012). CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 78, 1568–1575 10.1212/WNL.0b013e3182563bd0 [DOI] [PubMed] [Google Scholar]
- Shaw L. M., Vanderstichele H., Knapik-Czajka M., Clark C. M., Aisen P. S., Petersen R. C., et al. (2009). Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 10.1002/ana.21610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L. M., Vanderstichele H., Knapik-Czajka M., Figurski M., Coart E., Blennow K., et al. (2011). Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 121, 597–609 10.1007/s00401-011-0808-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Shaw C. L., Du Plessis D., Richardson A. M., Bailey K. L., Julien C., et al. (2005). Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 110, 501–512 10.1007/s00401-005-1079-4 [DOI] [PubMed] [Google Scholar]
- Sjogren M., Davidsson P., Tullberg M., Minthon L., Wallin A., Wikkelso C., et al. (2001). Both total and phosphorylated tau are increased in Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 70, 624–630 10.1136/jnnp.70.5.624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren M., Minthon L., Davidsson P., Granerus A. K., Clarberg A., Vanderstichele H., et al. (2000a). CSF levels of tau, beta-amyloid(1-42) and GAP-43 in frontotemporal dementia, other types of dementia and normal aging. J. Neural Transm. 107, 563–579 10.1007/s007020070079 [DOI] [PubMed] [Google Scholar]
- Sjogren M., Rosengren L., Minthon L., Davidsson P., Blennow K., Wallin A. (2000b). Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology 54, 1960–1964 [DOI] [PubMed] [Google Scholar]
- Snowden J., Neary D., Mann D. (2007). Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 114, 31–38 10.1007/s00401-007-0236-3 [DOI] [PubMed] [Google Scholar]
- Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292 10.1016/j.jalz.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinacker P., Hendrich C., Sperfeld A. D., Jesse S., Von Arnim C. A., Lehnert S., et al. (2008). TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 65, 1481–1487 10.1001/archneur.65.11.1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapiola T., Alafuzoff I., Herukka S. K., Parkkinen L., Hartikainen P., Soininen H., et al. (2009). Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 66, 382–389 10.1001/archneurol.2008.596 [DOI] [PubMed] [Google Scholar]
- Toledo J. B., Brettschneider J., Grossman M., Arnold S. E., Hu W. T., Xie S. X., et al. (2012). CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol. 124, 23–35 10.1007/s00401-012-0983-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski J. Q., Growdon J. H. (1998). A new consensus report on biomarkers for the early antemortem diagnosis of Alzheimer disease: current status, relevance to drug discovery, and recommendations for future research. J. Neuropathol. Exp. Neurol. 57, 643–644 [DOI] [PubMed] [Google Scholar]
- Trojanowski J. Q., Vandeerstichele H., Korecka M., Clark C. M., Aisen P. S., Petersen R. C., et al. (2010). Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimers Dement. 6, 230–238 10.1016/j.jalz.2010.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R. S., Kenyon L. C., Trojanowski J. Q., Gonatas N., Grossman M. (1996). Clinical, neuroimaging, and pathologic features of progressive nonfluent aphasia. Ann. Neurol. 39, 166–173 10.1002/ana.410390205 [DOI] [PubMed] [Google Scholar]
- Van Deerlin V. M., Sleiman P. M., Martinez-Lage M., Chen-Plotkin A., Wang L. S., Graff-Radford N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239 10.1038/ng.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Harten A. C., Kester M. I., Visser P. J., Blankenstein M. A., Pijnenburg Y. A., Van Der Flier W. M., et al. (2011). Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin. Chem. Lab. Med. 49, 353–366 10.1515/CCLM.2011.086 [DOI] [PubMed] [Google Scholar]
- Vanmechelen E., Vanderstichele H., Davidsson P., Van Kerschaver E., Van Der Perre B., Sjogren M., et al. (2000). Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci. Lett. 285, 49–52 10.1016/S0304-3940(00)01036-3 [DOI] [PubMed] [Google Scholar]
- Verwey N. A., Kester M. I., Van Der Flier W. M., Veerhuis R., Berkhof H., Twaalfhoven H., et al. (2010). Additional value of CSF amyloid-beta 40 levels in the differentiation between FTLD and control subjects. J. Alzheimers Dis. 20, 445–452 10.3233/JAD-2010-1392 [DOI] [PubMed] [Google Scholar]
- Wang L. S., Leung Y. Y., Chang S. K., Leight S., Knapik-Czajka M., Baek Y., et al. (2012). Comparison of xMAP and ELISA assays for detecting cerebrospinal fluid biomarkers of Alzheimer's disease. J. Alzheimers Dis. 31, 439–445 10.3233/JAD-2012-120082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M. W., Aisen P. S., Jack C. R., Jr., Jagust W. J., Trojanowski J. Q., Shaw L., et al. (2010). The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 6, 202-211 e207. 10.1016/j.jalz.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J. L., Weigand S. D., Boeve B. F., Senjem M. L., Gunter J. L., Dejesus-Hernandez M., et al. (2012). Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135, 794–806 10.1093/brain/aws001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V., Sundarraj S., Mann D., Sjogren M., Blenow K., Clark C. M., et al. (2003). Selective reduction of soluble tau proteins in sporadic and familial frontotemporal dementias: an international follow-up study. Acta Neuropathol. 105, 469–476 10.1007/s00401-002-0668-8 [DOI] [PubMed] [Google Scholar]
- Zhukareva V., Vogelsberg-Ragaglia V., Van Deerlin V. M., Bruce J., Shuck T., Grossman M., et al. (2001). Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann. Neurol. 49, 165–175 [DOI] [PubMed] [Google Scholar]