

Abstract
Neural tube defects (NTDs) are common birth defects of complex etiology. Family and population-based studies have confirmed a genetic component to NTDs. However, despite more than three decades of research, the genes involved in human NTDs remain largely unknown. We tested the hypothesis that rare copy number variants (CNVs), especially de novo germline CNVs, are a significant risk factor for NTDs. We used array-based comparative genomic hybridization (aCGH) to identify rare CNVs in 128 Caucasian and 61 Hispanic patients with non-syndromic lumbar-sacral myelomeningocele. We also performed aCGH analysis on the parents of affected individuals with rare CNVs where parental DNA was available (42 sets). Among the eight de novo CNVs that we identified, three generated copy number changes of entire genes. One large heterozygous deletion removed 27 genes, including PAX3, a known spina bifida-associated gene. A second CNV altered genes (PGPD8, ZC3H6) for which little is known regarding function or expression. A third heterozygous deletion removed GPC5 and part of GPC6, genes encoding glypicans. Glypicans are proteoglycans that modulate the activity of morphogens such as Sonic Hedgehog (SHH) and bone morphogenetic proteins (BMPs), both of which have been implicated in NTDs. Additionally, glypicans function in the planar cell polarity (PCP) pathway, and several PCP genes have been associated with NTDs. Here, we show that GPC5 orthologs are expressed in the neural tube, and that inhibiting their expression in frog and fish embryos results in NTDs. These results implicate GPC5 as a gene required for normal neural tube development.
INTRODUCTION
Neural tube defects (NTDs) arise from a failed or disordered closure of the neural tube during embryogenesis. These congenital disorders occur with a frequency of 1/1000–1/5000 worldwide and therefore, are among the most common birth defects (together with congenital heart abnormalities and craniofacial anomalies) (1–4). While environmental factors have been implicated in NTDs, these factors alone fail to explain most cases. Studies in mice have described over 200 (5) monogenic mutant lines with NTDs and have shown that the genes involved play multiple distinct roles in neurulation (1,6). Mouse studies also show that NTDs may be caused by multigenic influences, as revealed by the differing severities produced when genetic defects are combined (7). However, despite decades of research and hundreds of studies, we do not yet understand the genetic contribution to human NTDs. In humans, genetic etiologies for NTDs were first identified 30 years ago, when it was shown that the family members of a person with an NTD have an increased risk (from 2 to 5%) of having an NTD themselves (8–12). In addition, NTDs have been linked to several genes in the folate-homocysteine metabolic axis, consistent with epidemiological evidence that between 30 and 70% of NTDs can be prevented by prenatal folate (2–4). Polymorphisms in these genes, however, are not significantly associated with NTDs in all populations, suggesting that polygenic and environmental factors are important. Our groups and others have described NTDs in chromosomal and inherited syndromes, including trisomy 13 (13–16), trisomy 18 (16–19), trisomy 21, and 22q11.2 and 13q deletion syndromes (20–23). In rare cases, syndromes that variably present with NTDs have been associated with a mutation in a single gene (24–36). Using traditional linkage disequilibrium, patient studies of individual (non-folate-related) candidate genes, including jumonji (JMJ) (37), apolipoprotein E (ApoE), apolipoprotein B (ApoB) (38), bone morphogenetic protein-4 (BMP4) (39), CITED2 (40), transcription factor AP2 (TFAP2) (41), multiple sequence homeobox-2 (MSX2) (41), PAX3 (42) and noggin (NOG) (43), have only rarely succeeded in ascribing attributable risk for NTDs to specific genes.
More recently, several genes in the planar cell polarity (PCP) signaling pathway have been implicated in NTDs. PCP, also called tissue polarity, is the process by which cells become polarized within the plane of an epithelium. This form of polarization has been well studied in the adult epithelial tissues of Drosophila where phenotypes are observed in the form of altered orientation of hairs and bristles (44), among other defects. In the fly, genetic studies of a wide range of mutations affecting these highly organized structures identified a group of so-called ‘core’ PCP genes, required for PCP signaling in all tissues. These include Frizzled (Fz), Dishevelled (Dsh), Strabismus/Van Gogh (Stbm/Vang), Flamingo (Fmi), Prickle (Pk) and Diego (Dgo) (44–46). Members of the PCP pathway are highly conserved in vertebrates where they are also implicated in controlling tissue polarity in several epithelia. Additionally, many of these proteins regulate cell adhesion or cytoskeletal organization in non-polarized cells to regulate morphogenesis. PCP regulators are essential for controlling convergent extension (CE) of tissues during gastrulation and neurulation in the embryo (47–49), a process that requires cell shape changes and cell intercalation. In this context, regulation of core PCP components occurs through β-catenin-independent Wnt signaling (47,48), whereas PCP in Drosophila is not controlled by Wnt/Wg ligands. The importance for CE in neural tube closure was first demonstrated by molecular disruption of CE following expression of dominant-negative Dishevelled (Dvl) in the frog embryo (50). In these studies, failure of CE in the midline caused the neural folds to form too widely apart, preventing them from fusing at the midline (50). Genetic studies in mouse have further implicated core PCP pathway components in NTDs (e.g., Vangl2 (51–53), Celsr1 [an Fmi ortholog] (54), Scribble (Scrib) (55) and combined mutations of either Dvl1/Dvl2 (56) or Fz3/Fz6 (57)). A recent paper demonstrated that two key morphogenetic processes required for neural tube closure (CE and neural plate apical constriction) are linked by PCP signaling (58). PCP components are also likely to contribute to NTDs in humans. Genetic studies of human craniorachischisis suggest an association with mutations in CELSR1 and SCRIB (59). Recently, PRICKLE1 and PRICKLE2 mutations were implicated as predisposing factors or risk modifiers for human spina bifida (60,61). Two reviews summarize the compelling evidence for PCP gene involvement in NTDs (62,63).
The glypican family of heparin sulfate proteoglycan core proteins has recently been implicated in regulating PCP signaling and CE in vertebrates. Zebrafish knypek/gpc4 mutants exhibit abnormal convergence and extension during gastrulation and embryonic cells have abnormal PCP (64). Additionally, knockdown of gpc4 in frog embryos generates NTDs (65,66). Glypicans can modulate the activity of bone morphogenetic proteins (BMPs) and Sonic Hedgehog (SHH) (67,68), morphogens that regulate the bending of neural folds during closure of the neural tube (69–71). During mouse embryogenesis, high levels of Glypican 5 are expressed in the neural tube (72). Consistent with a potential role of glypicans in spina bifida, patients with 13q deletions that sometimes remove GPC5 and GPC6 can present with NTDs (23,73). Collectively, these results implicate the glypican gene family in promoting normal neural tube development.
An extensive body of literature now shows that changes in copy number variants (CNVs) play a substantial role in human disease (74–79) including birth defects (80,81). However, with the exception of a few small-scale genomic rearrangement studies (20,82,83), almost all association studies regarding human NTDs have focused on either single-nucleotide polymorphisms or gene mutations. To address this deficit, we identified three independent cohorts of non-syndromic myelomeningocele, the most commonly observed form of spina bifida, and performed whole-genome comparative genomic hybridization analysis. This study aimed to (1) identify rare copy number events directly affecting coding genes in cases; (2) determine whether any of these rare CNVs were de novo events and (3) test whether candidate genes affected by such CNVs were required for neural tube development in frogs and zebrafish. These analyses identified GPC5 as a human spina bifida candidate gene and demonstrated that reducing the expression of GPC5 orthologs in frogs or fish resulted in NTDs consistent with spina bifida. This work demonstrates that birth-defect-associated genes can be identified by CNV analysis and functionally validated in vertebrate model systems.
RESULTS
We performed array-based comparative genomic hybridization (aCGH on 189 spina bifida patients (128 Caucasians, 61 Mexicans) to identify rare CNVs. Only variants that affected coding sequences of genes were considered for further analysis (Supplementary Material, Table S1). For these cases, parental DNA was obtained when available and the variants were analyzed to determine whether they were de novo. Any variants that affected entire genes were considered for further functional analysis in vertebrate model systems.
Rare copy number variations
We defined a CNV in affected cohorts to be ‘rare’ if the segment has no overlap with any of the CNVs in the Database of Genomic Variants, or if the overlap with such variants was less than or equal to 50% of its genomic length. To identify coding regions of genes within those events, we further refined boundaries of the rare CNVs. The length and boundaries of a given CNV reflect the resolution of the array. Two CNV intervals from different individuals may have the same boundaries. Frequently, however, CNV intervals have varying extents of overlap with other CNV intervals in a given population. We define the ‘maximum frequency’ of any given CNV as the number of overlaps with other CNVs, and used the ‘minimal overlapping region’ to be the boundaries of rare events.
We identified a total of 1976 rare genomic variant events containing at least one exon of a coding gene, giving an average of 10 rare events per individual (all the events are detailed individually in Supplementary Material, Table S1). In these 1976 CNVs, a total of 2269 genes were affected. Interestingly, a subset of such CNV-altered genes have previously been implicated in neural tube formation; for instance, JMJD5 (‘jumonji’ domain containing 5 (37), HES1 and HES3 (84); PAX3 (27,85); and both BRD3 and BRD4 (bromodomain-containing 3 and 4) (86). Although it is reasonable to assume that this subset of CNVs contributed to the NTD in these patients, the majority of CNVs identified in this study could not readily be associated with NTDs without further analysis. Hence, we tested the hypothesis that rare coding genes with altered number of copies may have a common functional impact using network and pathway analysis.
We used the reactome functional interaction (FI) network (which consists of curated pathways and high-confidence predicted interactions identified from non-curated sources by machine-learning techniques) to identify the significantly enriched pathways that are interconnected by FI of genes affected by rare CNV events. The combined network contains nearly 9400 SwissProt proteins and 210 000 FIs. Of the 2269 coding genes affected by rare copy number aberrations, 44% were projected onto the FI network. Using hierarchical clustering based on the network, we identified 567 genes that were more highly interconnected with each other than by chance (P < 0.001). A subnetwork was built from these 567 genes. Spectral partitioning clustering algorithm revealed 15 functionally related network modules (0–14) as illustrated in Figure 1, 8 of which contain 15 or more genes (Supplementary Material, Table S2 lists the genes forming the modules). Each of the functional modules is then annotated for pathways using KEGG, Panther, Reactome, NCI pathways, BioCarta and CellMap, and the 47 statistically significant pathways with a false discovery rate (FDR) < 0.05 are listed in Supplementary Material, Table S3 (note that some pathways are indicated multiple times; thus, 47 pathways is the coalesced number). It is interesting to note that a subset of the pathways contained within the modules have been previously implicated in NTDs, including integrin signaling (FDR < 3e−4), 3) cell cycle (FDR < 1e−3), Wnt signaling (FDR < 5e−4), Hedgehog signaling (FDR < 3e−4) and glypican pathway (FDR = 4.4e−3). Notably, although only 8 modules contained 15 or more genes, both the Wnt signaling and Glypican pathway modules were among them (see Supplementary Material, Figs S2 and S3). We further tested whether these pathways were truly enriched due to their disease association or whether they were simply the result of copy number variation normally affecting the genes in these pathways. We analyzed CNPs generated for 2026 healthy genomes as reported by Shaik et al. (87). We found 1938 genes overlapping CNPs, out of which 721 were highly interconnected. The FI network analysis revealed a total of 174 statistically significant pathways (after coalescing pathways listed multiple times; FDR < 0.05; Supplementary Material, Table S4), which is in contrast to the much smaller group (47) of statistically significant and functionally relevant pathways observed in our spina bifida cohort (Supplementary Material, Table S3). This suggests that CNPs in healthy populations do not selectively target particular functional pathways, while ‘Rare CNVs’ in the spina bifida cohort specifically target pathways involved in neural tube development.
Figure 1.

Network analysis of coding genes overlapping rare CNVs. Module annotations are done with false discovery rate (FDR) < 0.05. Solid line—annotated interaction; dashed line—predicted interaction; arrow end—activating interaction; straight end—inhibiting interaction. Sources for annotations come from: K, KEGG, P, Panther, R, Reactome, N, NCI Pathways, B, BioCarta, C, CellMap.
We then asked how the functional gene pathways associated with recurrent CNPs compared with the spina bifida functional pathways. We defined a CNP to be recurrent if it was observed in >0.5% of the population. FI network analysis on 476 recurrent genes revealed three statistically significant modules consisting of eight pathways which can be categorized into three functionally independent pathways (Supplementary Material, Table S4). While two of the functional pathways, olfactory signaling (module 2) and immune response-related (module 6), may be targeted due to normal genomic variation, Wnt and Cadherin signaling (module 3) overlaps one of the pathways identified in the spina bifida cohort. However, the annotated genes within the Wnt signaling pathway do not overlap between the two datasets, suggesting that a specific subset of Wnt pathway genes may be more tightly associated with the disease process. In summary, the set of genes contained in the CNVs found in patients with NTDs, in comparison to a random set of genes or genes overlapping recurrent copy number polymorphisms, is enriched for those that participate in cellular processes important for neural tube formation. This supports the hypothesis that the rare CNVs we have identified in the disease cohorts are pathogenic in at least a subset of patients.
De novo variations
To test the hypothesis that a proportion of the rare CNVs in the cases are de novo germline variations, we obtained parental DNA from 42 cases and performed CGH analysis. We identified eight rare CNVs that were present in a case but not in either parent (i.e. de novo; Table 1). Out of the eight, three completely encompass at least one gene in the region. One heterozygous deletion removed 27 genes, including PAX3, a gene known to be in the deleted region in Wardenburg syndrome type 1 patients with spina bifida (although the patient in this case did not have other features of Wardenburg syndrome type 1) (88). Additionally, mouse model evidence implicates Pax3 in NTDs (89). A second CNV altered genes whose function or expression is mostly unknown (RGPD8, ZC3H6). These genes were not pursued further in the present study. A third CNV, a heterozygous deletion, removed GPC5 and two exons of GPC6, both of which encode glypicans (Fig. 2). We identified the specific breakpoints of this deletion by a long-range PCR (chr13: 88,692,539 (build hg18) and chr13: 93,166,512; see Materials and Methods) and found 49 bp of a LINE element of the L1 family inserted between them. Given the multitude of connections between glypicans and neural tube development (see Introduction), we considered GPC5 and GPC6 as potential spina bifida candidate genes.
Table 1.
List of de novo CNVs observed in 42 cases based on trio analysis. Counts of genes that are (a) partially over-lapping and coding, over-lapping and coding, partially over-lapping and non-coding, over-lapping and non-coding genes are listed. Genes whose CDS are within the copy number altered regions are considered potential candidates associated with the disorder and are listed
| Family ID | Genomic location | Cyto-band | Genomic length (kb) | Copy number polarity | Number of coding, overlapping genes | Number of coding, partially overlapping genes | Number of non-coding, overlapping genes | Number of non-coding, partially overlapping genes | Coding genes |
|---|---|---|---|---|---|---|---|---|---|
| SB111 | 2:219889913-225539715 | 2q35-q36.2 | 5649.80 | Del | 27 | 1 | 2 | 0 | TMEM198,RESP18, EPHA4, SGPP2, AP1S3, SPEG, CHPF, FARSB, SLC4A3, GMPPA, OBSL, SERPINE2, MRPL44, KCNE4, STK11IP, DNPEP, ACSL3, DES, FAM124B, PAX3, MOGAT1, ACCN4, CUL3, CCDC140, WDFY, SCG2, INHA, DOCK10 |
| SB2223 | 2:142717141-142875928 | 2q22.2 | 158.79 | Dup | 0 | 0 | 0 | 0 | |
| SB2223 | 7:26162685-26180745 | 7p15.2 | 18.06 | Del | 0 | 1 | 0 | 0 | NFE2L3 |
| SB2223 | 13:88695918-93166192 | 13q31.2-q31.3 | 4470.27 | Del | 1 | 1 | 3 | 0 | GPC5, GPC6 |
| SB147 | 2:112809127-112922053 | 2q13 | 112.93 | Dup | 1 | 1 | 0 | 1 | RGPD8, ZC3H6 |
| SB147 | 14:32461626-32498630 | 14q13.1 | 37.00 | Dup | 0 | 1 | 0 | 0 | NPAS3 |
| SB147 | 19:37026054-37058521 | 19q12 | 32.47 | Dup | 0 | 0 | 0 | 0 | |
| SB287 | 10:128208296-128230163 | 10q26.2 | 21.87 | Del | 0 | 0 | 0 | 0 |
Figure 2.
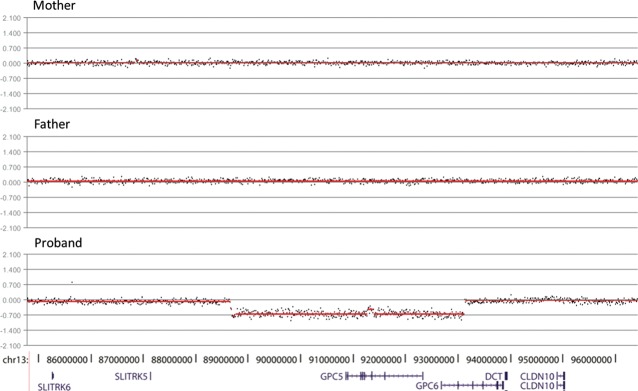
Identification of a de novo heterozygous deletion in a spina bifida proband which removes the polarity genes glypicans GPC5 and GPC6. Segmentation plots of aCGH data generated by Roche NimbleGen software of mother (top), father (middle) and proband (bottom). Genes within this region are indicated below the segmentation plots. Note that the entire GPC5 gene is removed by the deletion, whereas only the first two exons of GPC6 are removed.
Glypican 5 is required for neural tube closure in Xenopus embryos
To test whether GPC5 or GPC6 were reasonable spina bifida candidates, we examined their expression in the frog, Xenopus tropicalis. Using an organism resource database (www.xenbase.org) we identified a full-length expressed sequence tag (EST) for a X. tropicalis glypican 5-like gene. This transcript encodes a predicted protein with 40% identity/60% similarity to human GPC5 at the amino acid level. For gpc6, there is no available EST sequence data, although there is a predicted transcript present in the X. tropicalis genome. This putative transcript encodes a predicted protein with 87% similarity at the amino acid level. As shown in Figure 2, GPC5, GPC6 and DCT (dopachrome tautomerase) are sequentially organized on human chromosome 13. In the X. tropicalis genome, gpc5 is located on a separate genomic scaffold from the gpc6/dct pair. Together with analysis of fish (discussed below) and chick genomes, these observations reveal that there have been genomic rearrangements in this region in the course of evolution.
Using in situ hybridization on X. tropicalis embryos, we detected gpc5 expression diffusely throughout the neural plate during early neural-fold stage embryos (stage 15/16; Supplementary Material, Fig. S4). In mid-late neural-fold stage embryos, expression became enriched in the floorplate of the spinal cord, and by the early tailbud stage (∼stage 30), expression was prominent in the floorplate, and evident in the somites and optic vesicles (Supplementary Material, Fig. S4). Expression was not detectable by in situ hybridization during the cleavage or gastrula stages. These data are consistent with potential functions for Gpc5 during neurulation in this organism. In contrast, our efforts to detect expression of gpc6 at embryonic stages were unsuccessful, consistent with the lack of EST evidence for expression of this gene. Therefore, we pursued functional studies of gpc5. We designed antisense morpholino oligonucleotides (MOs) complementary to the gpc5 start codon to inhibit translation of this mRNA. We injected gpc5-MO, or a negative control MO, into fertilized X. tropicalis eggs and analyzed the resulting embryos for NTDs. Embryos injected with a 10-ng gpc5-MO developed normally through cleavage and gastrulation with no overt defects in early morphogenetic movements. However, more than half of these embryos went on to show neural tube closure defects (Fig. 3 and Table 2). Phenotypic inspection of gross morphology showed an open neural tube (spina bifida) particularly in the posterior neural tube. This effect was substantiated by in situ hybridization against pax6, to delineate the dorsal margin of the neural tube. Affected embryos showed gaps in the fusion of the dorsal neural tube margins along the midline, particularly in the posterior neural tube (Fig. 3F and G). These data support a role of Gpc5 in tetrapod neurulation.
Figure 3.

gpc5 morpholinos inhibit neural tube closure in X. tropicalis embryos. (A and B) Tailbud stage X. tropicalis embryos injected with gpc5-MOs (B) or left uninjected (A). (C and D) Stage 22/23 embryos injected with control morpholino (co-MO; C) or 10 ng gpc5-MO (D). Arrow in (D) indicates open neural tube. (E–G) Embryos injected with a co-MO (E) or gpc5-MO (F and G) were stained for pax6 by in situ hybridization. Arrow in (G) indicates open neural tube. Dorsal views, anterior is toward the top in (C, D) and toward the left in (A, B; E–G).
Table 2.
Summary of NTDs in gpc5-MO-injected X. tropicalis embryos
| Normal (%) | NTDs (%) | Other/blastopore closure defects (%) | |
|---|---|---|---|
| Uninjected/control MO (20 ng) | 47/47 (96) | 0/27 (0) | 0/27 (0) |
| gpc5-MO (10 ng + 10 ng co-MO)** | 22/69 (32) | 44/69 (64) | 3/69 (4) |
Summary of three experiments (**P < 0.0001, chi-square)
Glypican 5 is required for morphogenesis of the trunk in zebrafish embryos
To further test whether mutation in GPC5 or GPC6 might cause spina bifida, we examined expression of their orthologs in zebrafish (Danio rerio) embryos. In contrast to neurulation of the rostral central nervous system (CNS) in mammals and amphibians, in zebrafish the neuroectoderm initially forms a solid neural keel, which subsequently gains a lumen (90). Nonetheless, because morphogenesis of the neural tube in zebrafish embryos results from the folding of a neural epithelium, it is considered primary neurulation (91) rather than secondary neurulation, in which the neural rod derives from a condensation of a mesenchymal mass (92). Searching the zebrafish genome (Ensembl, Zv9), we identified an apparent GPC5 ortholog (i.e., gpc5) on chromosome 1 (see Methods). The predicted protein encoded by this transcript is 40% identical/58% similar to human GPC5. We also detected two orthologs of GPC6, gpc6a and gpc6b, which are 69% and 60% identical to human GPC6, respectively. Similar to the arrangement of GPC5 and GPC6 in the human genome, zebrafish gpc6a is adjacent to gpc5 (on chromosome 1), but in contrast to humans, no ortholog of dct is present nearby. Interestingly, gpc6b is adjacent to dct on chromosome 9, but no ortholog of gpc5 is nearby. We isolated RNA from 24 h post-fertilization (hpf) embryos and amplified pieces of gpc5 and gpc6a. We were unable to amplify gpc6b from complementary DNA (cDNA), similar to our experience with Xenopus gpc6.
We performed in situ hybridization to assess expression of gpc5 and gpc6a. In 12 hpf embryos (late somitogenesis stage) processed with an antisense gpc5 probe signal was detected broadly, including in the brain and spinal cord (Supplementary Material, Fig. S5A). In contrast, embryos hybridized with a sense gpc5 control probe exhibited no signal (Supplementary Material, Fig. S5B). At 36 hpf, expression of gpc5 was clearly present in the floor plate (Supplementary Material, Fig. S5C and D). In contrast, gpc6b expression at 12 hpf was undetected, and at 24 hpf was diffuse and barely detectable by in situ hybridization, even after extended developing (not shown). Based on the pattern of mRNA expression, Gpc5 was considered a good candidate to participate in zebrafish neurulation.
To inhibit gpc5 expression, we injected zebrafish embryos with an antisense MO that overlaps the junction of intron 5 and exon 6 in gpc5 mRNA (hereafter, gpc5 MO). We confirmed by RT–PCR that the levels of correctly spliced gpc5 are diminished in such embryos at 24 hpf (Supplementary Material, Fig. S6). In gpc5 MO-injected embryos at 30 hpf, we observed morphological phenotypes. Whereas in uninjected or control MO-injected embryos, the notochord was readily visible (Supplementary Material, Fig. S7A), in strongly affected gpc5 MO-injected embryos, the somite boundaries were indistinct and, remarkably, the notochord was not readily visible, especially in the tail (Supplementary Material, Fig. S7C and F). In less strongly affected embryos, the notochord appeared diminished in size and there was a downward bend at the end of the hindyolk (Supplementary Material, Fig. S7B and E). The fraction of injected embryos in these phenotypic categories correlated with the dose of MO injected (Supplementary Material, Fig. S7G).
To more clearly reveal the effects of gpc5 knockdown on the mesoderm, we fixed MO-injected embryos at 14 hpf (Fig. 4A and B) or 24 hpf (Fig. 4C and D) and processed them to reveal expression of myod, a marker of somatic mesoderm (Fig. 4A–D) or ntl (Fig. 4I and J), a marker of the notochord. In gpc5 MO-injected embryos at 14 hpf, somite boundaries were less distinct than normal; in addition, the length of the axis appeared shortened at this stage (Fig. 4A and B). At 24 hpf, the somite boundaries in gpc5 MO injected are much less distinct than normal, and the length of the embryos was clearly shorter than normal (Fig. 4C and D). Processing embryos to reveal ntl expression confirmed the observation that in strongly affected gpc5 MO-injected embryos, the notochord is absent (Fig. 4I and J) or fragmented (not shown). To highlight the CNS, we processed gpc5 MO-injected embryos to reveal elavl3, a marker of differentiated neurons (93). The CNS of gpc5 MO-injected embryos was clearly abnormally shaped and mispatterned in comparison to control MO-injected embryos (hereafter, controls) (Fig. 4E and F). The sections of these embryos showed widening of the neural tube and loss of neural tube organization following gpc5 knockdown (Fig. 4G and H). To confirm the specificity of this phenotype, we tested a second MO targeting the splice site of exon 1 and intron 1 of gpc5. This MO (gpc5 MO e1i1) also disrupted morphogenesis of somites, notochord and neural tube, although with lower penetrance than the gpc5 MO (Table 3). These phenotypes were not observed in embryos injected with a negative control MO (Table 3). In summary, inhibition of Gpc5 expression is sufficient to perturb morphogenesis of the trunk in zebrafish, resulting in an abnormally shaped neural tube which is significantly widened, reducing the likelihood of forming a properly closed neural tube. Taken together with the frog studies described above, these findings support the hypothesis that decreased expression of GPC5 predisposes human patients to spina bifida.
Figure 4.

gpc5 MO disrupt morphogenesis of the trunk in zebrafish embryos. Abnormal morphogenesis of the neural tube and surrounding tissue in gpc5 MO-injected embryos compared with controls, (A–J). Embryos fixed at 14 hpf (A and B) or 24 hpf (C–J) were stained for myod (A–D), a marker of somatic mesoderm; elavl3 (E–H), a marker of differentiated neurons; or ntl (I,J), a marker of the notochord. In comparison to (E) control MO-injected embryos, the hindbrain is broader and neurons appear disorganized in (F) gpc5 MO-injected embryos. This is highlighted in transverse trunk cross sections of (G) control and (H) gpc5 MO-injected embryos. In (A) control MO-injected embryos, somites exhibit a classic chevron shape, while in (B) gpc5 MO-injected embryos, myod expression is compressed. (I and J) In lateral view, the notohord is clear in (I) control MO-injected embryos, while it is absent in (J) gpc5 MO-injected embryos.
Table 3.
Summary of NTDs in gpc5-MO-injected D. rerio embryos
| Normal (%) | Trunk Defects (%) | Gastrulation failure (%) | |
|---|---|---|---|
| Control MO (2 mg/ml) + p53 (1.5 mg/ml) | 99/104 (95) | 0/104 (0) | 5/104 (5) |
| gpc5-MO (2 mg/ml) + p53 (1.5 mg/ml)* | 131/410 (32) | 243/410 (59) | 36/410 (9) |
| gpc5 MO e1i1 (2 mg/ml) + p53 (1.5 mg/ml)* | 156/258 (61) | 196/258 (37) | 6/258 (2) |
Summary of two to four experiments for each group (*P < 0.0001, chi-square)
DISCUSSION
Here, we have used aCGH analysis to screen 189 patients with spina bifida for potentially etiologic CNVs that are not found in a database of common CNVs (or have minimal overlap with CNVs found there). Our principal findings were 2-fold: first, we found that the list of 2269 genes contained within the boundaries of these CNVs is enriched for those that participate in regulatory pathways involved in neurulation, relative to a random list of genes of this length. This finding supports the notion that the CNVs contribute to the etiology in at least a subset of spina bifida patients. Second, we identified eight CNVs that are present in patients and not parents; such ‘de novo’ mutations are particularly likely to be involved in disease pathogenesis (94) and are thus worthy of special consideration. Of the eight de novo variants we discovered, only five altered gene exons (see Table 1). As discussed below, one of these removed a known spina bifida gene (PAX3), three contained genes with no known role in neural tube formation, and the final one contained two genes that seemed good candidates to contribute to this process (GPC5 and GPC6). We subjected these genes to functional tests in vertebrate model systems and found evidence that GPC5 is important for neural tube formation. Together this work has served both to identify a new spina bifida candidate gene and to provide a pipeline for future CNV analyses where confirmation by replication is not readily feasible due to small cohort sizes or the rarity of the etiologic chromosomal aberration.
Network-based pathway analysis suggests CNVs contribute to spina bifida
Our pathway annotation analysis of genes found within the CNV boundaries in the spina bifida patients revealed enrichment for key biological functional groups. Interestingly, six out of the eight high-confidence functional modules (represented by ≥15 genes) identified in our network analysis contain pathways that have been hypothesized or proven to be essential for neural tube closure (95) (for example, cell cycle, extra cellular matrix–receptor interaction, Wnt and Hedgehog signaling, calcium signaling pathway and glypican pathway). The two remaining functional modules were (1) signaling via Rho GTPases and (2) transcription, spliceosome, and formation and maturation of mRNA transcript. Although not explicitly hypothesized to be involved in neural tube closure, the first of these pathways is nonetheless represented in the NTD literature since the mice mutant for p190 RhoGAP, a Rho GTPase-activating protein, present with NTDs (96). The second of these pathways is generic for transcriptional processes and thus cannot persuasively be ruled in or out for its involvement in neural tube closure. Although further studies will be required to explain the contributions of the genes in these pathways to the development of this disorder, the results of our pathway analysis support the model that multiple susceptibility genes are functionally convergent and that alteration in copy numbers in any one of them can predispose humans to spina bifida. Importantly, these results underscore the utility of pathway analysis to uncover candidate genes for complex, multigenic disorders.
Analysis of de novo CNVs identifies potential genes that may contribute to spina bifida
We analyzed a subset of spina bifida patients for de novo CNVs, given that this class of CNV is especially likely to etiologic when both parents are unaffected. Of the five we found that disrupted genes (either in their entirety or in part), one affected PAX3 (a gene required for neural tube closure in mice and humans (97)), supporting the validity of our approach. Three of the other de novo CNVs altered genes for which little is known regarding function, or for which evidence suggests that they are less likely to be involved in neural tube development (see Table 1). For example, NFE2L3 is a cap ‘n’ collar transcription factor that, when knocked out in the mouse, does not display any obvious phenotypes (98). Furthermore, two genes (RGPD8 and ZC3H6) have protein domains with predicted functions (Golgi-targeted Ran GTPase-binding protein and zinc finger transcription factor, respectively) but no reported function in development. Additionally, genomic alterations in the NPAS3 gene have been associated with schizophrenia and mental retardation, but no NTDs were reported in these patients (99). In summary, we identified several de novo CNVs in patients with spina bifida that contain genes not previously implicated in neural tube closure. Whether these genes play a role in this process awaits further study.
Knockdown of gpc5 in frog and fish support the model that GPC5 is a spina bifida gene
The remaining de novo CNV directly altering a coding gene deleted GPC5 and part of GPC6 (Fig. 2), both of which encode glypicans. Substantial evidence implicates glypicans in neural tube development. First, in the mouse embryo, Gpc5 is prominently expressed in the neural tube (as well as in limbs and kidney) during embryogenesis (72). Second, the activity of morphogens implicated in NTDs (such as SHH and BMPs) is known to be modulated by glypicans (67,68,100), which are localized to the outer surface of the plasma membrane of cells and are thought to work by stabilizing the interaction of morphogens with their receptors (67). Third, glypican 4 morpholino knockdowns in frogs resulted in NTDs (65,66), and a zebrafish gpc4 mutant exhibits defects in convergence and extension consistent with disruption of the PCP pathway (64). Fourth, glypicans are classified as cell polarity genes, and several genes in this class have already been implicated in NTDs and neural tube closure (including Vangl2, Celsr1, Dishevelled, and Prickle1 and 2; see Introduction for references). These published findings, coupled with the identification of a de novo deletion in a spina bifida case that altered two glypican genes, prompted us to investigate the expression and role in neurulation of gpc5 and gpc6 orthologs in two vertebrate models, X. tropicalis and D. rerio. We did not detect expression of gpc6 orthologs in either species, suggesting that this gene does not play an obvious role in neurulation. On the other hand, we discovered that at neurulation stages in embryos of both species, gpc5 is expressed broadly and with clear expression in the presumptive neural ectoderm (where it is maintained at later stages, mainly in the ventral spinal cord). There were differences in the phenotype of embryos of the two species after gpc5 knockdown; for instance, a strong effect on notochord formation was only observed in zebrafish. Nonetheless, in both species morphogenesis of the neural tube was clearly abnormal upon depletion of gpc5 expression. The mechanistic basis of the requirement for GPC5 in spinal cord morphogenesis awaits further investigation; however, it is intriguing that GPC5 is specifically required for the binding of Shh (an important morphogen in the developing CNS) to its receptor Ptc1 (100). In summary, identification of a de novo deletion in a spina bifida patient that affects GPC5, as well as functional validation in two vertebrate model systems, supports the hypothesis that mutation of GPC5 predisposes humans to spina bifida. Intriguingly, our FI network analysis of genes altered by rare CNVs (Fig. 1) identified multiple statistically enriched pathways which are directly linked to glypicans (Wnt signaling, glypican pathway, Hedgehog signaling, TGF-beta receptor signaling), further underscoring the connection between GPC5 and spina bifida.
Final confirmation that GPC5 contributes to the pathogenesis of spina bifida awaits the identification of additional GPC5 mutations in spina bifida patients. In the cohort of 189 such patients, we identified only a single patient with a GPC5 deletion. This observation, combined with the dramatic effect of gpc5 knockdown in frogs and fish, suggests that GPC5 mutations are rare but of high impact, the class of variant that may help explain some of the missing heritability in polygenic complex disorders (101,102). However, it should be noted that GPC5 mutations do not necessarily lead to spina bifida; for example, a recent report identified two Feingold syndrome patients with heterozygous deletions of one or more exons of GPC5 which did not present with NTDs (103). In these cases, deletion of a microRNA cluster (miR-17–92) immediately upstream of GPC5 was shown to be the likely cause of the syndrome (which presents with microcephaly, short stature and digital anomalies (103)). Conversely, the patient with the GPC5 deletion reported here had no apparent Feingold syndrome phenotypes, even though this deletion also removes the miR-17–92 cluster. These observations underscore the significant role played by the genetic background in promoting the disease state in these cases.
MATERIALS AND METHODS
Clinical cohort
We used three independent non-syndromic myelomeningocele spina bifida cohorts assembled in three different centers. DNA samples from spina bifida patients and parents were collected following Internal Review Board approved protocols from the Northwestern University (Dr Bassuk), Washington University St Louis (Dr Gurnett) and the University of Texas Health Sciences Center at Houston (UTHSC; Dr Northrup) and were isolated using standard commercially available genomic DNA purification kits. One hundred and twenty-eight patients were Caucasians from the Northwestern University, Washington University and UTHSC, while 61 were Hispanics from the UTHSC (104). All patients were examined by a neurosurgeon, and a neurologist or geneticist, and all patients had evidence of non-syndromic lumbar–sacral myelomeningocele. All patient samples were obtained with informed consents following the local Internal Review Board criteria and the Declaration of Helsinki criteria. All samples were de-identified as per these criteria.
Comparative genomic hybridization
Copy number profiles were generated in all affected cases and, when available, unaffected parents, using an aCGH technology from Roche NimbleGen following the protocol recommended by the manufacturer. The array used is the 2.1 million feature whole-genome human tiling array with a median probe spacing of 1169 bp (catalog number 05541921001, Human CGH 2.1M Whole-Genome Tiling v2.0D Array). In brief, the sample cohorts were labeled with Cy3 and the reference genome was labeled with Cy5. All samples were hybridized with a population-matched male reference genome. Additional information can be accessed in the Supplementary Material section.
Array reuse procedure
We employed an array reuse procedure for this study, which we developed in-house. In short, we used a glycerol–salt denaturation buffer to strip arrays of labeled, hybridized DNA fragments (105), followed by hybridization with another sample; up to five uses were employed per array. A detailed protocol of the reuse procedure is provided in Supplementary Material, Figure S8. In order to qualitatively assess the data quality and reproducibility from the reuse procedure, we performed aCGH experiments using the same pair of ‘Reuse Test’ and ‘Reuse Reference’ human genomic DNA on seven different arrays for uses 1, 3 and 5, and various experimental and reference samples from other studies for uses 2 and 4; these intermediate hybridizations were performed to eliminate the possibility that the same hybridization patterns generated by the Reuse Test and Reference samples were carried over from one use to another. To estimate the statistical significance of repeatability, we compared CNV calls for array uses 1, 3 and 5. Microarray hybridization data underwent a series of steps including ‘normalization’ and ‘segmentation’ to detect break points of copy number changes. The third use of array number 7 was not included due to a mixer leak during hybridization. The details of the CNV detection algorithm are described in the following section.
CNVs from 20 experiments were clustered using single linkage hierarchical clustering. Because of the greedy nature of this algorithm, the Jaccard index is used to separate loosely linked nodes of large clusters into tightly linked sub-clusters (106). The Jaccard index is a measure of the similarity of two object sets, in this case the observed number of shared branches between two nodes of a cluster in relation to the number of possible shared branches. All intact clusters of CNVs are thus obligated to show a minimum degree of similarity among all members. We identified 250 gain and 148 deletion clusters. Repeatability measures for all array hybridizations (whether comparisons are made for the same or different array usage) are then estimated based on 398 CNV events using a three-way ANOVA statistical test for three independent variables, namely ‘usage’, ‘arrays’ and ‘copy number type’. The CNV type was included as an independent variable in order to separate the effect due to gains and losses. Each event is weighted as inversely proportional to its genomic length. The results of the analysis, shown in Supplementary Material, Table S5, yielded statistically insignificant P-values of 0.82, 0.99 and 0.99 for both the main and interaction effects. The results suggest a very high reproducibility of detecting CN events across arrays of the same use or across arrays of different uses. To reconfirm, we further calculated Spearman's rank correlation coefficient, ρ, for all possible pair-wise combinations which is shown in Supplementary Material, Figure S9. Eighteen out of 20 experiments gave a Spearman rho of >0.95, strongly suggesting a high correlation amongst all datasets. One array from reuse 2 and one array from reuse 3 produced a rho of 0.92. This is due to a moderate reduction in signal to noise as observed by an increase in median absolute deviation by 15% compared with the rest of the data. In summary, we conclude that, using our reuse strategy, Roche NimbleGen microarrays can generate qualitatively similar data across at least five array uses.
Data analysis of spina bifida hybridizations
Microarray hybridization data underwent two major steps before it was interpreted as demonstrating copy number changes (Supplementary Material, Fig. S1): (a) quality control and (b) normalization. We have employed quality measures at various stages of the process to achieve uniform data quality. We extracted 40 different parameters associated with every hybridization in order to identify any scores beyond acceptable thresholds. Additionally, all scores are stored in a database to facilitate a comprehensive review and analysis of trends and batch effects. These parameters include measure of DNA quality, various signal and noise measures, estimation of statistical parameters on the copy neutral state using an expectation–maximization algorithm, which is further used to define ‘Copy Number Altered’ state. The results obtained with poor quality DNA manifest themselves as a trend (low frequency components) in the ratio of intensities in the two channels, which can result in a low-amplitude false signal. We have developed a mathematical algorithm to detect the trends based on the integrated power of the Fourier-transformed signal. The experiments with a trend measure higher than a threshold value were repeated. Quality-controlled data are subjected to normalization procedures to adjust for uneven hybridization, unbalanced fluorescent signals in two channels and other systemic variations with no biological relevance. We employed two normalization procedures: local and LOWESS normalization. Local normalization corrects for nonuniform intensities across the array due to experimental procedures and LOWESS normalization adjusts for the expectation that the genome is diploid.
The normalized ratios of intensities (referred to as LRR) are readily modeled as segmental deletions and duplications or amplifications. The method utilizes minimization of variance with a Kolmogorov–Smirnov (KS) test to determine significance; segments are defined as non-overlapping, genomic regions where the copy number has changed from the baseline. The algorithm consists of two components. The first part is based on the minimization of variance. Initially, we search for breakpoints that may be boundaries of segments. The profile is divided into 100 probe-long windows. Within a window, the first local minimum of variance is identified and accepted as a new breakpoint. Whenever a new breakpoint is found, the algorithm recursively breaks the said region until no breakpoints can be found therein. If no breakpoints are found, the window is shifted by half its size, and the recursive procedure continues until the end of the chromosome is reached. Once the initial breakpoints are identified, each breakpoint is validated for statistical significance by comparing the distributions of segments on either side of the breakpoint using the KS null hypothesis test. If the P-value is <10−5, then the breakpoint is accepted. If not, the segments are merged. For each hybridization, the LRR is modeled as three modes corresponding to ‘No-Copy change’, ‘Gain’ or ‘Loss’ and estimates the modal parameters using an expectation–maximization algorithm. A CNV segment is then defined to be a ‘Duplication’ if LRR > (mu + sigma) of two-copy state and LRR ≥ 0.32 and as ‘Deletion’ if LRR < (mu − sigma) of two-copy state) and LRR ≤ −0.6. For each segment of gain (or loss), both boundaries are further tested for statistical significance using the KS null hypothesis test and then an estimate of the likelihood that a segment of gain (loss) deviates from a two-copy state is calculated.
Sequencing of glypican deletion breakpoints
To sequence the breakpoints of the deletion, GFF segmentation files were generated for data from sample SB2223 using the default CGH settings and then loaded into SignalMap v1.9 (Roche NimbleGen). The breakpoints of the deletion were then estimated based on the 5′-most and 3′-most positions of the array probes that generated a deletion shift value. Primers (obtained from Integrated DNA Technologies) were designed (using Primer3; http://frodo.wi.mit.edu/) 500, 1000, 1500 and 2000 bp outside the estimated breakpoints of the deletion (CACCAGTAACAAGTTTAAGCCTCT, GTCAAAAATTCCAAGACACTGAATAAT, CAAGAGCAGTAGCAAGGATTTACC, TCTAAGGGCTAATACATACACTTCGTT, ATCTCATGTGTCCTGTTTTTATACCA, TAGCAATATCAGCTCTACCTACAACA, AAAAGACAGTAGCTTAGAACAAGATTCC, ATTTACAATGGGTAGAGGTTGCAG). A 10 µl PCR was performed using DNA from sample 22.23 and the Takara LA Taq kit (0.1 µl LA Taq, 2.5 µl LA PCR buffer, 1.6 µl dNTPs, 5.9 µl PCR grade water) at 98°C for 10 s, 66°C for 15 min and 72°C for 10 min for 30 cycles. PCR products were run on a 1% gel at 100 V for 90 min. Primers 500 bp outside the estimated location of the deletion yielded a 6 kb fragment. Additional primers were designed (Primer3) to move closer to the breakpoints (CACCAGTAACAAGTTTAAGCCTCTG, ATTATCTCTCGATACCACAGACACA, TTCTAACAATTCATCCCTTCTCTTAGT, GCAATTTACTCAAGGACACACAAC, TTAACTGCACAAACTGTAGAGCAT, CATTTTTCTTAGCATCACATAGCACT, CTCCCAGGCTTAGGAAAAGGA, CCAAACCAAGAAGAAGTCAAATCC, AATATCTTCAAATCTCCCCTTAAAT, AGAGAAATAAATAAAGCATGTTCAAA, AAACAGCCCGTATAGCCAAAACAG, ATCATGGGCAGATTTACTGCTTGCT). A 10 µl PCR was performed using DNA from the 6 kb PCR product and the Takara LA Taq kit. PCR products were run on a 1% gel at 100 V for 90 min. Primers CACCAGTAACAAGTTTAAGCCTCTG and AAACAGCCCGTATAGCCAAAACAG yielded a 3 kb fragment. More primers were designed (Primer 3) and a 10 µl PCR was performed using DNA from the 3 kb PCR fragment and the Takara LA Taq kit. PCR products were run on a 1% gel at 100 V for 75 min. Primers GAAAGGAGGGTAGGAGTAGGTCAG and TTCAAGGACATTCAGGTAGATTTTATT yielded a 1 kb fragment. This fragment was purified using a PCR purification kit (Qiagen), and A tails were added with Taq DNA polymerase (Roche). A TOPO TA cloning kit (Invitrogen) was used to clone and transform the 1 kb DNA fragment into competent E. coli cells. DNA from competent cells was purified using a QIAprep Miniprep kit (Qiagen). DNA was subjected to Sanger sequencing (CCG) using a primer 125 bp from the deletion on the left side (TATAACAAGGGCTATGGGTGTTAGG) and a primer 170 bp from the deletion on the right side (GGCCTACACCTGGTCAGAATTATC). The leftmost breakpoint was determined to occur just after chr13: 88 692 539 (build hg18) to the left of GPC5, and the rightmost breakpoint was determined to occur just before chr13: 93 166 512, removing exons 1 and 2 of GPC6. In between both breakpoints, 49 bp of a LINE element of the L1 family was identified.
Expression and functional analysis of frog gpc5 and gpc6 orthologs
Genomic sequences for X. tropicalis gpc5 (GenBank: XM 002933375) and gpc6 (XM 002939284) were identified, and a gpc5 plasmid was obtained commercially (GenBank: ES678878, Open Biosystems, Huntsville, AL). Morpholino oligo 25 mers were designed against the start codons and obtained from GeneTools (start codons are italics/underlined):
gpc5-MO: 5′-AGCAAGAGAGAGCGGCTCATTTCAC-3′ (nt 176–200)
gpc6-MO: 5′-TAACTGTGATCTCCCTTAAGCACAT-3′ (nt 1–25)
Morpholinos were dissolved in nuclease-free water to a stock concentration of 1 mm and stored at 4°C. Prior to injection, MOs were diluted to 1 µg/µl and centrifuged. A fluorescein-labeled control MO (GeneTools, LLC, Philomath, OR, USA) was added to bring the total MO dose to 20 ng per embryo. Frog husbandry was performed essentially as described (107). X. tropicalis eggs were obtained from females stimulated with human chorionic gonadotropin (200 U per frog) and fertilized using a testis homogenized in L15 medium. Embryos were rinsed in 1/9× Marc's modified Ringers (MMR) (1× MMR: 0.1 m NaCl, 1.8 mm KCl, 2.0 mm CaCl2, 1.0 mm MgCl2, 15.0 mm HEPES, pH 7.6), dejellied in 3% cysteine in 1/9× MMR and rinsed extensively in 1/9× MMR. For injection, embryos at the one- to two-cell stage were placed in 2% Ficoll in 0.3× MMR and injected with oligo. After several hours, the embryos were transferred to 1/9× MMR and then to 1/20× MMR (with 1 µg/ml gentamicin) for overnight culture. Vitelline membranes were removed and the embryos fixed in MEMFA at the neurula stage and processed for in situ hybridization as described (108).
Expression and functional analysis of zebrafish gpc5 and gpc6 orthologs
We searched EST databases for close homologs of human GPC5 and GPC6. We detected what appeared to be two halves of a gpc5 ortholog, one mapped to chromosome 1 (ENSDART00000129557, chr1: 2 809 161–2 821 811) and the other to an unassembled contig (ENSDART00000125684, NA503:37 227–160 006). We isolated mRNAs from wild-type zebrafish embryos at 24 hpf, synthesized first-strand cDNA as described (109) and conducted PCR with primers in these two pieces (f—CGATCCGTGGAATTTTGTTT, r—ACCCAGTTGGCTGACGTTAC) and amplified a band of 1695 bases. This confirms that the two pieces are indeed part of a single transcript, presumably found on chromosome 1. We cloned this PCR product, made a DIG-labeled antisense RNA probe and conducted RNA in situ hybridization using standard methods as described (110).
We detected two apparent orthologs of GPC6 (i.e. gpc6a and gpc6b). Like gpc5, the two halves of gpc6a map to different genomic positions. Interestingly, the first half of gpc6a (ENSDART00000126093) is located on chromosome 1 (chr1: 2641199–2673283), within 200 kb of gpc5. The second half of gpc6a maps to chromosome 12 (ENSDART00000090338, chr12: 50004532–50075404). We designed primers to these two regions (f—CAACTCGCAGTTCACCTTCA, r—CAGTGCAGGGATGCTCAGTA) and amplified from 24 hpf first-strand cDNA a band of 1131 bp, confirming that the two regions contribute to a single transcript. The most likely interpretation of these findings is that gpc6a lies within a region of chromosome 1 that is difficult to assemble, leading to mis-assignment of half of it to chromosome 12. Of note, human GPC5 is also ∼200 kb from GPC6. Thus, allowing for the errors in genome assembly, it appears that gpc6a has shared synteny with human GPC6. The other predicted ortholog of GPC6, gpc6b, maps to chromosome 9 (chr9: 55 348 132–55 406 016), adjacent to dct. Thus, gpc6b has shared synteny with the gpc6 ortholog we examined in X. tropicalis. Similar to Xenopus gpc6, there are no EST data to indicate that gpc6b is expressed. We tested several primer pairs and were not able to amplify it from cDNA prepared from embryos at embryonic stages. This gene was not pursued further.
To test the efficacy of the gpc5 MO (which targets the intron 5/exon 5 boundary, sequence below), we injected embryos at the one-cell stage with ∼10 ng of gpc5 MO. When MO-injected embryos reached 24 hpf, we harvested RNA, and conducted RT–PCR with primers in exons 4 and 7 of gpc5 (gpc5 RT–PCR primers: forward, gaggagcacagcgaggatac; reverse, attaaagcgcaggctgaaaa). RNA loading was assessed comparably by RT–PCR to beta actin (beta actin primer sequences: forward, GAGATGATGCCCCTCGTG; reverse, GCTCAATGGGGTATTTGAGG). This analysis confirmed that in such embryos, the levels of correctly spliced gpc5 mRNA were strongly reduced in comparison to uninjected embryos. The negative control MO, supplied by Gene-Tools (Philomath), has no known target in the zebrafish genome.
To test the specificity of the gpc5 MO, we initially attempted to reverse its effects by co-injecting gpc5 mRNA but were not successful. However, we found that embryos injected with gpc5 mRNA without a gpc5 MO developed with abnormal morphogenesis of the trunk, similar to gpc5 MO-injected embryos. This result suggests that morphogenesis of the trunk is sensitive to overexpression of gpc5, as well as deficiency of expression, although this issue merits further investigation. As an alternate test of specificity, we ordered a second gpc5 MO, gpc5 e1i1 MO (sequence below), and found it to elicit the same phenotypes as gpc5 MO.
gpc5 MO: TGCACCTGCAACACACCAACACACA
gpc5 e1i1 MO: CCAGCACATCACATTCTTACCTGTT
negative control MO: CCTCTTACCTCAGTTACAATTTATA
p53 MO: GCGCCATTGCTTTGCAAGAATTG
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (1R01DE021071-01 to J.R.M., GM083999 to D.W.H.) and the National Science Foundation (grant number IOS-1147221 to R.A.C.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jacek Topczewski for alerting us to the fact that the apparent zebrafish gpc5 orthologs of chromosome 1 and an unassembled contig were potentially part of the same gene. We are grateful to Greg Bonde for excellent technical assistance in the fish experiments. We also thank Irina Kalatskaya for helpful discussions on network analysis.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Copp A.J., Greene N.D., Murdoch J.N. The genetic basis of mammalian neurulation. Nat. Rev. Genet. 2003;4:784–793. doi: 10.1038/nrg1181. doi:10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- 2.Wald N.J., Bower C. Folic acid, pernicious anaemia, and prevention of neural tube defects. Lancet. 1994;343:307. doi: 10.1016/s0140-6736(94)91156-8. doi:10.1016/S0140-6736(94)91156-8. [DOI] [PubMed] [Google Scholar]
- 3.Wald N.J., Law M.R., Jordan R. Folic acid food fortification to prevent neural tube defects. Lancet. 1998;351:834–835. doi: 10.1016/S0140-6736(05)78966-2. doi:10.1016/S0140-6736(05)78966-2. [DOI] [PubMed] [Google Scholar]
- 4.Wald N.J., Law M.R., Morris J.K., Wald D.S. Quantifying the effect of folic acid. Lancet. 2001;358:2069–2073. doi: 10.1016/s0140-6736(01)07104-5. doi:10.1016/S0140-6736(01)07104-5. [DOI] [PubMed] [Google Scholar]
- 5.Harris M.J., Juriloff D.M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. A. 2010;88:653–669. doi: 10.1002/bdra.20676. doi:10.1002/bdra.20676. [DOI] [PubMed] [Google Scholar]
- 6.Harris M.J., Juriloff D.M. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res. A. 2007;79:187–210. doi: 10.1002/bdra.20333. doi:10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- 7.Lakkis M.M., Golden J.A., O'Shea K.S., Epstein J.A. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Dev. Biol. 1999;212:80–92. doi: 10.1006/dbio.1999.9327. doi:10.1006/dbio.1999.9327. [DOI] [PubMed] [Google Scholar]
- 8.Carter C.O., Roberts J.A. The risk of recurrence after two children with central nervous system malformations. Lancet. 1967;1:306–308. doi: 10.1016/s0140-6736(67)91240-8. doi:10.1016/S0140-6736(67)91240-8. [DOI] [PubMed] [Google Scholar]
- 9.Holmes L.B. Current concepts in genetics: Congential malformations. N. Engl. J. Med. 1976;295:204–207. doi: 10.1056/NEJM197607222950406. doi:10.1056/NEJM197607222950406. [DOI] [PubMed] [Google Scholar]
- 10.Holmes L.B., Driscoll S.G., Atkins L. Etiologic heterogeneity of neural tube defects. N. Engl. J. Med. 1976;294:365–369. doi: 10.1056/NEJM197602122940704. doi:10.1056/NEJM197602122940704. [DOI] [PubMed] [Google Scholar]
- 11.Lynch S.A. Non-multifactorial neural tube defects. Am. J. Med. Genet. C. 2005;135C:69–76. doi: 10.1002/ajmg.c.30055. doi:10.1002/ajmg.c.30055. [DOI] [PubMed] [Google Scholar]
- 12.Manning S.M., Jennings R., Madsen J.R. Pathophysiology, prevention, and potential treatment of neural tube defects. Ment. Retard. Dev. Disabil. Res. Rev. 2000;6:6–14. doi: 10.1002/(SICI)1098-2779(2000)6:1<6::AID-MRDD2>3.0.CO;2-B. doi:10.1002/(SICI)1098-2779(2000)6:1<6::AID-MRDD2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 13.Helali N., Lafolla A.K., Kahler S.G., Qumsiyeh M.B. A case of duplication of 13q32–> qter and deletion of 18p11.32 –> pter with mild phenotype: Patau syndrom and duplication of 13q revisited. J. Med. Genet. 1996;33:600–602. doi: 10.1136/jmg.33.7.600. doi:10.1136/jmg.33.7.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman T.E., Siegel M.J. Special imaging casebook. Trisomy 13 and occult dysraphism with tethered cord. J. Perinatol. 1997;17:172–174. [PubMed] [Google Scholar]
- 15.Kjaer I., Keeling J.W., Smith N.M., Hansen B.F. Pattern of malformations in the axial skeleton in human triploid fetuses. Am. J. Med. Genet. 1997;72:216–221. doi: 10.1002/(sici)1096-8628(19971017)72:2<216::aid-ajmg17>3.0.co;2-q. doi:10.1002/(SICI)1096-8628(19971017)72:2<216::AID-AJMG17>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 16.Nolting D., Hansen B.F., Keeling J.W., Reintoft I., Kjaer I. Prenatal malformed lumbar vertebral corpora in trisomies 21, 18, and 13, evaluated radiographically and histologically. Acta Path. Micro. Im. 2000;108:422–428. doi: 10.1034/j.1600-0463.2000.d01-78.x. [DOI] [PubMed] [Google Scholar]
- 17.Budorick N.E., Pretorius D.H., McGahan J.P., Grafe M.R., James H.E., Slivka J. Cephaloceledection in utero: sonographic and clinical features. Ultrasound Obst. Gyn. 1995;5:77–85. doi: 10.1046/j.1469-0705.1995.05020077.x. doi:10.1046/j.1469-0705.1995.05020077.x. [DOI] [PubMed] [Google Scholar]
- 18.O'Reilly G.C., Shields L.E. Karyotyping for isolated neural tube defects: a report of two cases. J. Reprod. Med. 2000;45:950–952. [PubMed] [Google Scholar]
- 19.Shields L.E., Carpenter L.A., Smith K.M., Nghiem H.V. Ultrasonographic diagnosis of trisomy 18: is it practical in the early second trimester? J. Ultras. Med. 1998;17:327–331. doi: 10.7863/jum.1998.17.5.327. [DOI] [PubMed] [Google Scholar]
- 20.Bassuk A.G., Craig D., Jalali A., Mukhopadhyay A., Kim F., Charrow J., Gulbu U., Epstein L.G., Bowman R., McLone D., et al. The genetics of tethered cord syndrome. Am. J. Med. Genet. A. 2005;132:450–453. doi: 10.1002/ajmg.a.30439. [DOI] [PubMed] [Google Scholar]
- 21.Nickel R.E., Magenis R.E. Neural tube defects and deletions of 22q11. Am. J. Med. Genet. 1996;66:25–27. doi: 10.1002/(SICI)1096-8628(19961202)66:1<25::AID-AJMG6>3.0.CO;2-V. doi:10.1002/(SICI)1096-8628(19961202)66:1<25::AID-AJMG6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 22.Nickel R.E., Pillers D.A., Merkens M., Magenis R.E., Driscoll D.A., Emanual B.S., Zonana J. Velocardio-facial and DiGeorge syndromes with meningomyelocele and deletions of the 22Q11 region. Eur. J. Pediatr. Surg. 1993;3(Suppl 1):27–28. [PubMed] [Google Scholar]
- 23.Luo J., Balkin N., Stewart J.F., Sarwark J.F., Charrow J., Nye J.S. Neural tube defects and the 13q deletion syndrome: evidence for a critical region in 13q33–34. Am. J. Med. Genet. 2000;91:227–230. doi: 10.1002/(sici)1096-8628(20000320)91:3<227::aid-ajmg14>3.0.co;2-i. doi:10.1002/(SICI)1096-8628(20000320)91:3<227::AID-AJMG14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 24.Beltran-Valero de Bernabe D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W.B., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am. J. Hum. Genet. 2002;71:1033–1043. doi: 10.1086/342975. doi:10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen M.M., Knobloch W.H., Gorlin R.J. A dominantly inherited syndrome of hyaloidopathy, rentinal degeneration, cleft palate and maxillary hypoplasia (Cervenka syndrome) Birth Defects. 1971;7:83–86. [PubMed] [Google Scholar]
- 26.Cook G.R., Knobloch W.H. Autosomal recessive vitreoretinopathy and encephaloceles. Am. J. Ophthalmol. 1982;94:18–25. doi: 10.1016/0002-9394(82)90185-4. [DOI] [PubMed] [Google Scholar]
- 27.Hol F.A., Hamel B.C., Geurds M.P., Mullaart R.A., Barr F.G., Macina R.A., Mariman E.C. A frameshift mutation in the gene for PAX3 in a girl with spina bifida and mild signs of Waardenburg syndrome. J. Med. Genet. 1995;32:52–56. doi: 10.1136/jmg.32.1.52. doi:10.1136/jmg.32.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kliemann S.E., Waetge R.T., Suzuki O.T., Passos-Bueno M.R., Rosemberg S. Evidence of neuronal migration disorders in Knobloch syndrome: clinical and molecular analysis of two novel families. Am. J. Med. Genet. A. 2003;119A:15–19. doi: 10.1002/ajmg.a.20070. doi:10.1002/ajmg.a.20070. [DOI] [PubMed] [Google Scholar]
- 29.Knobloch W.H., Layer J.M. Clefting syndromes associated with retinal detachment. Am. J. Ophthalmol. 1972;73:517–530. doi: 10.1016/0002-9394(72)90003-7. [DOI] [PubMed] [Google Scholar]
- 30.Menzel O., Bekkeheien R.C., Reymond A., Fukai N., Boye E., Kosztolanyi G., Aftimos S., Deutsch S., Scott H.S., Olsen B.R., et al. Knobloch syndrome: novel mutations in COL18A1, evidence for genetic heterogeneity, and functionally impaired polymorphism in endostatin. Hum. Mutat. 2004;23:77–84. doi: 10.1002/humu.10284. doi:10.1002/humu.10284. [DOI] [PubMed] [Google Scholar]
- 31.Mintz-Hittner H.A., Semina E.V., Frishman L.J., Prager T.C., Murray J.C. VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology. 2004;111:828–836. doi: 10.1016/j.ophtha.2003.07.006. doi:10.1016/j.ophtha.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Oh S.P., Warman M.L., Seldin M.F., Cheng S.D., Knoll J.H., Timmons S., Olsen B.R. Cloning of cDNA and genomic DNA encoding human type XVIII collagen and localization of the alpha 1(XVIII) collagen gene to mouse chromosome 10 and human chromosome 21. Genomics. 1994;19:494–499. doi: 10.1006/geno.1994.1098. doi:10.1006/geno.1994.1098. [DOI] [PubMed] [Google Scholar]
- 33.Passos-Bueno M.R., Marie S.K., Monteiro M., Neustein I., Whittle M.R., Vainzof M., Zatz M. Knobloch syndrome in a large Brazilian consanguineous family: confirmation of autosomal recessive inheritance. Am. J. Med. Genet. 1994;52:170–173. doi: 10.1002/ajmg.1320520209. doi:10.1002/ajmg.1320520209. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki O.T., Sertie A.L., Der Kaloustian V.M., Kok F., Carpenter M., Murray J.C., Czeizel A.E., Kliemann S.E., Rosemberg S., Monteiro M., et al. Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am. J. Hum. Genet. 2002;71:1320–1329. doi: 10.1086/344695. doi:10.1086/344695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Esch H., Poirier K., de Zegher F., Holvoet M., Bienvenu T., Chelley J., Devriendt K., Fryns J.P. ARX mutation in a boy with transsphenoidal encephalocele and hypopituitarism. Clin. Genet. 2004;65:503–505. doi: 10.1111/j.1399-0004.2004.00256.x. doi:10.1111/j.1399-0004.2004.00256.x. [DOI] [PubMed] [Google Scholar]
- 36.Ylikarppa R., Eklund L., Sormunen R., Kontiola A.I., Utriainen A., Maatta M., Fukai N., Olsen B.R., Pihlajaniemi T. Lack of type XVIII collagen results in anterior ocular defects. FASEB J. 2003;17:2257–2259. doi: 10.1096/fj.02-1001fje. [DOI] [PubMed] [Google Scholar]
- 37.Volcik K.A., Zhu H., Finnell R.H., Shaw G.M., Canfield M., Lammer E.J. Evaluation of the jumonji gene and risk for spina bifida and congenital heart defects. Am. J. Med. Genet. A. 2004;126A:215–217. doi: 10.1002/ajmg.a.20574. doi:10.1002/ajmg.a.20574. [DOI] [PubMed] [Google Scholar]
- 38.Volcik K.A., Zhu H., Shaw G.M., Lammer E.J., Finnell R.H. Apolipoprotein E and apolipoprotein B genotypes and risk of spina bifida. Teratology. 2002;66:257–259. doi: 10.1002/tera.10097. doi:10.1002/tera.10097. [DOI] [PubMed] [Google Scholar]
- 39.Felder B., Stegmann K., Schultealbert A., Geller F., Strehl E., Ermert A., Koch M.C. Evaluation of BMP4 and its specific inhibitor NOG as candidates in human neural tube defects (NTDs) Eur. J. Hum. Genet. 2002;10:753–756. doi: 10.1038/sj.ejhg.5200875. doi:10.1038/sj.ejhg.5200875. [DOI] [PubMed] [Google Scholar]
- 40.Volcik K.A., Zhu H., Finnell R.H., Shaw G.M., Canfield M., Lammer E.J. Evaluation of the cited2 gene and risk for spina bifida and congenital heart defects. Am. J. Med. Genet. A. 2004;126A:324–325. doi: 10.1002/ajmg.a.20578. doi:10.1002/ajmg.a.20578. [DOI] [PubMed] [Google Scholar]
- 41.Stegmann K., Boecker J., Richter V., Capra V., Finnell R.H., Ngo E.T., Strehl E., Ermert A., Koch M.C. A screen for mutations in human homologues of mice exencephaly genes Tfap2alpha and Msx2 in patients with neural tube defects. Teratology. 2001;63:167–117. doi: 10.1002/tera.1031. doi:10.1002/tera.1031. [DOI] [PubMed] [Google Scholar]
- 42.Volcik K.A., Blanton S.H., Kruzel M.C., Townsend I.T., Tyerman G.H., Mier R.J., Northrup H. Testing for genetic associations with the PAX gene family in a spina bifida population. Am. J. Med. Genet. 2002;110:195–202. doi: 10.1002/ajmg.10434. doi:10.1002/ajmg.10434. [DOI] [PubMed] [Google Scholar]
- 43.Bauer K.A., George T.M., Enterline D.S., Stottmann R.W., Melvin E.C., Siegel D., Samal S., Hauser M.A., Klingensmith J., Nye J.S., et al. A novel mutation in the gene encoding noggin is not causative in human neural tube defects. J. Neurogenet. 2002;16:65–71. [PubMed] [Google Scholar]
- 44.Wang Y., Nathans J. Tissue/planar cell polarity in vertebrates: new insights and new questions. Development. 2007;134:647–658. doi: 10.1242/dev.02772. doi:10.1242/dev.02772. [DOI] [PubMed] [Google Scholar]
- 45.Jenny A., Mlodzik M. Planar cell polarity signaling: a common mechanism for cellular polarization. Mt. Sinai J. Med. 2006;73:738–750. [PubMed] [Google Scholar]
- 46.Strutt D. Frizzled signalling and cell polarisation in Drosophila and vertebrates. Development. 2003;130:4501–4513. doi: 10.1242/dev.00695. doi:10.1242/dev.00695. [DOI] [PubMed] [Google Scholar]
- 47.Wallingford J.B. Neural tube closure and neural tube defects: studies in animal models reveal known knowns and known unknowns. Am. J. Med. Genet. C. 2005;135C:59–68. doi: 10.1002/ajmg.c.30054. doi:10.1002/ajmg.c.30054. [DOI] [PubMed] [Google Scholar]
- 48.Barrow J.R. Wnt/PCP signaling: a veritable polar star in establishing patterns of polarity in embryonic tissues. Semin. Cell Dev. Biol. 2006;17:185–193. doi: 10.1016/j.semcdb.2006.04.002. doi:10.1016/j.semcdb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Montcouquiol M., Crenshaw E.B., Kelley M.W. Noncanonical Wnt signaling and neural polarity. Annu. Rev. Neurosci. 2006;29:363–386. doi: 10.1146/annurev.neuro.29.051605.112933. doi:10.1146/annurev.neuro.29.051605.112933. [DOI] [PubMed] [Google Scholar]
- 50.Wallingford J.B., Harland R.M. Neural tube closure requires Dishevelled-dependent convergent extension of the midline. Development. 2002;129:5815–5825. doi: 10.1242/dev.00123. doi:10.1242/dev.00123. [DOI] [PubMed] [Google Scholar]
- 51.Murdoch J.N., Doudney K., Paternotte C., Copp A.J., Stanier P. Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum. Mol. Genet. 2001;10:2593–2601. doi: 10.1093/hmg/10.22.2593. doi:10.1093/hmg/10.22.2593. [DOI] [PubMed] [Google Scholar]
- 52.Kibar Z., Vogan K.J., Groulx N., Justice M.J., Underhill D.A., Gros P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant loop-tail. Nat. Genet. 2001;28:251–255. doi: 10.1038/90081. doi:10.1038/90081. [DOI] [PubMed] [Google Scholar]
- 53.Kibar Z., Gauthier S., Lee S.-H., Vidal S., Gros P. Rescue of the neural tube defect of loop-tail mice by a BAC clone containing the Ltap gene. Genomics. 2003;82:397–400. doi: 10.1016/s0888-7543(03)00113-7. doi:10.1016/S0888-7543(03)00113-7. [DOI] [PubMed] [Google Scholar]
- 54.Curtin J.A., Quint E., Tsipouri V., Arkell R.M., Cattanach B., Copp A.J., Henderson D.J., Spurr N., Stanier P., Fisher E.M., et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003;13:1129–1133. doi: 10.1016/s0960-9822(03)00374-9. doi:10.1016/S0960-9822(03)00374-9. [DOI] [PubMed] [Google Scholar]
- 55.Murdoch J.N., Henderson D.J., Doudney K., Gaston-Massuet C., Phillips H.M., Paternotte C., Arkell R., Stanier P., Copp A.J. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum. Mol. Genet. 2003;12:87–98. doi: 10.1093/hmg/ddg014. doi:10.1093/hmg/ddg014. [DOI] [PubMed] [Google Scholar]
- 56.Hamblet N.S., Lijam N., Ruiz-Lozano P., Wang J., Yang Y., Luo Z., Mei L., Chien K.R., Sussman D.J., Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. doi:10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y., Guo N., Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 2006;26:2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. doi:10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimura T., Honda H., Takeichi M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell. 2012;149:1084–1097. doi: 10.1016/j.cell.2012.04.021. doi:10.1016/j.cell.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 59.Robinson A., Escuin S., Doudney K., Vekemans M., Stevenson R.E., Greene N.D., Copp A.J., Stanier P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012;33:440–447. doi: 10.1002/humu.21662. doi:10.1002/humu.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bosoi C.M., Capra V., Allache R., Trinh V.Q.-H., de Marco P., Merello E., Drapeau P., Bassuk A.G., Kibar Z. Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum. Mutat. 2011;32:1371–1375. doi: 10.1002/humu.21589. doi:10.1002/humu.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen S., Zhu H., Lu W., Mitchell L.E., Shaw G.M., Lammer E.J., Finnell R.H. Planar cell polarity pathway genes and risk for spina bifida. Am. J. Med. Genet. A. 2010;152A:299–304. doi: 10.1002/ajmg.a.33230. doi:10.1002/ajmg.a.33230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu G., Xupei H., Hua Y., Mu D. Roles of planar cell polarity pathways in the development of neutral tube defects. J. Biomed. Sci. 2011;18:66–76. doi: 10.1186/1423-0127-18-66. doi:10.1186/1423-0127-18-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zohn I.E., Chesnutt C.R., Niswander L. Cell polarity pathways converge and extend to regulate neural tube closure. Trends Cell Biol. 2003;13:451–454. doi: 10.1016/s0962-8924(03)00173-9. doi:10.1016/S0962-8924(03)00173-9. [DOI] [PubMed] [Google Scholar]
- 64.Topczewski J., Sepich D.S., Myers D.C., Walker C., Amores A., Lele Z., Hammerschmidt M., Postlethwait J., Solnica-Krezel L. The zebrafish glypican knypek controls cell polarity during gastrulation movements of convergent extension. Dev. Cell. 2001;1:251–264. doi: 10.1016/s1534-5807(01)00005-3. doi:10.1016/S1534-5807(01)00005-3. [DOI] [PubMed] [Google Scholar]
- 65.Galli A., Roure A., Zeller R., Dono R. Glypican 4 modulates FGF signalling and regulates dorsoventral forebrain patterning in Xenopus embryos. Development. 2003;130:4919–4929. doi: 10.1242/dev.00706. doi:10.1242/dev.00706. [DOI] [PubMed] [Google Scholar]
- 66.Ohkawara B., Yamamoto T.S., Tada M., Ueno N. Role of glypican 4 in the regulation of convergent extension movements during gastrulation in Xenopus laevis. Development. 2003;130:2129–2138. doi: 10.1242/dev.00435. doi:10.1242/dev.00435. [DOI] [PubMed] [Google Scholar]
- 67.Filmus J., Capurro M., Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. doi:10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan D., Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb Perspect Biol. 2009;1:a002493. doi: 10.1101/cshperspect.a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juriloff D.M., Harris M.J. Mouse models for neural tube closure defects. Hum. Mol. Genet. 2000;9:993–1000. doi: 10.1093/hmg/9.6.993. doi:10.1093/hmg/9.6.993. [DOI] [PubMed] [Google Scholar]
- 70.Stottmann R.W., Berrong M., Matta K., Choi M., Klingensmith J. The BMP antagonist Noggin promotes cranial and spinal neurulation by distinct mechanisms. Dev. Biol. 2006;295:647–663. doi: 10.1016/j.ydbio.2006.03.051. doi:10.1016/j.ydbio.2006.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ybot-Gonzalez P., Gaston-Massuet C., Girdler G., Klingensmith J., Arkell R.M., Greene N.D.E., Copp A.J. Neural plate morphogenesis during mouse neurulation is regulated by antagonism of Bmp signalling. Development. 2007;134:3203–3211. doi: 10.1242/dev.008177. doi:10.1242/dev.008177. [DOI] [PubMed] [Google Scholar]
- 72.Saunders S., Paine-Saunders S., Lander A.D. Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb and brain. Dev. Biol. 1997;190:78–93. doi: 10.1006/dbio.1997.8690. doi:10.1006/dbio.1997.8690. [DOI] [PubMed] [Google Scholar]
- 73.Chen C.-P., Su Y.-N., Tsai F.-J., Lin M.-H., Wu P.-C., Chern S.-R., Lee C.-C., Pan C.-W., Wang W. Partial monosomy 13q (13q21.32→qter) and partial trisomy 8p (8p12→pter) presenting with anencephaly and increased nuchal translucency: array comparative genomic hybridization characterization. Taiwan. J. Obstet. Gynecol. 2011;50:205–211. doi: 10.1016/j.tjog.2010.04.001. doi:10.1016/j.tjog.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 74.Ballif B.C., Hornor S.A., Jenkins E., Madan-Khetarpal S., Surti U., Jackson K.E., Asamoah A., Brock P.L., Gowans G.C., Conway R.L., et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. doi:10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 75.Kallioniemi A. CGH microarrays and cancer. Curr. Opin. Biotechnol. 2008;19:36. doi: 10.1016/j.copbio.2007.11.004. doi:10.1016/j.copbio.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 76.Lenz G., Wright G.W., Emre N.C., Kohlhammer H., Dave S.S., Davis R.E., Carty S., Lam L.T., Shaffer A.L., Xiao W., et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl Acad. Sci. USA. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. doi:10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharp A.J., Mefford H.C., Li C., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., de Gregori M., Ciccone R., et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. doi:10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A., et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. doi:10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 79.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P., et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. doi:10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu X.-Y., Phung M.T., Shaw C.A., Pham K., Neil S.E., Patel A., Sahoo T., Bacino C.A., Stankiewicz P., Kang S.-H.L., et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. doi:10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D., et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. doi:10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Epstein L.G., Jalali A., Chary A.N., Khan S., Ross J., Coppinger J., Carlson K., Charrow J., Burton B., Zimmerman D., et al. Neuroimaging findings in children with rare or novel de novo chromosomal anomalies. Birth Defects Res. A. 2008;82:200–210. doi: 10.1002/bdra.20443. doi:10.1002/bdra.20443. [DOI] [PubMed] [Google Scholar]
- 83.Gustavsson P., Schoumans J., Staaf J., Borg Å., Nordenskjöld M., Annerén G. Duplication 16q12.1–q22.1 characterized by array CGH in a girl with spina bifida. Eur. J. Med. Genet. 2007;50:237–241. doi: 10.1016/j.ejmg.2007.01.004. doi:10.1016/j.ejmg.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 84.Harris M.J., Juriloff D.M. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 1999;60:292–305. doi: 10.1002/(SICI)1096-9926(199911)60:5<292::AID-TERA10>3.0.CO;2-6. doi:10.1002/(SICI)1096-9926(199911)60:5<292::AID-TERA10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 85.Burren K.A., Savery D., Massa V., Kok R.M., Scott J.M., Blom H.J., Copp A.J., Greene N.D. Gene-environment interactions in the causation of neural trube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum. Mol. Genet. 2008;17:3675–3685. doi: 10.1093/hmg/ddn262. doi:10.1093/hmg/ddn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nagashima T., Maruyama T., Furuya M., Kajitani T., Uchida H., Masuda H., Ono M., Arase T., Ozato K., Yoshimura Y. Histone acetylation and subcellular localization of chromosomal protein BRD4 during mouse oocyte meiosis and mitosis. Mol. Hum. Reprod. 2007;13:141–148. doi: 10.1093/molehr/gal115. doi:10.1093/molehr/gal115. [DOI] [PubMed] [Google Scholar]
- 87.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M., et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. doi:10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kromberg J.G., Krause A. Waardenburg syndrome and spina bifida. Am. J. Med. Genet. 1993;45:536–537. doi: 10.1002/ajmg.1320450430. doi:10.1002/ajmg.1320450430. [DOI] [PubMed] [Google Scholar]
- 89.Greene N.D.E., Massa V., Copp A.J. Understanding the causes and prevention of neural tube defects: Insights from the splotch mouse model. Birth Defects Res. A. 2009;85:322–330. doi: 10.1002/bdra.20539. doi:10.1002/bdra.20539. [DOI] [PubMed] [Google Scholar]
- 90.Schmitz B., Papan C., Campos-Ortega J.A. Neurulation in the anterior trunk region of the zebrafish Brachydanio rerio. Dev. Genes Evol. 1993;202:250–259. doi: 10.1007/BF00363214. [DOI] [PubMed] [Google Scholar]
- 91.Lowery L.A., Sive H. Strategies of vertebrate neurulation and a re-evaluation of teleost neural tube formation. Mech. Dev. 2004;121:1189–1197. doi: 10.1016/j.mod.2004.04.022. doi:10.1016/j.mod.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 92.Schoenwolf G.C. Histological and ultrastructural studies of secondary neurulation in mouse embryos. Am. J. Anat. 1984;169:361–376. doi: 10.1002/aja.1001690402. doi:10.1002/aja.1001690402. [DOI] [PubMed] [Google Scholar]
- 93.Kudoh T., Tsang M., Hukriede N.A., Chen X., Dedekian M., Clarke C.J., Kiang A., Schultz S., Epstein J.A., Toyama R., et al. A gene expression screen in zebrafish embryogenesis. Genome Res. 2001;11:1979–1987. doi: 10.1101/gr.209601. doi:10.1101/gr.209601. [DOI] [PubMed] [Google Scholar]
- 94.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A., et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. doi:10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Copp A.J., Greene N.D. Genetics and development of neural tube defects. J. Pathol. 2010;220:217–230. doi: 10.1002/path.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brouns M.R., Matheson S.F., Hu K.Q., Delalle I., Caviness V.S., Silver J., Bronson R.T., Settleman J. The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development. 2000;127:4891–4903. doi: 10.1242/dev.127.22.4891. [DOI] [PubMed] [Google Scholar]
- 97.Au K.S., Ashley-Koch A., Northrup H. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev. Disabil. Res. Rev. 2010;16:6–15. doi: 10.1002/ddrr.93. doi:10.1002/ddrr.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nouhi Z., Chevillard G., Derjuga A., Blank V. Endoplasmic reticulum association and N-linked glycosylation of the human Nrf3 transcription factor. FEBS Lett. 2007;581:5401–5406. doi: 10.1016/j.febslet.2007.10.041. doi:10.1016/j.febslet.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 99.Macintyre G., Alford T., Xiong L., Rouleau G.A., Tibbo P.G., Cox D.W. Association of NPAS3 exonic variation with schizophrenia. Schizophr. Res. 2010;120:143–149. doi: 10.1016/j.schres.2010.04.002. doi:10.1016/j.schres.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 100.Li F., Shi W., Capurro M., Filmus J. Glypican-5 stimulates rhabdomyosarcoma cell proliferation by activating Hedgehog signaling. J. Cell Biol. 2011;192:691–704. doi: 10.1083/jcb.201008087. doi:10.1083/jcb.201008087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schork N.J., Murray S.S., Frazer K.A., Topol E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. doi:10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bansal V., Libiger O., Torkamani A., Schork N.J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 2010;11:773–785. doi: 10.1038/nrg2867. doi:10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Pontual L., Yao E., Callier P., Faivre L., Drouin V., Cariou S., Van Haeringen A., Genevieve D., Goldenberg A., Oufadem M., et al. Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat. Genet. 2011;43:1026–1030. doi: 10.1038/ng.915. doi:10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Au K.S., Tran P.X., Tsai C.C., O'Byrne M.R., Lin J.I., Morrison A.C., Hampson A.W., Cirino P., Fletcher J.M., Ostermaier K.K., et al. Characteristics of a spina bifida population including North American Caucasian and Hispanic individuals. Birth Defects Res. A. 2008;82:692–700. doi: 10.1002/bdra.20499. doi:10.1002/bdra.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akat H., Aytac N. Renaturation of DNA (Deoxyribonucleic Acid) in ethylene glycol and glycerol. JFS (E.U.F.F., Turkey) 2006;29:1–15. [Google Scholar]
- 106.Greenacre M. Correspondence Analysis in Practice. Chapman and Hall/CRC Press, London; 2007. [Google Scholar]
- 107.Khokha M.K., Yeh J., Grammer T.C., Harland R.M. Depletion of three BMP antagonists from Spemann's organizer leads to a catastrophic loss of dorsal structures. Dev. Cell. 2005;8:401–411. doi: 10.1016/j.devcel.2005.01.013. doi:10.1016/j.devcel.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 108.Kerr T.C., Cuykendall T.N., Luettjohann L.C., Houston D.W. Maternal Tgif1 regulates nodal gene expression in Xenopus. Dev. Dyn. 2008;237:2862–2873. doi: 10.1002/dvdy.21707. doi:10.1002/dvdy.21707. [DOI] [PubMed] [Google Scholar]
- 109.O'Brien E.K., d'Alençon C., Bonde G., Li W., Schoenebeck J., Allende M.L., Gelb B.D., Yelon D., Eisen J.S., Cornell R.A. Transcription factor Ap-2α is necessary for development of embryonic melanophores, autonomic neurons and pharyngeal skeleton in zebrafish. Dev. Biol. 2004;265:246–261. doi: 10.1016/j.ydbio.2003.09.029. doi:10.1016/j.ydbio.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 110.Li W., Cornell R.A. Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev. Biol. 2007;304:338–354. doi: 10.1016/j.ydbio.2006.12.042. doi:10.1016/j.ydbio.2006.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.