

Abstract
Fragile X premutation-associated disorders, including Fragile X-associated Tremor Ataxia Syndrome, result from unmethylated CGG repeat expansions in the 5′ untranslated region (UTR) of the FMR1 gene. Premutation-sized repeats increase FMR1 transcription but impair rapid translation of the Fragile X mental retardation protein (FMRP), which is absent in Fragile X Syndrome (FXS). Normally, FMRP binds to RNA and regulates metabotropic glutamate receptor (mGluR)-mediated synaptic translation, allowing for dendritic synthesis of several proteins. FMRP itself is also synthesized at synapses in response to mGluR activation. However, the role of activity-dependent translation of FMRP in synaptic plasticity and Fragile X-premutation-associated disorders is unknown. To investigate this question, we utilized a CGG knock-in mouse model of the Fragile X premutation with 120–150 CGG repeats in the mouse Fmr1 5′ UTR. These mice exhibit increased Fmr1 mRNA production but impaired FMRP translational efficiency, leading to a modest reduction in basal FMRP expression. Cultured hippocampal neurons and synaptoneurosomes derived from CGG KI mice demonstrate impaired FMRP translation in response to the group I mGluR agonist 3,5-dihydroxyphenylglycine. Electrophysiological analysis reveals enhanced mGluR-mediated long-term depression (mGluR-LTD) at CA3–CA1 synapses in acute hippocampal slices prepared from CGG KI mice relative to wild-type littermates, similar to Fmr1 knockout mice. However, unlike mGluR-LTD in mice completely lacking FMRP, mGluR-LTD in CGG knock-in mice remains dependent on new protein synthesis. These studies demonstrate partially overlapping synaptic plasticity phenotypes in mouse models of FXS and Fragile X premutation disorders and support a role for activity-dependent synthesis of FMRP in enduring forms of synaptic plasticity.
INTRODUCTION
Fragile X Syndrome (FXS) is the most common known monogenic cause of autism and intellectual disability, affecting upwards of 1 in 4000 boys and 1 in 8000 girls (1,2). FXS results from the expansion of a CGG microsatellite repeat in the 5′ untranslated region (UTR) of the FMR1 gene on the X chromosome. In humans, this sequence is normally <45 CGG repeats. Expansions to >200 repeats trigger hypermethylation of the repeat and FMR1 promoter, resulting in transcriptional silencing of the FMR1 gene and the absence of the Fragile X mental retardation protein (FMRP), (3–7).
FMRP is an RNA-binding protein that regulates activity-dependent translation of associated transcripts at the synapse (8). Mice lacking FMRP (Fmr1 KO mice) exhibit specific defects in synaptic signaling mediated through group I metabotropic glutamate receptors [mGluRs; (9)]. At CA3–CA1 synapses in the hippocampus, mGluR activation normally leads to a long-term depression (LTD) of synaptic efficacy that requires new dendritic protein synthesis (10–13). mGluR agonists trigger rapid FMRP dephosphorylation and degradation, which allows synaptic translation of FMRP-associated transcripts (14–16). In mice lacking FMRP, mGluR-LTD is enhanced and no longer requires new protein synthesis, and mGluR agonists fail to trigger the translation of FMRP target mRNAs (9,13,15,17,18). The absence of FMRP is thought to decouple mGluR 1/5 activity from protein translation, such that basal dendritic translation of these target mRNAs is increased, but mGluR-coupled dendritic translation is lost (19).
One of the dendritically localized transcripts whose translation is regulated by mGluR signaling is FMRP itself (16,17,20,21). Although the function of this newly synthesized FMRP is unknown, it has been proposed to act as a brake on local protein production, hence constraining LTD by limiting the new translation of LTD effector proteins (19,22). A critical prediction of this model is that the magnitude of LTD should be enhanced by diminished mGluR-dependent translation of FMRP. Despite its appeal, and its consistency with studies using the Fmr1 knockout mouse as an experimental model, this idea has never been directly tested.
‘Premutation’ expansions at the FMR1 locus to between 55 and 200 CGG repeats are associated with the age-related neurodegenerative condition Fragile X-associated Tremor Ataxia Syndrome (FXTAS) (23–26). This disorder, characterized clinically by gait ataxia, action tremor, dementia and neuropsychiatric symptoms, occurs in ∼40% of male premutation carriers over the age of 50 (27). However, premutation range repeats are relatively common in the population [estimates upwards of 1:813 males and 1:259 females (28,29)], and have the potential to significantly influence the risk of other human diseases. Recent studies in young premutation carriers demonstrate higher rates of autism and attention deficit hyperactivity disorder (ADHD)-like symptoms in the absence of FXTAS symptoms (30–36) and FXS phenotypes have been reported in larger premutation and unmethylated full mutation carriers who produce FMR1 mRNA but inefficiently translate FMRP (37–43).
Unlike full mutation expansions, premutation-sized repeats are unmethylated and over-transcribed, leading to a 2–8-fold elevation in the production of FMR1 mRNA (44–46). However, the CGG repeat expansion forms a hairpin loop in the 5′ UTR of the RNA transcript that impairs ribosomal scanning and induces significant translational inefficiency (47–50). This leads to low-normal or decreased basal FMRP expression in Fragile X-premutation carriers, depending on the repeat size (47,51). The neurodegeneration seen in FXTAS and other age-related premutation phenotypes are thought to result primarily from an RNA gain-of-function mechanism (52–54). In contrast, work in two independently generated FMR1 premutation mouse models suggests an additional role for FMRP insufficiency in aspects of the premutation phenotype, especially in younger animals that do not yet demonstrate neurodegenerative sequelae (55–58). Defects in these mice include alterations in neuronal migration, dendritic branching, synaptic activity in cultured neurons, and behavioral defects including altered performance on measures of anxiety and social interaction (58–60).
Given the known critical roles for FMRP in synaptic function and the translational inefficiency induced by CGG repeat expansions, we hypothesized that mice with large unmethylated CGG repeat expansions would exhibit a specific defect in their ability to rapidly translate FMRP at synapses. A defect in activity-dependent synthesis of FMRP would allow for the analysis of the function of newly produced synaptic FMRP, including its role in long-lasting forms of synaptic plasticity. We therefore evaluated dendritic FMRP synthesis and synaptic function in a premutation mouse model where a CGG repeat expansion has been knocked into the mouse Fmr1 locus (58,60).
Here, we show that mice with 120–150 CGG repeats in the mouse Fmr1 5′ UTR have modestly reduced basal FMRP expression despite elevated Fmr1 mRNA levels, consistent with a robust impairment in translational efficiency. Strikingly, these animals exhibit impaired mGluR-dependent translation of dendritic FMRP. Young CGG KI mice exhibit normal basal synaptic properties, but enhanced mGluR-LTD, as in Fmr1 KO mice (9). However, the mechanism underlying this functional alteration is distinct from that in Fmr1 KO animals, as mGluR-LTD in CGG KI mice remains dependent on new protein synthesis. Our results provide a link between local FMRP synthesis and mGluR-dependent synaptic plasticity, and raise the possibility that some aspects of the cognitive defects observed in premutation carriers and unmethylated FXS patients may result from altered activity-dependent translation of FMRP.
RESULTS
Reduced FMRP translational efficiency in premutation model mice
To evaluate the neurobiological effects of ‘premutation’ range CGG repeats in the Fmr1 gene, we utilized a mouse model of the Fragile X premutation which contains ∼120–150 CGG repeats knocked-in to the endogenous mouse Fmr1 5′ UTR [CGG KI, (60), Fig. 1A]. Similar to human premutation patients, the expression of Fmr1 mRNA is significantly increased in cortical tissue [Fmr1 2/4 exon junction wild-type (WT) 1 ± 0.27, KI 5.24 ± 0.98 P < 0.05; Fmr1 16/17 exon junction WT 1 ± 0.24, KI 4.46 ± 0.74; P < 0.05, n = 5; Fig. 1B], as well as hippocampus (Fmr1 2/4 exon junction WT 1 ± 0.07, KI 4.04 ± 1.11 P < 0.05; Fmr1 16/17 exon junction WT 1 ± 0.07, KI 4.40 ± 1.99, data not shown) in CGG KI mice at 1 month of age (P28–37) compared with littermate controls (Fig. 1B and data not shown). Despite this increase in mRNA, FMRP expression is significantly reduced in both CGG KI cortex (P28–37, WT 100 ± 10.09%, KI 37.50 ± 4.37%, P < 0.05, n = 5; Fig. 1C) and hippocampus (P35–60, WT 100 ± 17.00%, KI 44.93 ± 14.71%, P < 0.05, n = 5; Fig. 1D) from young animals compared with littermate controls. To determine the relative translational efficiency of Fmr1 mRNA in cortical tissues, we created a ratio of total FMRP/relative Fmr1 mRNA from the same animals. Using this analysis, we find that the efficiency of Fmr1 mRNA translation is dramatically reduced in young CGG KI mice compared with littermate controls (FMRP CTX/Fmr1 mRNA; WT 100 ± 21.26%, KI 7.60 ± 0.99%, P < 0.05, n = 5; Fig. 1E).
Figure 1.

Elevated cortical Fmr1 mRNA and decreased Fragile X mental retardation protein (FMRP) in the fragile X premutation mouse. (A) PCR genotyping of CGG KI male mice and WT littermates showing the expanded CGG repeat. KI band corresponds to ∼120 repeats; WT band corresponds to 8 CGG repeats. (B) Fmr1 mRNA levels in the cortex of p28–37 fragile X premutation male mice by qPCR using two different sets of primers against Fmr1. The bar graph summarizes three experiments, n = 5. (C) Representative immunoblot to FMRP (1C3 1:1000) in p28–37 male mouse cortices from the indicated genotypes. Below: Summary of three experiments. Mean (±SEM) cortical FMRP in 1-month-old (p28–38; n = 5) and 6-month-old (p177–181; n = 3) CGG KI mice is decreased compared with littermate controls. The relative decrease between genotypes is greater in older animals. (D) Representative immunoblot against FMRP (17722 1:1000) in hippocampi of p35 CGG KI animals compared with WT littermate controls. Below: Mean (±SEM) hippocampal FMRP in p35–p60 male CGG KI mice compared with WT littermate controls. n = 5. (E) Translational efficiency of cortical Fmr1 RNA expressed as the ratio of FMRP to Fmr1 RNA levels in each individual animal, plotted on log10 scale; n = 5. *P < 0.05, Student's t-test.
Consistent with previous reports (58,60), FMRP is also reduced in the cortex of older (6-month-old) CGG KI mice (WT 100 ± 17.57%, KI 18.57 ± 2.68%, P < 0.05, n = 3; Fig. 1C) and, interestingly, when compared with WT littermates, the reduction in FMRP expression is greater in older CGG KI animals than in younger animals (1 month: KI 37.50 ± 4.37%, n = 5; 6 month: KI 18.57 ± 2.68%, n = 3; P < 0.05). This may reflect either a relatively greater decrease in FMR1 transcription in CGG KI versus WT mice with age or could result from somatic instability that is known to occur in these mice (61,62).
Activity-dependent synaptic translation of FMRP is impaired in CGG KI mice
To examine the sub-cellular distribution of FMRP in CGG KI neurons, we generated dissociated hippocampal neurons from CGG KI and WT littermate controls (P1–3). Neurons were probed with antibodies to FMRP on day in vitro (DIV) 14–17 (Fig. 2A), and FMRP expression in somatic and dendritic regions was assessed. FMRP expression was reduced in both the cell soma and proximal dendrite by similar amounts (soma: WT 100 ± 5.74%, KI 49.82 ± 2.69%, P < 0.05; dendrite: WT 100 ± 4.63%, KI 66.87 ± 2.94%, P < 0.05, n = 23–24 neurons from 2 animals; Fig. 2B–D), suggesting that, while FMRP expression is lower, what FMRP is expressed in CGG KI neurons is appropriately distributed.
Figure 2.
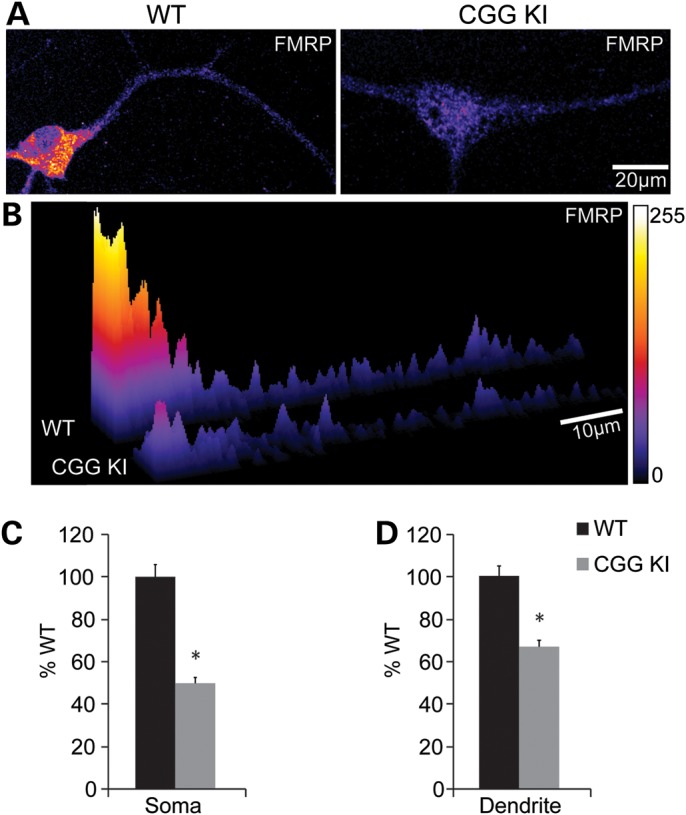
Reduced FMRP is distributed throughout dendrites in cultured CGG KI neurons. (A) DIV 14–17 cultured hippocampal neurons from male P1–3 CGG KI and littermate WT animals stained for FMRP (1C3 1:500). (B) 3D surface plot of the relative pixel intensity for the linearized images shown in A demonstrating reduced FMRP expression throughout the soma and dendrite. (C) Total non-zero FMRP fluorescence intensity was quantified in soma, revealing CGG KI neurons have 50% of WT FMRP levels. (D) Summary of fluorescence intensity studies in dendrites (0–40 µm), showing reduced FMRP in CGG KI neurons compared with WT neurons; n = 23–24 neurons from two animals in each group. *P < 0.05, Student's t-test.
The reduced efficiency of Fmr1 mRNA translation in CGG KI mice suggests that rapid, mGluR-dependent synthesis of FMRP might also be disrupted in the CGG KI mice. To address this question, we first examined changes in FMRP expression upon mGluR1/5 stimulation in synaptoneurosomes (SNs), a biochemical preparation enriched for synaptic components and often used as a means to examine protein synthesis at isolated synapses (18,20). SNs were prepared from the neocortex of P14–21 CGG KI mice and their WT littermates. In all experiments, we verified the appropriate enrichment of the synaptic scaffolding protein PSD-95 at different stages of SN preparation, and found that the enrichment of PSD-95 was similar between WT and CGG KI mice (Fig. 3A). PSD-95 expression in SNs were similar in WT and CGG KI mice (WT 100 ± 23.8%, CGG KI 99.2 ± 24.8%, n = 6). Consistent with our immunocytochemical results (Fig. 2), the expression of FMRP in unstimulated SNs was reduced in CGG KI, relative to WT mice (% WT, 42.92 ± 21.51%, P < 0.05, n = 5; Fig. 3B). We next examined changes in FMRP expression in response to mGluR stimulation: SNs were stimulated with the group 1 mGluR agonist, (RS)-3,5-dihydroxyphenylglycine (DHPG, 100 µM) for either 10 or 30 min at 37°C. Similar to effects seen previously in WT SNs (20), DHPG induced significant increases in FMRP at both 10 and 30 min time points relative to controls (10 min: WT 199.77 ± 56.97%; 30 min: WT 202.13 ± 54.83%, P < 0.05, n = 15; Fig. 3C and D). This increase was dependent on new protein synthesis (% 30 min untreated samples: 30 min DHPG: 162.5 ± 32.6%; 30 min DHPG + Anisomycin: 124.5 DHPG + Anisomycin 15.1%, expressed as % untreated n = 6). In contrast, SNs prepared from CGG KI mice did not show changes in FMRP expression in response to DHPG stimulation, consistent with impaired mGluR-dependent translation (Control: KI 42.92 ± 21.51%; 10 min: KI 31.16 ± 9.35%; 30 min: KI 40.84 ± 20.12%; NS, n = 5; Fig. 3C and D).
Figure 3.

CGG KI SNs do not respond to mGluR stimulation. SNs were prepared from WT and CGG KI cortical homogenates. (A) Verification of SN preparation was confirmed by PSD-95 enrichment between the initial homogenate (H), filtered sample (F), post-centrifugation supernatant (S) and final synaptoneurosome fraction (SN) in each WT and CGG KI preparation. (B) Representative immunoblot against FMRP (17 722 1:1000) in CGG KI SNs compared with littermate WT control. (C) SNs treated with 100 μM 3,5-dihydroxyphenylglycine (DHPG) for 10 or 30 min. Samples were immunoblotted for FMRP (17 722 1:1000) and actin (1:5000). (D) Quantification of FMRP immunoreactivity normalized to untreated samples. WT n = 15, CGG KI n = 5, *P < 0.05, Kruskal–Wallis one-way ANOVA.
To further assess mGluR-dependent FMRP translation in CGG KI neurons, we took advantage of mice expressing green fluorescent protein (GFP) on the X chromosome to generate hippocampal cultures where neurons harboring the premutation are intermingled with normal length CGG repeat WT neurons (Fig. 4A–C). This approach allows us to evaluate cell-autonomous roles of the premutation by comparing CGG KI neurons with neighboring WT neurons in the same culture, a strategy similar to that used previously for other X-linked mutations (15,63–65). Mice expressing GFP on the X chromosome were crossed with CGG KI mice to generate heterozygous XGFP/CGG KI females (Fig. 4A). This cross generates females possessing one WT X chromosome with a normal copy of Fmr1 and GFP and one X chromosome with a premutation range CGG repeat knocked-in to the Fmr1 allele, but no GFP. Owing to X-inactivation, roughly half the neurons will inactivate the CGG KI X chromosome and express normal Fmr1 mRNA along with GFP. The remaining neurons will inactivate the GFP-expressing chromosome and instead express the CGG KI Fmr1 allele. Analysis of dissociated neuronal cultures and histological staining of hippocampi shows roughly equal proportions of GFP+ and GFP− cells in both XGFP/WT and XGFP/KI female mice (Fig. 4B and data not shown).
Figure 4.

CGGKI/XGFP heterozygous cultures reveal selective DHPG induction of FMRP in WT neurons. (A) The breeding scheme used to generate mosaic female mice with one WT (GFP+) X chromosome, and one CGG KI (GFP−) X chromosome. (B) Fluorescent nuclei staining (DAPI 1:10 000) in coronal sections from an XGFP/WT female reveal GFP+ and GFP− cells in the hippocampus. (C) Primary hippocampal neurons from mosaic XGFP/CGG KI mice allow both WT (GFP+) and KI (GFP−) neurons in culture. (D) Quantitative analysis on soma from DIV 14–17 XGFP/CGG KI neurons stained for Map2 (Sigma 1:1000) and FMRP (17 722 1:500). CGG KI (GFP−) soma showed reduced basal FMRP fluorescence compared with WT (GFP−) neurons. (E) Basal FMRP expression is maintained in proximal and distal dendrites of CGG KI mice. WT n = 24, CGG KI n = 14, *P < 0.05, Student's t-test. (F) Cultures were treated with DHPG (100 µM for 20 min) prior to FMRP and Map2 staining. (G) Proximal dendrite segments showed selective FMRP immunofluorescence increases in WT (GFP+) neurons, but not in CGG KI (GFP−) neurons. (H) The effects of DHPG are mitigated by pretreatment with anisomycin (40 µM for 30 min) in WT proximal dendrites. There is no effect of DHPG or anisomycin on FMRP expression in the initial segment of CGG KI dendrites. WT n = 15–42 neurons from 1–2 animals, CGG KI n = 7–25 neurons from 1–2 animals. *P < 0.05, one-way ANOVA with Fisher's-LSD.
We first confirmed the effects of CGG repeat expansions on basal FMRP expression in XGFP/CGG KI cultures. GFP(−)/CGG KI(+) neurons exhibit reduced FMRP immunoreactivity in mixed XGFP/CGG KI cultured networks at DIV 14–17 compared with neighboring GFP(+)/FMR1 WT neurons (Fig. 4C–E). Consistent with studies in non-mosaic neuronal cultures (Fig. 2), these effects were seen both in the soma (WT 100 ± 4.79%, KI 30.07 ± 1.70%, P < 0.05, n = 14–24 neurons; Fig. 4D and E) and in both proximal and distal dendritic segments of CGG KI GFP neurons (0–40 µm: WT 100 ± 8.65%, KI 46.46 ± 7.03%; 40–80 µm: WT 100 ± 17.22, KI 51.47 ± 5.92%; 80–120 µm: WT 100 ± 15.05%, KI 55.05 ± 5.36%, P < 0.05, n = 13–23 neurons; Fig. 4D). The total amount of FMRP detected decreases with distance from the cell soma in both control and CGG KI neurons. However, the relative difference in expression of basal FMRP between WT and CGG KI neurons is smaller in proximal and distal dendritic compartments than in the cell soma, suggesting that decreases in FMRP reflect a primary failure in translational efficiency rather than a breakdown in FMRP transport into dendrites. We next examined whether the premutation had a cell-autonomous effect on mGluR-initiated translation of new FMRP. XGFP/CGG KI cultures were stimulated with DHPG (100 µM, 20 min) prior to FMRP and Map2 immunostaining. After mGluR activation, WT neurons showed a significant increase in dendritic FMRP immunoreactivity (Control: WT 100 ± 6.85%; DHPG: WT 133.74 ± 11.46%, P < 0.05; Fig. 4F–H) and this effect was blocked by pretreatment with the protein synthesis inhibitor anisomycin (40 µM, 30 min prior to and throughout DHPG application; Anisomycin + DHPG: WT 96.81 ± 7.75%; Anisomycin: WT 102.53 ± 7.50%; Fig. 4H). In contrast, DHPG did not alter FMRP expression in CGG KI neurons in the presence or absence of anisomycin (Control: KI 49.70 ± 4.33%; DHPG: KI 50.42 ± 6.20%; Anisomycin + DHPG: KI 38.73 ± 2.74%; Anisomycin: KI 42.04 ± 3.20%; NS; Fig. 4F–H). These data support the hypothesis that premutation range expanded CGG repeats impair mGluR-dependent synthesis of FMRP in a cell-autonomous fashion.
Enhanced mGluR-LTD in hippocampal slices prepared from CGG knock-in mice
Since mGluR-dependent translation is critical for certain forms of synaptic plasticity that are altered in FXS model mice, we next tested whether there was any overlap between the synaptic plasticity phenotypes in Fmr1 KO mice and CGG KI mice. We first examined basal synaptic properties at CA3–CA1 synapses in acute hippocampal slice preparations from young CGG KI mice with their WT littermates (P31–35). Field excitatory postsynaptic potentials (fEPSPs) were evoked by stimulating Schaffer collaterals and recording in stratum radiatum of area CA1. In response to a series of stimulation pulses of increasing intensity, we found that the corresponding increase in fEPSP slope was nearly identical in WT and CGG KI mice (Fig. 5A). These largely overlapping input/output curves show that CGG KI mice do not exhibit alterations in basal synaptic efficacy relative to WT mice. In addition, we tested whether paired pulse facilitation, a measure of short-term synaptic plasticity and presynaptic function, was altered in CGG KI mice. In response to pairs of stimulation pulses with varying inter-pulse intervals, WT and CGG KI mice exhibited similar robust facilitation of the second synaptic response at all intervals (Fig. 5B), suggesting that the neurotransmitter release probability is largely similar between the two genotypes. Hence, basal synaptic function is similar between CGG KI mice and their WT littermates.
Figure 5.

Basal synaptic function is unchanged in CGG KI mice. (A) Hippocampal field excitatory postsynaptic potentials (fEPSPs) in response to Schaffer collateral stimulation of increasing strength show a similar input/output response curve in CGG KI animals compared with littermate WT controls. n = 19 (WT) and 19 (CGG KI). (B) No difference is detected in paired-pulse facilitation, a measure of basal neurotransmitter release probability, between CGG KI mice and littermate WT mice at any inter-stimulus interval, suggesting that the neurotransmitter release probability at CA3–CA1 synapses is not altered by the premutation. n = 8 (WT) and 8 (CGG KI).
Our results suggest that premutation range repeats impair FMRP translation even in young mice, raising the question of whether this loss of new FMRP synthesis might mimic aspects of the FXS phenotype. To test this idea, we next examined mGluR-dependent LTD at these CA3–CA1 synapses. After confirming that evoked fEPSPs were stable over time, LTD was induced by brief application of DHPG (100 µM, 10 min; Fig. 6). As previously described, DHPG treatment induced a sustained depression of fEPSPs in WT slices that persisted well beyond drug application (Fig. 6). Interestingly, we found that this mGluR-dependent LTD was significantly exaggerated in slices from CGG KI mice (Fig. 6A), a synaptic phenotype that is similar to Fmr1 KO mice (9). These results demonstrate that, even during early life, the expanded premutation CGG repeat in the Fmr1 gene leads to altered hippocampal synaptic plasticity.
Figure 6.

Exaggerated mGluR-LTD in CGG KI mice is protein synthesis dependent. (A) Field EPSPs were recorded in CA1 stratum radiatum in response to Schaffer collateral stimulation. Addition of the group 1 mGluR agonist DHPG (100 μM; 10 min) induced LTD at CA3–CA1 synapses; this mGluR-LTD was significantly enhanced in CGG KI mice. n = 9 (WT) and 13 (CGG KI). Inset: Shown are representative averages of four consecutive field potential waveforms from each group during the baseline period and 1 h after LTD induction. (B) mGluR-LTD in FMR1 KO mice persists in the presence of the protein synthesis inhibitor anisomycin (20 µM), as previously reported (9). n = 7 (control) and 8 (aniso). (C) In contrast, the enhanced mGluR-LTD in CGG KI mice remains sensitive to protein synthesis inhibitors. n = 13 (control) and 7 (aniso).
Enhancement of mGluR-LTD in premutation and FXS model mice are mechanistically distinct
Like young CGG KI mice, Fmr1 KO mice also exhibit enhanced mGluR-LTD (9). Since this exaggerated mGluR-LTD in FXS model mice is thought to contribute to intellectual disability and/or autistic features in FXS, it was of interest to determine to what extent the exaggerated LTD in each case was due to similar or distinct mechanisms. To explore this issue, we examined whether protein synthesis inhibitors would impair the induction of mGluR-LTD in CGG KI and Fmr1 KO mice. In WT mice, mGluR-LTD requires rapid dendritic protein synthesis for its induction (12), whereas mGluR-LTD in Fmr1 KO mice is completely resistant to protein synthesis inhibitors (13,16). Consistent with these findings, we found that the magnitude of mGluR-LTD in Fmr1 KO mice was not affected by blocking protein synthesis with anisomycin (Fig. 6B). In contrast, the enhanced mGluR-LTD seen in young CGG KI mice was significantly diminished with anisomycin (Fig. 6C), indicating that mGluR-LTD remains dependent on new protein synthesis in these mice, as in WT mice. Taken together, these results suggest that while young FXS and premutation model mice share the same exaggerated mGluR-LTD phenotype, the mechanism underlying this plasticity is distinct in the two mouse models.
DISCUSSION
The roles of FMRP in both normal and aberrant control of synaptic function have received considerable attention in the past two decades. This effort has been greatly facilitated by work in the Fmr1 KO mouse, which recapitulates several important features of FXS, and has been instrumental in the rapid development of novel therapeutic approaches (66). In addition, significant advances have been made in our understanding of the molecular consequences of premutation CGG repeat expansions, which enhance FMR1 transcription but impair FMRP translation and elicit toxicity directly as RNA (54). In contrast, considerably less is known about the impact of premutation range CGG repeat expansions on neuronal function. Premutation expansions do not typically lead to overt intellectual disability, but they are increasingly linked to a broad range of important clinical phenotypes in patients, including neuropsychiatric symptoms and autistic features earlier in life (23,24,30,32–34). These clinical features are recapitulated in Fragile X premutation model mice that exhibit altered social interactions and anxiety behaviors compared with littermate controls (58). We therefore examined neuronal function in young Fragile X premutation model mice, with a specific focus on the impact of the CGG repeat on activity-dependent FMRP translation.
Our results demonstrate that premutation model mice exhibit a dramatic decrease in the translational efficiency of Fmr1 mRNA that impairs rapid, activity-dependent synthesis of FMRP in dendrites. This defect in local FMRP synthesis is associated with exaggerated mGluR-dependent LTD, a phenotype first reported in Fmr1 KO mice. This shared synaptic phenotype, however, is mechanistically distinct between Fmr1 KO and premutation model mice, as mGluR-dependent LTD in CGG KI mice remains dependent on new protein synthesis (Fig. 6B and C). Coupled with data demonstrating altered dendritic spine morphology and development in CGG KI mice (56,58), our results reveal a shared defect in synaptic plasticity in FXS and premutation model mice and suggest an important role for activity-dependent FMRP synthesis at synapses in regulating the magnitude of synaptic strength.
FMRP is an RNA-binding protein found associated with stalled ribosomes (69), where it acts primarily as a translational suppressor (67–69). mGluR signaling induces dephosphorylation of FMRP, which then dissociates from polysome–transcript complexes and is rapidly degraded, leading to an activity-dependent burst of translation of FMRP target mRNAs (Fig. 7A) (14,70,71). Intriguingly, FMRP also binds and regulates the translation of its own mRNA in vitro and FMRP is rapidly synthesized at synapses in response to mGluR activation in vivo (16,17,20,68,72,73). The role of FMRP as a translation repressor, and the clear role of certain FMRP targets (e.g. activity-regulated cytoskeletal-associated protein; Arc) as mediators of mGluR-LTD (10,74), has bolstered the hypothesis that newly synthesized FMRP functions to provide negative feedback on further local translation, thus constraining the magnitude of LTD after mGluR activation (Fig. 7A) (8,19,22). This notion of newly synthesized FMRP as a ‘brake’ on local translation is consistent with observations that mGluR-LTD and other forms of mGluR-mediated plasticity require local protein synthesis in only a brief time window after induction (12,75). In the complete absence of FMRP, mGluR-LTD is enhanced but no longer requires new protein synthesis (9,13). This has been interpreted as resulting from an uncoupling of mGluR activation and synthesis of critical mGluR-LTD effector proteins (Fig. 7B) (8,19). Thus, whereas synaptic levels of Arc and other LTD mediator proteins are low basally in WT neurons and increase as a result of mGluR-dependent synthesis, Arc in FMR1 KO neurons is basally elevated, but is no longer synthesized in response to mGluR activation (Fig. 7B) (15).
Figure 7.

A working model of mGluR-LTD in WT, KO and CGG KI mice. Group I mGluR receptors are critical modulators of synaptic overactivity. (A) Normally, FMRP bound transcripts, including Fmr1 mRNA, exist in stalled polyribosomal complexes at synapses. (i) Activation of group I mGluRs triggers the internalization of AMPA receptors and the dissociation/clearance of FMRP from target mRNAs. (ii) This allows for the rapid translation of proteins required for the maintenance of AMPA receptor internalization (LTD proteins), leading to long-lasting changes in synaptic strength. In parallel, FMRP is itself synthesized at synapses. (iii) This new FMRP acts as a brake on further translation of mRNA targets. The end result is mGluR-LTD that requires a temporally constrained burst of local protein translation after receptor activation. (B) In FXS model mice, translation of FMRP target transcripts is uncoupled from mGluR signaling. (i) This results in a basal increase in production of LTD proteins. Upon mGluR activation, AMPA receptors are internalized normally but the presence of excess basal LTD effector proteins leads to the enhancement of mGluR-LTD. As the over-synthesis of LTD effector proteins is not tied to mGluR activation, induction of mGluR-LTD in FXS model mice does not require new protein synthesis. (C) In Fragile X premutation model mice, there is adequate basal expression of FMRP to allow for the localization of FMRP with associated transcripts at synapses. (i) mGluR activation triggers the dissociation of FMRP from these transcripts normally. (ii) However, the CGG repeat expansion blocks rapid FMRP synthesis. Without this new FMRP, there is no brake to prevent the ongoing synthesis of FMRP target transcripts. (iii) The result is overproduction of LTD effector proteins and enhanced mGluR-LTD. In contrast to FXS model mice, synaptic protein translation in premutation model mice remains coupled to mGluR activation and the mGluR-LTD is thus dependent on new protein synthesis. This working model makes a number of specific predictions which will be tested in future studies.
In CGG KI mice, our results demonstrate that mGluR-LTD is exaggerated as in Fmr1 KO mice, but that this enhanced mGluR-LTD remains dependent on new protein synthesis, as occurs typically in WT animals (Fig. 6C). We suggest that this protein synthesis-dependent enhancement of mGluR-LTD occurs because of a specific failure in activity-dependent FMRP production (Fig. 7C). Although basal FMRP levels are lower in CGG KI mice, FMRP is maintained in both proximal and distal dendritic compartments at levels that are 40–60% of normal, which is above the threshold at which alterations in mGluR-triggered AMPA receptor recycling occurs (76). This suggests that basal synthesis of FMRP, although inefficient, is adequate to achieve the suppression of translation of LTD effector proteins in the absence of mGluR activity (Fig. 7C). However, with mGluR activation, the rapid synthesis of dendritic FMRP is significantly impaired by the CGG repeat expansion. This means that there is inadequate new FMRP produced to halt the ongoing translation of FMRP target mRNAs, leading to an overproduction of these LTD effector proteins. This overproduction of LTD effector proteins presumably drives the enhanced LTD phenotype, but unlike FMR1 KO cultures, production of these proteins remains coupled to mGluR activity, as the release of FMRP cargo transcripts is still required to initiate the LTD (Fig. 7C). Within this framework, we propose that new translation of FMRP at synapses is critical for constraining mGluR-LTD, likely through limiting the sustained expression of LTD effectors by repressing their continued synaptic translation. However, some aspects of the effects observed here may also derive from either basal insufficiency of FMRP or from CGG repeat RNA-mediated toxic effects. Future experiments will be required to demonstrate altered synthesis of LTD effector proteins in CGG KI mice and to formally exclude contributions from these additional factors on synaptic function in CGG KI mice.
In humans, the consequences of premutation range CGG repeats are age-dependent. Of relevance, a recent study examined mGluR-dependent synaptic plasticity in aged animals (10–13-month-old), comparing WT animals and a different mouse model of the fragile X premutation (77). They found that aged premutation model mice exhibited weaker immediate synaptic depression following mGluR activation relative to their WT counterparts, but the level of sustained synaptic depression was similar across genotypes. In contrast, in younger animals, we find no difference in acute synaptic depression driven by mGluR activation, but a significant increase in the magnitude of enduring synaptic depression following mGluR stimulation. Although Hunsaker et al. (77) used a different Fmr1 premutation mouse model than the one employed here, these results raise the interesting possibility that the impact of the Fmr1 premutation may evolve as a function of age. One possibility is that the effects of enhanced mGluR-LTD on the development of childhood and early-adult-onset phenotypes in premutation carriers may be dissociable from the development of late-adult-onset FXTAS in premutation carriers, where RNA-mediated toxicity and neurodegeneration might be expected to have a greater impact.
In this work, we focused on the features of mGluR-LTD in young premutation model mice, given that exaggerated hippocampal mGluR-LTD in Fmr1 KO mice is widely considered relevant to the intellectual disability and autistic symptoms seen in FXS. However, it is likely that Fmr1 premutation repeats may have a broader impact on neural excitability. A recent series of in vitro studies demonstrated that neurons cultured from premutation mice develop abnormal firing properties (59). These neuronal networks exhibit clustered firing and increased Ca2+ oscillations, as well as disruptions in neurotransmitter transport machinery (59). Neurons derived from induced pluripotent stem cells generated from premutation carrier fibroblasts exhibit a similar increase in Ca2+ dynamics (78). The authors speculated that the functional deficits arise from an improper excitation/inhibition ratio created by the altered transport of glutamate and GABA. While changes in the ratio of excitation to inhibition would influence Ca2+ dynamics and thus the firing properties of neurons, we did not find evidence of altered basal synaptic transmission in our ex vivo experiments (Fig. 5).
Recent clinical evidence highlights potential points of confluence in symptoms found in young premutation carriers with FXS, suggesting that comparisons between FXS and premutation model mice may help to better identify specific behavioral and neurophysiological correlates of disease features. Specifically, work by a number of groups has demonstrated increased rates of autism and ADHD in premutation carriers, as well as neuropsychiatric symptoms, and executive and amygdala dysfunction (30,79–86). This amygdala dysfunction and structural changes in premutation carriers without FXTAS correlate with lower blood FMRP expression (82). Consistent with this, two CGG KI mouse models exhibit numerous behavioral defects that mirror those observed in Fmr1 KO animals (58,77,87). We find that FXS model mice and Fmr1 premutation model mice of similar ages share an important synaptic plasticity phenotype. Our data raise the intriguing possibility that neuropsychiatric abnormalities, autism and ADHD-like symptoms in young premutation patients may be linked to the mGluR-dependent plasticity deficits examined in mouse models of these disorders. However, it should be noted that the repeat sizes studied in CGG KI mice here and elsewhere are significantly larger than that seen in the average premutation carrier, as repeats become progressively less stable with expansions above 55 repeats. These findings are therefore more relevant to those rare patients who have >100 CGG repeats or who have an unmethylated full mutation. This model may be particularly relevant to this latter category, as recent data suggests that a significant (>30%) portion of FXS patients exhibit incomplete FMR1 DNA methylation and some FMR1 RNA transcription (41). Importantly, this epigenetic alteration correlates with clinical severity and response to some experimental therapies (41). As clinical trials proceed in this patient population with agents that either directly or indirectly target the mGluR pathway(41,66,88), it will be important to understand how mechanistic differences in different mutation states elicit altered mGluR-LTD, and incorporate this knowledge into better practice and drug development.
MATERIALS AND METHODS
Mice and cell culture
Animal use followed NIH guidelines and was in compliance with the University of Michigan Committee on Use and Care of Animals. DNA was extracted from tail biopsies and isolated with DirectPCR lysis reagent (Viagen) and proteinase K (0.2 µg/µl, Roche), incubated overnight at 55°C. Proteinase K was heat inactivated and DNA samples were genotyped first with primers against the Y chromosome (5′GTGAGAGGCACAAGTTGGC, 5′GTCTTGCCTGTATGTGATGG) to determine the sex of each animal using Platinum® PCR Supermix (Invitrogen). To amplify the knocked-in CGG repeat expansion, we targeted mouse specific Fmr1 allele (5′AGCCCCGCACTTCCACCACCAGCTCCTCCA, 5′GCTCAGCTCCGTTTCGGTTTCACTTCCGGT) in male hemizygous animals using the Expand High Fidelity PCR System (Roche) supplemented with 2 m Betaine (Sigma) and 5% dimethyl sulfoxide (DMSO; Fisher Scientific) as described previously (89). As genotyping was performed on tail samples early in life, small expansions in repeat length may have occurred due to somatic instability in older animals (61). Dissociated hippocampal neuron cultures were prepared from postnatal (P1–3) mice as previously described (90). Experiments were performed at 14–17 days in vitro (DIV).
Drugs
(RS)-3,5-DHPG (Tocris) was prepared fresh each day in sterile water, or artificial cerebrospinal fluid (aCSF, in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2 and 10 dextrose). Anisomycin (Sigma) was prepared as a 1000× stock in DMSO, stored at −20°C, and diluted to final concentration in aCSF or conditioned media.
Western blotting
Brain lysate samples were homogenized in RIPA buffer (50 mm Tris–HCl, 150 mm NaCl, 0.1% SDS, 1% NP−40, 0.5% deoxycholic acid-sodium salt, pH 7.4) containing Complete Mini protease inhibitor cocktail (Roche). Samples were sonicated and centrifuged, and total protein content of the supernatant measured using a DC Protein assay (Bio-Rad). Equal amounts of protein were mixed with 4× Laemmli buffer and boiled for 5 min before separation on 10 or 12% polyacrylamide gels. Gels were transferred to PVDF membranes and blocked with Tris-buffered saline containing 0.1% Triton-X (TBST) and 5% non-fat milk for 60 min at RT, and incubated with an antibody against FMRP (Millipore mouse monoclonal 1C3 1:1000 or Abcam rabbit polyclonal 17 722, 1:1000) or PSD-95 (Abcam, 6G6–1C9, 1:2000) overnight at 4°C. After washing with TBST, blots were incubated with a corresponding HRP-conjugated secondary antibody (anti-rabbit or anti-mouse 1:5000; Jackson Immunoresearch); this was followed by chemiluminescent detection (Western Lightning Plus-ECL, PerkinElmer). The same blots were reprobed with a mouse monoclonal antibody against β-tubulin (University of Iowa's Developmental Studies Hybridoma Bank E7, 1:5000) or β-actin (Sigma 1:5000) to confirm equal loading. Band intensity was quantified in the linear range with densitometry using NIH ImageJ.
Quantitative polymerase chain reaction
Dissected cortex or hippocampi from P28–60 male mice were flash-frozen and stored at −80°C. RNA was extracted using TRIzol Reagent (Invitrogen), following the manufacturer's guidelines. Equal amounts of extracted RNA (1 µg) were used to generate cDNA (iScript™ cDNA synthesis kit, Bio-Rad). Quantitative polymerase chain reaction (QPCR) was performed using iQ™ SYBR© Green Supermix (Bio-Rad) and primers against the 2/4 (5′CATGAAGATTCAATAACAGTTGC, 5′CACTTTAGCTAACCACCAACAG) or 16/17 (5′CCGAACAGATAATCGTCCACG, 5′ACGCTGTCTGGCTTTTCCTTC) exons of mouse Fmr1, and actin (5′GGCATCCTCACCCTGAAGTA, 5′AGAGGCGTACAGGGATAGCA). Samples were run in triplicate, and Fmr1 expression data normalized to actin expression for each sample.
Translational efficiency calculation
The translational efficiency ratio was calculated by deriving FMR1 mRNA expression levels determined by qRT–PCR from one cortex while total protein lysates were prepared from the contralateral cortex from the same animal. For each animal, cortical FMR1 mRNA expression (relative to actin) was normalized to the mean FMR1 mRNA expression in control cortices. Similarly, cortical FMRP levels were expressed as a ratio to actin expression and then normalized to the mean FMRP expression in control cortices. These numbers were then expressed as a ratio of normalized FMRP expression/normalized FMR1 mRNA expression. Finally, the mean value of this ratio in WT animals was set at 100 and all individual animal values were expressed as a percentage of this number.
Synaptoneurosomes
SNs were prepared from male P14–21 WT and CGG KI mice as described previously (18, 91). Briefly, cortices were homogenized in 3 ml of homogenization buffer [containing (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 2.5 CaCl2, 1.53 KH2PO4, 212.7 glucose and 1 DTT, pH 7.4], supplemented with Complete Mini protease inhibitor cocktail (Roche) on ice. Samples were passed through a 100 μm nylon mesh filter, followed by two 10 μm nylon mesh filters (Millipore), followed by centrifugation at 1000 g for 15 min at 4°C. The pellets were suspended in 1.1 ml homogenization buffer per cortex. SN preparations were divided into 10 × 100 µl aliquots for technical duplicates, and pre-warmed for 10 min at 37°C before stimulation with (RS)-3,5-DHPG (Tocris, 100 µM). After incubation with DHPG at 37°C, samples were passed through a 28 gauge needle, and processed for western blotting as above. Expression of all samples was normalized to unstimulated samples maintained at 37°C for 60 min and statistical significance was determined using a Kruskal–Wallis one-way analysis of variance. Similar results were observed when comparisons were done with pre-stimulated samples (i.e. samples from the same SN prep that were never warmed to 37°C, data not shown).
Immunohistochemistry
Animals were anesthetized with 0.2 mg Ketamine/20 µg Xylazine per kilogram prior to transcardial perfusion (2 ml per min) with 5–10 ml of ice-cold sterile phosphate-buffered saline (PBS) and 5–10 ml of 4% paraformaldehyde (PFA) followed by brain dissection. Brains were sunk in 30% sucrose in PBS at 4°C prior to sectioning with a vibratome at 30 µm. Free-floating sections were stored in cryostorage (30% sucrose, 33.33% ethylene glycol, 0.05 m PB pH 7.4) at −20°C. Sections were removed from cryostorage by rotating in PBS at 4°C overnight. Sections were permeabilized in 0.1% Triton X in PBS for 5 min, followed by staining with DAPI (1:10 000) for 15 min at room temperature. Sections were washed 2× with PBS, and mounted on slides in ProLong® Gold Antifade Reagent with DAPI (Invitrogen).
Immunocytochemistry and microscopy
All experiments were conducted at 37°C. Neurons were treated with anisomycin (40 µM) or vehicle (DMSO 1:1000) for 30 min in conditioned media. Cultures were then stimulated with DHPG (100 µM) for 20 min in the presence of anisomycin, or left as controls with vehicle, or with anisomycin alone. After treatment, neurons were fixed with warmed 4% PFA/4% sucrose in PBS with 1 mm MgCl2 and 0.1 mm CaCl2 (PBS-MC), permeabilized (0.1% Triton X in PBS-MC, 5 min), blocked with 5% normal goat serum in PBS-MC for 1 h and labeled with an antibody against FMRP (Millipore 1C3 1:200 or Abcam 17722 1:500). For co-labeling of dendrites, we used antibodies against Map2 (Sigma M4403 1:1000, Millipore AB5622 1:1000) for 60 min at RT, or overnight at 4°C. Secondary detection was achieved with Alexa 488-, 555- or 635-conjugated goat anti-rabbit or goat anti-mouse antibodies (1:500 or 1:1000) for 60 min at RT.
All imaging was performed on an inverted Olympus FV1000 laser scanning confocal microscope with identical acquisition parameters for each treatment condition. Image analysis was performed on maximal intensity z-projected images using custom-written analysis routines for ImageJ. Statistical analysis utilized a one-way analysis of variance (ANOVA) to detect differences across conditions within genotype. N≈ 20–40/condition across multiple individual experiments for each genotype.
Electrophysiology
Hippocampal slices were prepared from P35–42 male CGG KI mice and their male WT littermates. Mice were lightly anesthetized with isoflurane before decapitation. Then, the brain and hippocampal lobules were rapidly removed and placed in ice cold artificial cerebrospinal fluid [aCSF, containing in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, 10 dextrose (pH 7.4) saturated with 95% O2, 5% CO2]. Transverse slices (400 µm) of the hippocampus were cut using a tissue chopper (Stoelting, Wood Dale, IL, USA) and CA3 was surgically isolated from CA1 with a scalpel. Slices recovered for 2–5 h at room temperature in a submersion chamber containing aCSF prior to recording. For recording, hippocampal slices were transferred to a recording chamber and continuously perfused at 32°C with aCSF at a rate of 1–2 ml/min.
Recording electrodes were pulled from borosilicate capillary glass (G150-4, Warner) and filled with aCSF. The recording pipette was placed in the middle of stratum radiatum of CA1. Synaptic responses were elicited using cluster stimulation electrodes (FHC, Bowdoin, ME, USA) placed in CA1stratum radiatum, lateral to the recording electrode. Current was delivered for 100 µs with an ISO-flex stimulator (AMPI, Jerusalem, Israel). Stable baseline responses were collected every 30 s (0.033 Hz) by using a stimulation intensity (20–140 μA) yielding ∼50% of the maximal synaptic response. The fEPSP signal was amplified 1000 times with a DAM-50 DC differential amplifier (WPI) and filtered at 3 kHz. Recordings were collected at 10 kHz using Clampex 10.2 and analyzed using Clampfit 10.2 (Molecular Devices, Sunnyvale, CA, USA). For all experiments, the initial slope of each fEPSP was expressed as the percentage of the baseline average. Pooled data represent the mean fEPSP slope (±SEM). Statistical significance was determined using an independent t-test, P < 0.05.
FUNDING
This work was supported by the National Institutes of Mental Health (grant number RO1MH085798 to M.A.S.); the National Institutes of Neurological Disorders and Stroke (grant number F31NS073372 to A.J.I.); the National Institutes of Health (grant number T32GM008322 to A.J.R., K08NS069809 to P.K.T.); the PEW Biomedical Scholars Program to M.A.S.; and the Harris Professorship to P.K.T.
ACKNOWLEDGEMENTS
We thank Sundeep Kalantry for his kind gift of XGFP mice and Cara Westmark for providing us with FMR1 KO mice. We are grateful to Cynthia Carruthers and Christian Althaus for their assistance with culture maintenance and preparation and Grace Van Hyfte for assistance with imaging analysis. We also thank Hank Paulson for his insights and comments during the preparation of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Rogers S.J., Wehner D.E., Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. doi:10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez R.N., Feinberg R.L., Vaurio R., Passanante N.M., Thompson R.E., Kaufmann W.E. Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am. J. Med. Genet. A. 2009;149A:1125–1137. doi: 10.1002/ajmg.a.32848. doi:10.1002/ajmg.a.32848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P., et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. doi:10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 4.Kremer E.J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S.T., Schlessinger D., Sutherland G.R., Richards R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. doi:10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- 5.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. doi:10.1016/0092-8674(91)90125-I. [DOI] [PubMed] [Google Scholar]
- 6.Bell M.V., Hirst M.C., Nakahori Y., MacKinnon R.N., Roche A., Flint T.J., Jacobs P.A., Tommerup N., Tranebjaerg L., Froster-Iskenius U., et al. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64:861–866. doi: 10.1016/0092-8674(91)90514-y. doi:10.1016/0092-8674(91)90514-Y. [DOI] [PubMed] [Google Scholar]
- 7.Oberle I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., Boue J., Bertheas M., Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. doi:10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 8.Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. doi:10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber K.M., Gallagher S.M., Warren S.T., Bear M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl Acad. Sci. USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. doi:10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park S., Park J.M., Kim S., Kim J.A., Shepherd J.D., Smith-Hicks C.L., Chowdhury S., Kaufmann W., Kuhl D., Ryazanov A.G., et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. doi:10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepherd J.D., Rumbaugh G., Wu J., Chowdhury S., Plath N., Kuhl D., Huganir R.L., Worley P.F. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. doi:10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber K.M., Kayser M.S., Bear M.F. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. doi:10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 13.Nosyreva E.D., Huber K.M. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. doi:10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- 14.Nalavadi V.C., Muddashetty R.S., Gross C., Bassell G.J. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J. Neurosci. 2012;32:2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012. doi:10.1523/JNEUROSCI.5057-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niere F., Wilkerson J.R., Huber K.M. Evidence for a fragile x mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered arc translation and long-term depression. J. Neurosci. 2012;32:5924–5936. doi: 10.1523/JNEUROSCI.4650-11.2012. doi:10.1523/JNEUROSCI.4650-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou L., Antion M.D., Hu D., Spencer C.M., Paylor R., Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. doi:10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Todd P.K., Mack K.J., Malter J.S. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl Acad. Sci. USA. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. doi:10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muddashetty R.S., Kelic S., Gross C., Xu M., Bassell G.J. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. doi:10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bear M.F., Huber K.M., Warren S.T. The mGluR theory of fragile X mental retardation. Trends. Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. doi:10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Weiler I.J., Irwin S.A., Klintsova A.Y., Spencer C.M., Brazelton A.D., Miyashiro K., Comery T.A., Patel B., Eberwine J., Greenough W.T. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl Acad. Sci. USA. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. doi:10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antar L.N., Afroz R., Dictenberg J.B., Carroll R.C., Bassell G.J. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. doi:10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todd P.K., Malter J.S. Fragile X mental retardation protein in plasticity and disease. J. Neurosci. Res. 2002;70:623–630. doi: 10.1002/jnr.10453. doi:10.1002/jnr.10453. [DOI] [PubMed] [Google Scholar]
- 23.Bourgeois J.A., Coffey S.M., Rivera S.M., Hessl D., Gane L.W., Tassone F., Greco C., Finucane B., Nelson L., Berry-Kravis E., et al. A review of fragile X premutation disorders: expanding the psychiatric perspective. J. Clin. Psychiatry. 2009;70:852–862. doi: 10.4088/JCP.08m04476. doi:10.4088/JCP.08r04476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berry-Kravis E., Abrams L., Coffey S.M., Hall D.A., Greco C., Gane L.W., Grigsby J., Bourgeois J.A., Finucane B., Jacquemont S., et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord. 2007;22:2018–2030. doi: 10.1002/mds.21493. quiz 2140 doi:10.1002/mds.21493. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan A.K., Marcus M., Epstein M.P., Allen E.G., Anido A.E., Paquin J.J., Yadav-Shah M., Sherman S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 2005;20:402–412. doi: 10.1093/humrep/deh635. doi:10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 26.Greco C.M., Berman R.F., Martin R.M., Tassone F., Schwartz P.H., Chang A., Trapp B.D., Iwahashi C., Brunberg J., Grigsby J., et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. doi:10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 27.Jacquemont S., Hagerman R.J., Leehey M.A., Hall D.A., Levine R.A., Brunberg J.A., Zhang L., Jardini T., Gane L.W., Harris S.W., et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. J. Am. Med. Assoc. 2004;291:460–469. doi: 10.1001/jama.291.4.460. doi:10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 28.Jacquemont S., Hagerman R.J., Hagerman P.J., Leehey M.A. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet. Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. doi:10.1016/S1474-4422(06)70676-7. [DOI] [PubMed] [Google Scholar]
- 29.Seltzer M.M., Baker M.W., Hong J., Maenner M., Greenberg J., Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012;159B:589–597. doi: 10.1002/ajmg.b.32065. doi:10.1002/ajmg.b.32065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farzin F., Perry H., Hessl D., Loesch D., Cohen J., Bacalman S., Gane L., Tassone F., Hagerman P., Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. doi:10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 31.Loesch D.Z., Litewka L., Churchyard A., Gould E., Tassone F., Cook M. Tremor/ataxia syndrome and fragile X premutation: diagnostic caveats. J. Clin. Neurosci. 2007;14:245–248. doi: 10.1016/j.jocn.2006.01.015. doi:10.1016/j.jocn.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Clifford S., Dissanayake C., Bui Q.M., Huggins R., Taylor A.K., Loesch D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007;37:738–747. doi: 10.1007/s10803-006-0205-z. doi:10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 33.Grigsby J., Brega A.G., Jacquemont S., Loesch D.Z., Leehey M.A., Goodrich G.K., Hagerman R.J., Epstein J., Wilson R., Cogswell J.B., et al. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS) J. Neurol. Sci. 2006;248:227–233. doi: 10.1016/j.jns.2006.05.016. doi:10.1016/j.jns.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Loesch D.Z., Huggins R.M., Hagerman R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard Dev. Disabil. Res. Rev. 2004;10:31–41. doi: 10.1002/mrdd.20006. doi:10.1002/mrdd.20006. [DOI] [PubMed] [Google Scholar]
- 35.Loesch D.Z., Bui Q.M., Grigsby J., Butler E., Epstein J., Huggins R.M., Taylor A.K., Hagerman R.J. Effect of the fragile X status categories and the fragile X mental retardation protein levels on executive functioning in males and females with fragile X. Neuropsychology. 2003;17:646–657. doi: 10.1037/0894-4105.17.4.646. doi:10.1037/0894-4105.17.4.646. [DOI] [PubMed] [Google Scholar]
- 36.Loesch D.Z., Huggins R.M., Bui Q.M., Epstein J.L., Taylor A.K., Hagerman R.J. Effect of the deficits of fragile X mental retardation protein on cognitive status of fragile x males and females assessed by robust pedigree analysis. J. Dev. Behav. Pediatr. 2002;23:416–423. doi: 10.1097/00004703-200212000-00004. doi:10.1097/00004703-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Allen E.G., Sherman S., Abramowitz A., Leslie M., Novak G., Rusin M., Scott E., Letz R. Examination of the effect of the polymorphic CGG repeat in the FMR1 gene on cognitive performance. Behav. Genet. 2005;35:435–445. doi: 10.1007/s10519-005-2792-4. doi:10.1007/s10519-005-2792-4. [DOI] [PubMed] [Google Scholar]
- 38.Feng Y., Zhang F., Lokey L.K., Chastain J.L., Lakkis L., Eberhart D., Warren S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. doi:10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 39.Tassone F., Hagerman R.J., Chamberlain W.D., Hagerman P.J. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am. J. Med. Genet. 2000;97:195–203. doi: 10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R. doi:10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 40.Tassone F., Hagerman R.J., Loesch D.Z., Lachiewicz A., Taylor A.K., Hagerman P.J. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am. J. Med. Genet. 2000;94:232–236. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h. doi:10.1002/1096-8628(20000918)94:3<232::AID-AJMG9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 41.Jacquemont S., Curie A., des Portes V., Torrioli M.G., Berry-Kravis E., Hagerman R.J., Ramos F.J., Cornish K., He Y., Paulding C., et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 2011;3:64ra61. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- 42.Hagerman R.J., Staley L.W., O'Conner R., Lugenbeel K., Nelson D., McLean S.D., Taylor A. Learning-disabled males with a fragile X CGG expansion in the upper premutation size range. Pediatrics. 1996;97:122–126. [PubMed] [Google Scholar]
- 43.Chonchaiya W., Schneider A., Hagerman R.J. Fragile X: a family of disorders. Adv. Pediatr. 2009;56:165–186. doi: 10.1016/j.yapd.2009.08.008. doi:10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tassone F., Hagerman R.J., Taylor A.K., Gane L.W., Godfrey T.E., Hagerman P.J. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000;66:6–15. doi: 10.1086/302720. doi:10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Todd P.K., Oh S.Y., Krans A., Pandey U.B., Di Prospero N.A., Min K.T., Taylor J.P., Paulson H.L. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010;6:e1001240. doi: 10.1371/journal.pgen.1001240. doi:10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., Glover K., Bentley D., Hagerman P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. doi:10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaufmann W.E., Abrams M.T., Chen W., Reiss A.L. Genotype, molecular phenotype, and cognitive phenotype: correlations in fragile X syndrome. Am. J. Med. Genet. 1999;83:286–295. doi:10.1002/(SICI)1096-8628(19990402)83:4<286::AID-AJMG10>3.0.CO;2-H. [PubMed] [Google Scholar]
- 48.Zumwalt M., Ludwig A., Hagerman P.J., Dieckmann T. Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol. 2007;4:93–100. doi: 10.4161/rna.4.2.5039. doi:10.4161/rna.4.2.5039. [DOI] [PubMed] [Google Scholar]
- 49.Primerano B., Tassone F., Hagerman R.J., Hagerman P., Amaldi F., Bagni C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8:1482–1488. [PMC free article] [PubMed] [Google Scholar]
- 50.Ludwig A.L., Hershey J.W., Hagerman P.J. Initiation of translation of the FMR1 mRNA Occurs predominantly through 5′-end-dependent ribosomal scanning. J. Mol. Biol. 2011;407:21–34. doi: 10.1016/j.jmb.2011.01.006. doi:10.1016/j.jmb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tassone F., Hagerman R.J., Garcia-Arocena D., Khandjian E.W., Greco C.M., Hagerman P.J. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J. Med. Genet. 2004;41:e43. doi: 10.1136/jmg.2003.012518. doi:10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y., Jin P. RNA-mediated neurodegeneration in fragile X-associated tremor/ataxia syndrome. Brain Res. 2012;1462:112–117. doi: 10.1016/j.brainres.2012.02.057. doi:10.1016/j.brainres.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagerman P.J. Current Gaps in Understanding the Molecular Basis of FXTAS. Tremor Other Hyperkinet. Mov. (N Y) 2012;2:pii: 63. doi: 10.7916/D80C4TH0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renoux A.J., Todd P.K. Neurodegeneration the RNA way. Prog. Neurobiol. 2012;97:173–189. doi: 10.1016/j.pneurobio.2011.10.006. doi:10.1016/j.pneurobio.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunningham C.L., Martinez Cerdeno V., Navarro Porras E., Prakash A.N., Angelastro J.M., Willemsen R., Hagerman P.J., Pessah I.N., Berman R.F., Noctor S.C. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum. Mol. Genet. 2011;20:64–79. doi: 10.1093/hmg/ddq432. doi:10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y., Tassone F., Berman R.F., Hagerman P.J., Hagerman R.J., Willemsen R., Pessah I.N. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010;19:196–208. doi: 10.1093/hmg/ddp479. doi:10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berman R.F., Willemsen R. Mouse models of fragile x-associated tremor ataxia. J. Investig. Med. 2009;57:837–841. doi: 10.231/JIM.0b013e3181af59d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qin M., Entezam A., Usdin K., Huang T., Liu Z.H., Hoffman G.E., Smith C.B. A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol. Dis. 2011;42:85–98. doi: 10.1016/j.nbd.2011.01.008. doi:10.1016/j.nbd.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao Z., Hulsizer S., Tassone F., Tang H.T., Hagerman R.J., Rogawski M.A., Hagerman P.J., Pessah I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum. Mol. Genet. 2012;21:2923–2935. doi: 10.1093/hmg/dds118. doi:10.1093/hmg/dds118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G.E., Grabczyk E., Nussbaum R.L., Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. doi:10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lokanga R.A., Entezam A., Kumari D., Yudkin D., Qin M., Smith C.B., Usdin K. Somatic expansion in mouse and human carriers of Fragile X premutation alleles. Hum. Mutat. 2012 doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh K., Gaur P., Prasad S. Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol. Biol. Rep. 2007;34:173–181. doi: 10.1007/s11033-006-9032-8. doi:10.1007/s11033-006-9032-8. [DOI] [PubMed] [Google Scholar]
- 63.Hanson J.E., Madison D.V. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J. Neurosci. 2007;27:4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. doi:10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalantry S., Purushothaman S., Bowen R.B., Starmer J., Magnuson T. Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature. 2009;460:647–651. doi: 10.1038/nature08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hadjantonakis A.K., Gertsenstein M., Ikawa M., Okabe M., Nagy A. Non-invasive sexing of preimplantation stage mammalian embryos. Nat. Genet. 1998;19:220–222. doi: 10.1038/893. doi:10.1038/893. [DOI] [PubMed] [Google Scholar]
- 66.Bhakar A.L., Dolen G., Bear M.F. The pathophysiology of fragile X (and what it teaches us about synapses) Annu. Rev. Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. doi:10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. doi:10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z., Zhang Y., Ku L., Wilkinson K.D., Warren S.T., Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. doi:10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laggerbauer B., Ostareck D., Keidel E.M., Ostareck-Lederer A., Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. doi:10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 70.Narayanan U., Nalavadi V., Nakamoto M., Pallas D.C., Ceman S., Bassell G.J., Warren S.T. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. doi:10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narayanan U., Nalavadi V., Nakamoto M., Thomas G., Ceman S., Bassell G.J., Warren S.T. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 2008;283:18478–18482. doi: 10.1074/jbc.C800055200. doi:10.1074/jbc.C800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Todd P.K., Malter J.S., Mack K.J. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res. Mol. Brain Res. 2003;110:267–278. doi: 10.1016/s0169-328x(02)00657-5. doi:10.1016/S0169-328X(02)00657-5. [DOI] [PubMed] [Google Scholar]
- 73.Siomi H., Choi M., Siomi M.C., Nussbaum R.L., Dreyfuss G. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 1994;77:33–39. doi: 10.1016/0092-8674(94)90232-1. doi:10.1016/0092-8674(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 74.Waung M.W., Pfeiffer B.E., Nosyreva E.D., Ronesi J.A., Huber K.M. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. doi:10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Merlin L.R., Bergold P.J., Wong R.K. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J. Neurophysiol. 1998;80:989–993. doi: 10.1152/jn.1998.80.2.989. [DOI] [PubMed] [Google Scholar]
- 76.Nakamoto M., Nalavadi V., Epstein M.P., Narayanan U., Bassell G.J., Warren S.T. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl Acad. Sci. USA. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. doi:10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hunsaker M.R., Kim K., Willemsen R., Berman R.F. CGG trinucleotide repeat length modulates neural plasticity and spatiotemporal processing in a mouse model of the fragile X premutation. Hippocampus. 2012;12:2260–2275. doi: 10.1002/hipo.22043. doi:10.1002/hipo.22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu J., Koscielska K.A., Cao Z., Hulsizer S., Grace N., Mitchell G., Nacey C., Githinji J., McGee J., Garcia-Arocena D., et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet. 2012;21:3795–3805. doi: 10.1093/hmg/dds207. doi:10.1093/hmg/dds207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cornish K., Kogan C., Turk J., Manly T., James N., Mills A., Dalton A. The emerging fragile X premutation phenotype: evidence from the domain of social cognition. Brain Cogn. 2005;57:53–60. doi: 10.1016/j.bandc.2004.08.020. doi:10.1016/j.bandc.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 80.Kogan C.S., Turk J., Hagerman R.J., Cornish K.M. Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: a controlled study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:859–872. doi: 10.1002/ajmg.b.30685. doi:10.1002/ajmg.b.30685. [DOI] [PubMed] [Google Scholar]
- 81.Hocking D.R., Kogan C.S., Cornish K.M. Selective spatial processing deficits in an at-risk subgroup of the fragile X premutation. Brain Cogn. 2012;79:39–44. doi: 10.1016/j.bandc.2012.02.005. doi:10.1016/j.bandc.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 82.Hessl D., Wang J.M., Schneider A., Koldewyn K., Le L., Iwahashi C., Cheung K., Tassone F., Hagerman P.J., Rivera S.M. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biol. Psychiatry. 2011;70:859–865. doi: 10.1016/j.biopsych.2011.05.033. doi:10.1016/j.biopsych.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hessl D., Rivera S., Koldewyn K., Cordeiro L., Adams J., Tassone F., Hagerman P.J., Hagerman R.J. Amygdala dysfunction in men with the fragile X premutation. Brain. 2007;130:404–416. doi: 10.1093/brain/awl338. doi:10.1093/brain/awl338. [DOI] [PubMed] [Google Scholar]
- 84.Hessl D., Tassone F., Loesch D.Z., Berry-Kravis E., Leehey M.A., Gane L.W., Barbato I., Rice C., Gould E., Hall D.A., et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am. J. Med. Genet B Neuropsychiatr. Genet. 2005;139B:115–121. doi: 10.1002/ajmg.b.30241. doi:10.1002/ajmg.b.30241. [DOI] [PubMed] [Google Scholar]
- 85.Hunter J.E., Allen E.G., Abramowitz A., Rusin M., Leslie M., Novak G., Hamilton D., Shubeck L., Charen K., Sherman S.L. Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav. Genet. 2008;38:493–502. doi: 10.1007/s10519-008-9214-3. doi:10.1007/s10519-008-9214-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Loesch D.Z., Huggins R.M., Bui Q.M., Taylor A.K., Pratt C., Epstein J., Hagerman R.J. Effect of fragile X status categories and FMRP deficits on cognitive profiles estimated by robust pedigree analysis. Am. J. Med. Genet. A. 2003;122A:13–23. doi: 10.1002/ajmg.a.20214. doi:10.1002/ajmg.a.20214. [DOI] [PubMed] [Google Scholar]
- 87.Hunsaker M.R., Wenzel H.J., Willemsen R., Berman R.F. Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav. Neurosci. 2009;123:1315–1324. doi: 10.1037/a0017616. doi:10.1037/a0017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagerman R., Lauterborn J., Au J., Berry-Kravis E. Fragile X syndrome and targeted treatment trials. Results Probl. Cell Differ. 2012;54:297–335. doi: 10.1007/978-3-642-21649-7_17. doi:10.1007/978-3-642-21649-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tassone F., Pan R., Amiri K., Taylor A.K., Hagerman P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. doi:10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jakawich S.K., Nasser H.B., Strong M.J., McCartney A.J., Perez A.S., Rakesh N., Carruthers C.J., Sutton M.A. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. doi:10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hollingsworth E.B., McNeal E.T., Burton J.L., Williams R.J., Daly J.W., Creveling C.R. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J. Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]