

Abstract
Hirschsprung disease (HSCR) is a human congenital disorder, defined by the absence of ganglia from variable lengths of the colon. These ganglia comprise the enteric nervous system (ENS) and are derived from migratory neural crest cells (NCCs). The inheritance of HSCR is complex, often non-Mendelian and characterized by variable penetrance. Although extensive research has identified many key players in the pathogenesis of Hirschsprung disease, a large number of cases remain genetically undefined. Therefore, additional unidentified genes or modifiers must contribute to the etiology and pathogenesis of Hirschsprung disease. We have discovered that Tcof1 may be one such modifier. Haploinsufficiency of Tcof1 in mice results in a reduction of vagal NCCs and their delayed migration along the length of the gut during early development. This alone, however, is not sufficient to cause colonic aganglionosis as alterations in the balance of NCC proliferation and differentiation ensures NCC colonize the entire length of the gut of Tcof1+/− mice by E18.5. In contrast, Tcof1 haploinsufficiency is able to sensitize Pax3+/− mice to colonic aganglionosis. Although, Pax3 heterozygous mice do not show ENS defects, compound Pax3;Tcof1 heterozygous mice exhibit cumulative apoptosis which severely reduces the NCC population that migrates into the foregut. In addition, the proliferative capacity of these NCC is also diminished. Taken together with the opposing effects of Pax3 and Tcof1 on NCC differentiation, the synergistic haploinsufficiency of Tcof1 and Pax3 results in colonic aganglionosis in mice and may contribute to the pathogenesis of Hirschsprung disease.
INTRODUCTION
One of the many challenges during embryonic development is to ensure complete formation of a nervous system along the entire length of the gastrointestinal tract. This enteric nervous system (ENS) is part of the peripheral nervous system that is required to regulate the secretory and peristaltic activity of the gut (1) and the enteric ganglia are derived from migratory progenitor cells called the neural crest. Neural crest cells (NCCs), originating predominantly within the vagal region (somites 1–7) of the neural tube (2), migrate extensively throughout the embryo and along the length of the gut wall during which their survival, proliferation and differentiation are intrinsically co-ordinated and integrated with the growth of the gut tube (1,3). Failure to form a complete ENS results in the absence of enteric ganglia (aganglionosis), which is the underlying cause of the human disorder termed Hirschsprung disease (HSCR) [for review see (4)].
HSCR is a complex multigenic disease that affects 1:5000 live births (5). Mutations have been identified in over a dozen different genes with the receptor tyrosine kinase, RET being the central player, responsible for 50% of familial and around 20% of spontaneous cases (6). RET and endothelin signaling pathway components, SOX10, ZEB2 and PHOX2B transcription factors, NRG1, KBP and L1-CAM have also been implicated in the disease (1,3,4,7–10). In addition, colonic aganglionosis has been associated with another disorder of NCC development called Waardenburg syndrome (WS), (11). The paired-box containing transcription factor, PAX3 is involved in type I and type III WS (12–14) and homozygous loss of Pax3 in Splotch mice results in the absence of enteric ganglia along the intestinal wall, an early feature of HSCR (15). The effects of Pax3 loss-of-function in these mice have been attributed to its regulation of the main HSCR-associated gene, RET (15–17).
Despite the identification of many of the key players in HSCR, the genetic basis of many HSCR cases remains unknown. The inheritance of HSCR is complex, not always Mendelian and often characterized by variable penetrance (5). Therefore, other genes or modifiers must also be involved in the etiology and pathogenesis of the disease (10). We recently discovered in mouse models that haploinsufficiency of Tcof1, which encodes the nucleolar protein Treacle, mimics many of the early features of HSCR (18). p53-dependent neuroepithelial cell death reduces the numbers of NCCs that delaminate from the neural tube and migrate toward and into the foregut in these mice (19). Pax3 loss-of-function also results in p53-dependent neuroepithelial apoptosis and subsequent pathogenesis of intestinal aganglionosis. This common mechanism implied Tcof1 and Pax3 may function synergistically in the formation of the ENS. Consequently, we discovered in compound Pax3;Tcof1 heterozygous mice that Tcof1 acts as a modifier of Pax3 during NCC development and in the pathogenesis of colonic aganglionosis, which has important implications for our understanding of HSCR in humans.
RESULTS
Haploinsufficiency of TCOF1 is associated with the craniofacial neurocristopathy known as Treacher Collins syndrome (20). In mouse models of this disorder, Tcof1 loss-of-function has been shown to induce p53-dependent cell death within the neuroepithelium (19,21). This apoptotic elimination diminishes the pool of NCCs which are needed to generate most of the bone, cartilage and connective tissues of the head and face. However, NCCs are born along nearly the entire length of the neuraxis, and the vagal and trunk NCCs are particularly important for organogenesis and peripheral nervous system development. Consistent with this observation, we recently identified a reduction in the vagal NCC progenitor cell pool within Tcof1+/− mice, together with a delay in their migration along the length of the gut which affects ENS development and mimics many of the early features of HSCR (18). Loss of Tcof1 alone, however, is insufficient to cause colonic aganglionosis as a complete ENS forms along the entire length of the gut by birth (18). Similar to Tcof1+/− haploinsufficiency, Pax3 loss-of-function also results in p53-mediated neuroepithelial cell death, but in contrast to Tcof1+/− mice, this does result in intestinal aganglionosis in Pax3−/− mice (19,22). In humans, mutations in PAX3 are typically associated with WS and although no direct connection to the rarer Waardenburg-Shah syndrome (WS in combination with Hirschsprung disease) has been identified to date, this raised the question of whether TCOF1 loss-of-function could act as a modifier in the etiology of HSCR. Therefore, we explored whether Tcof1 interacts synergistically with Pax3 in the pathogenesis of colonic aganglionosis.
Pax3 homozygous (Splotch) mutants die during mid-gestation and show complete intestinal aganglionosis similar to that observed in Ret mutant animals (23). In contrast Pax3+/− heterozygous mice are viable and normal except for minor pigmentation defects as evidenced by the presence of a white belly spot (24–26). We intercrossed Pax3 Splotch heterozygous (Pax3+/−) and Tcof1+/− animals to investigate the synergistic effects of compound haploinsufficiency of these genes on the development of the ENS. For simplicity, animals that contain wild-type Pax3 and Tcof1 alleles herein will be referred to as Pax3+/+, those that are wild-type for Tcof1 but heterozygous for Pax3 will be denoted Pax3+/−. Animals that are heterozygous for Tcof1 but wild-type at the Pax3 locus are named Tcof1+/−. Mice that are heterozygous at both loci will be described as Pax3;Tcof1+/− and animals containing two mutant copies of Pax3 will be labeled Pax3−/−.
E9.5–14.5 Tcof1+/− and Pax3;Tcof1+/− embryos were typically slightly smaller than wild-type and Pax3+/− embryos, however, they did not exhibit any developmental delay. To ensure that any changes in ENS formation were not due to embryonic size differences, we size matched all of the guts used in the experiments described below. To analyze the ENS formation, embryonic day (E) 12.5 size-matched whole guts were co-immunostained with the neural crest marker, p75 and βIII tubulin (TuJ1) to identify neuroblasts and differentiated neurons (Fig. 1). At this stage, NCCs were present within the stomach, small intestine (SI) and cecum, and had advanced along an average of 30% of the total length of the colon in Pax3+/+ and Pax3+/− embryos. NCC numbers and the extent of differentiation as measured by TuJ1 immunostaining appeared equivalent (Fig. 1 and Supplementary Material, Fig. S1). The position of the NCC migration wavefront was co-incident with the rostral most extent of TuJ1 immunostaining but not necessarily the most advanced TuJ1+ cell body. In contrast, in Tcof1+/− embryos, NCCs had only migrated along the length of the SI and were typically halted at the border with the cecum (n = 10/14, Fig. 1 and Supplementary Material, Fig. S1). Furthermore, in almost a third of the guts examined, NCCs had not even reached the end of the SI (n = 4/14). Close examination of these guts revealed that whilst the number of p75+ cells in the stomach and SI appeared similar to those within Pax3+/+ and Pax3+/−embryos, TuJ1 immunostaining revealed fewer differentiated neuronal cell bodies in Tcof1+/− embryos (Fig. 1). Concomitant with reduced neuronal differentiation, there was increased fasciculation of nerve fibers in the SI of Tcof1+/− embryos.
Figure 1.

Delayed NCC migration in E12.5 Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− guts. p75 and TuJ1 immunostaining of E12.5 size-matched whole guts shows NCC have colonized an average of 30% of the colon in Pax3+/+ and Pax3+/− guts; however, there is a successive reduction in the colonization of the gut length from Tcof1+/− to Pax3;Tcof1+/− to Pax3−/− guts. Reduced neuronal differentiation is apparent in Tcof1+/− guts in comparison with other genotypes as seen by fewer TuJ1+ cell bodies. White arrowhead denotes the NCC migration wave-front in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− guts. P, proximal.
The combined haploinsufficiency of Pax3 and Tcof1 (Pax3;Tcof1+/−) resulted in a greater proportion of guts (50 versus 33% for Tcof1+/− embryos) where the NCCs had not advanced to the end of the SI (n = 6/12, Fig. 1 and Supplementary Material, Fig. S1). In addition, fewer NCCs were present within the gut wall of Pax3;Tcof1+/− embryos in comparison with individual Tcof1+/−, Pax3+/− and Pax3+/+ embryos (Fig. 1). Surprisingly though, despite the exacerbated reduction in NCCs, more TuJ1+ cells were observed within the stomach and SI in contrast to Tcof1+/− guts. Immunostaining of E12.5 guts from independent intercrosses of Pax3 splotch heterozygous mice showed that the complete loss of Pax3 (Pax3−/−) resulted in an extensive migration delay of NCCs such that only the stomach contained p75+ cells as has previously been described [Fig. 1 and (15)]. Reduced neuronal differentiation was also apparent in these preparations and the vast majority of TuJ1 immunostaining was restricted to the extrinsic innervation of the stomach (Fig. 1). Whilst the ENS phenotype of the Pax3;Tcof1+/− embryos was not as severe as Pax3−/−, there was a significant increase in the proportion of guts that showed delayed colonization of the SI in comparison with Tcof1+/− embryos. Therefore, a synergistic interaction between Tcof1 and Pax3 increased the severity of ENS defects in comparison with individual Tcof1+/− or Pax3+/− mutants. Interestingly, the mechanism by which these two genes affect NCCs within the gut wall appears to be different as seen by their opposing effects on NCC differentiation.
Examination of E14.5 embryos using TuJ1 revealed that the entire length of the colon contained neural crest-derived cells in Pax3+/+ and Pax3+/− embryos (n = 20 and 22, respectively, Supplementary Material, Fig. S2). In contrast, the migration wavefront was delayed in almost half of the Tcof1+/− guts (n = 9/19, Supplementary Material, Fig. S2). The neuronal cell bodies and nerve fibers were organized into a characteristic network along the SI that appeared to be equivalent to that seen in Pax3+/+ and Pax3+/− guts (Supplementary Material, Fig. S2). Immunostaining of the guts of Pax3;Tcof1+/− embryos, however, revealed a significant delay in the extent of colonization by neural crest derived-cells such that only one of the guts analyzed had cells present at the end of the colon. All of the other guts examined showed a reduction in the migration of neural crest-derived cells (n = 20/21, Supplementary Material, Fig. S2). Indeed, TuJ1+ cells had not even advanced into the colon in some of the guts. Despite this extensive migration delay along the length of the gut, the neuronal network present in the SI was similar to that observed in guts from all of the genotypes analyzed, whilst, the cecum often contained regions devoid of TuJ1 immunostaining. Therefore, as development proceeded, the perturbation of ENS development in Pax3;Tcof1+/− guts became more pronounced such that there was an increase in the incidence and extent of migration delay detected in these embryos compared with Tcof1+/− embryos.
Cell death within the NCC progenitor pool is increased in compound mutants
Reduced colonization of the gut wall by NCCs could result from altered survival or proliferation of the initial progenitor cell pool. Therefore, we co-immunostained 10 µm cryosections of the vagal neural tube (somites 1–7) in E9.5–10.5 embryos from Pax3+/− and Tcof1+/− intercrosses and independent matings of Pax3+/− mice with p75 to mark NCCs and either phosphoHistone H3 or TUNEL to identify mitotic or apoptotic cells, respectively. Transverse sections clearly showed that the neural tube of Tcof1+/− embryos is smaller than that of Pax3+/+, Pax3+/− and Pax3−/− embryos, whilst the neural tube of compound Pax3;Tcof1 heterozygotes were even further reduced in size (Fig. 2). Despite this size difference, no developmental delay was observed in these embryos. Analysis of the p75+ cells that had migrated toward and into the foregut demonstrated that there was no statistical difference in NCC numbers between Pax3+/+ and Pax3+/− embryos (Fig. 3 and Supplementary Material, Table S1). However, there was a progressive 40, 67 and 87% reduction of NCCs in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos, respectively (Fig. 3 and Supplementary Material, Table S1). In addition, there was a significant decrease in the proportion of mitotic NCCs in Pax3;Tcof1+/− embryos when compared with Pax3+/+ counterparts (8.1 ± 1.9 versus 11.1 ± 1.4%, P = 0.02; Figs 2 and 3 and Supplementary Material, Table S1) as measured through dual p75, phosphoHistone H3 immunostaining. The reduction in mitotic NCCs was even more pronounced in Pax3−/− embryos (4.3 ± 1 versus 11.1 ± 1.4%, P = 3×10−5; Figs 2 and 3 and Supplementary Material, Table S1). This demonstrates the relative importance of both Tcof1 and Pax3 in regulating vagal NCC proliferation.
Figure 2.

Reduced proliferation within the NCC that migrate toward and into the foregut in Pax3;Tcof1+/− and Pax3−/− embryos. The mitotic index of NCC was determined by counting pHH3+/p75 double-positive cells in cryosections at the vagal neural tube level (somites 1–7) of Pax3+/+, Pax3+/−, Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos. There is an incremental reduction in the numbers of NCC around the foregut in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos, respectively. White asterisk denotes the severe reduction of p75 immunostaining around the foregut in Pax3;Tcof1+/− embryos and the absence of p75+ NCC around the foregut in Pax3−/− embryos. Nuclei are labeled with DAPI. pHH3, phospho Histone H3.
Figure 3.

Histograms showing smaller progenitor cell pool sizes in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos as a result of reduced proliferative capacity and increased apoptosis. NCC proliferation and apoptosis were determined in 10 μm cryosections of embryos at the vagal neural tube level (somites 1–7). Specifically pHH3+/p75 or TUNEL+/p75 double-positive cells were scored within these embryos. The number of NCC numbers were reduced by 40% in Tcof1+/−, 67% in Pax3;Tcof1+/− and 87% in Pax3−/− embryos, respectively. These reductions were associated with a smaller proliferative capacity and increased apoptosis in Pax3;Tcof1+/− and Pax3−/− embryos only. pHH3, phospho Histone H3.
TUNEL immunostaining revealed extensive apoptosis in the neuroepithelium of Tcof1+/− and Pax3;Tcof1+/− embryos compared with Pax3+/+, Pax3+/− and Pax3−/− embryos (Fig. 4 and Supplementary Material, Table S1). We also identified a small number of TUNEL+ NCCs that had migrated toward and into the foregut. The degree of apoptotic NCCs was equivalent between Pax3+/+, Pax3+/− and Tcof1+/− embryos (Figs 3 and 4 and Supplementary Material, Table S1). In contrast, the percentage of apoptotic NCCs was increased in Pax3;Tcof1+/− embryos (5.9 ± 0.6 versus 2.7 ± 0.8%, P = 8.6×10−5; Fig. 3 and Supplementary Material, Table S1) and further exacerbated in Pax3−/− embryos (7.2 ± 1 versus 2.7 ± 0.8%, P = 1.3×10−6; Fig. 3 and Supplementary Material, Table S1). Taken together, these data show that diminishment of the NCC progenitor cell pool migrating toward and into the gut is caused by neuroepithelial apoptosis in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos. However, this is then further exacerbated by decreased proliferation and a concomitant increase in apoptosis in migrating NCCs within Pax3;Tcof1+/− and Pax3−/− embryos. Indeed, these changes reflect the incremental severity of the effect of loss of these genes on ENS formation observed within the guts of Tcof1+/−, Pax3;Tcof1 compound heterozygote and Pax3−/− mutant embryos.
Figure 4.
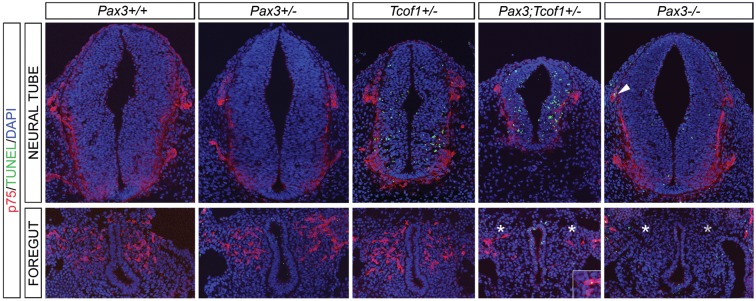
Apoptosis reduces the NCC numbers that migrate toward and into the foregut in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos. Apoptosis was measured in these embryos using TUNEL and p75 immunostaining of cryosections at the vagal neural tube level (somites 1–7). TUNEL-positive cells were detected in the neural tube of Tcof1+/−, Pax3;Tcof1+/− and to a lesser extent in Pax3−/− embryos. In addition increased apoptosis was seen in the NCCs that had migrated toward and into the foregut in Pax3;Tcof1+/− and Pax3−/− embryos, seen in the higher magnification insert in figure and also the arrowhead in the Pax3−/− NT. White asterisk denotes the severe reduction of p75 immunostaining around the foregut in Pax3;Tcof1+/− embryos and the absence of p75+ NCC around the foregut in Pax3−/− embryos. Nuclei are detected with DAPI staining.
Haploinsufficiency of Tcof1 alters the commitment of the progenitor cell pool that migrates into and along the gut
Pax3 has previously been demonstrated to physically interact with the transcription factor Sox10 to regulate the expression of the receptor tyrosine kinase gene, RET (15–17), the major effector gene of HSCR (4,6). Sox10 is expressed in early migrating NCCs and maintains ENS progenitors in a multipotential state (27–30). In contrast, RET expression is initially detected within the NCCs as they reach the foregut and is up-regulated within the cells that become committed to a neuronal fate (29–31). To investigate whether the loss of Pax3 and/or Tcof1 has an effect on the identity of the ENS progenitor cell pool, we co-immunostained 10 µm cryosections of E10 embryos from Pax3+/− and Tcof1+/− intercrosses with Sox10 and p75 and determined that the vast majority of the p75+ NCCs that migrate toward and into the foregut express Sox10 in embryos of all genotypes examined (Supplementary Material, Fig. S3). We then co-immunostained the cryosections with either RET and Sox10 or RET and p75 to determine the proportion of multipotential NCCs within each of the different genotypes. Examination of the sections co-immunostained with Sox10 and RET revealed that haploinsufficiency of Tcof1 reduced the percentage of Sox10+ cells that were co-expressing RET [69.7 ± 10 versus 98 ± 1%, P = 0.02; Fig. 5 and (18)]. We also identified a small but statistically significant reduction in the proportion of RET+ cells-expressing Sox10 that had migrated toward the foregut in Pax3;Tcof1+/− embryos in comparison with their wild-type counterparts (94.4 ± 2.7 versus 98 ± 1%, P = 0.03; Fig. 5). Pax3−/− embryos were not included in these analyses since very few NCCs were seen around the foregut at these stages where RET expression is first detected.
Figure 5.

The ENS progenitor cell pool in Tcof1+/− embryos is less committed than in the other genotypes. Cryosections of E10 foregut immunostained with the ENS progenitor marker Sox10 and the neuronal commitment marker, RET revealed fewer cells had initiated RET expression in Tcof1+/− embryos. Immunostaining of E11.5 cryosections of SIs showed a reduction in the number of p75+ NCC present in the guts of Tcof1+/− embryos expressing RET at both the migration wavefront and along the length of the SI. Nuclei are stained with DAPI.
Since we identified a large reduction in the fraction of NCCs expressing RET in Tcof1+/− embryos compared with wild-type embryos (Fig. 5) and we also observed reduced differentiation of neural crest-derived cells within the gut of these embryos at E12.5 as determined by TuJ1 immunostaining (Fig. 1), we examined the expression of RET within the p75+ NCCs present in the SI. For this analysis, we performed co-immunostaining of 10 µm cryosections of E11.5 guts with p75 and RET (Fig. 5). The percentage of RET+ NCCs was quantified both at the migration wavefront and along the SI. There were reduced numbers of NCCs expressing RET at the migration wavefront in the guts of Tcof1+/− embryos (86.8 ± 8 versus 98.2 ± 1%, P = 0.002; Fig. 5) and to a lesser extent along the SI (95 ± 3 versus 99 ± 1%, P = 0.007; Fig. 5). However, this effect was abolished when Tcof1 haploinsufficiency was combined with the loss of Pax3 (Fig. 5) since similar proportions of RET+/p75+ NCCs were counted within Pax3;Tcof+/− and Pax3+/+ guts. These data support the equivalent extent of NCC differentiation observed within the intestines of Pax3;Tcof1+/− mice at E11.5 compared with their wild-type counterparts (Fig. 5).
In all, Pax3;Tcof1+/− and Pax3−/− embryos contain fewer NCCs that migrate toward and into the gut than Tcof1+/− embryos. Whilst we observed a small reduction in the proportion of Sox10+ cells expressing RET that had migrated toward the foregut in Pax3;Tcof1+/− embryos, the extent of differentiation of NCCs within the intestines was similar to wild-type controls. In contrast, the ENS progenitor cell pool in Tcof1+/− embryos was consistently less committed than in the other genotypes which could account for the reduced neuronal differentiation observed in these guts. These results suggest that Pax3 functions to maintain the proliferative capacity of NCCs, whilst Tcof1 plays a role in regulating the differentiation of these cells since the combined loss of one copy of each of the genes in Pax3;Tcof1+/− embryos balances out these two individual processes to the levels observed in wild-type embryos.
Loss of Tcof1 sensitizes Pax3+/− mice to colonic aganglionosis
To ascertain whether the early reduction in the progenitor cell pool together with delayed NCC migration along the gut wall we observed at E12.5 and E14.5 in Tcof1+/− and Pax3;Tcof1+/− embryos resulted in terminal aganglionosis later in development, we stained E18.5 guts with TuJ1. An ENS was present along the entire length of the gut in Pax3+/+, Pax3+/− and Tcof1+/−embryos (Fig. 6), while variable lengths (between 5 and 100%) of the colon were aganglionic in nearly all Pax3;Tcof1+/− embryos (n = 12/15; Fig. 6). Pax3−/− animals die around E13 and thus could not be included in these studies. Nonetheless, since individual Pax3+/− and Tcof1+/− heterozygous mutations do not result in colonic aganglionosis, but compound Pax3;Tcof1+/− mutations do, this indicates that Tcof1 acts synergistically with Pax3 in ENS formation and may act as a modifier in the pathogenesis of HSCR.
Figure 6.
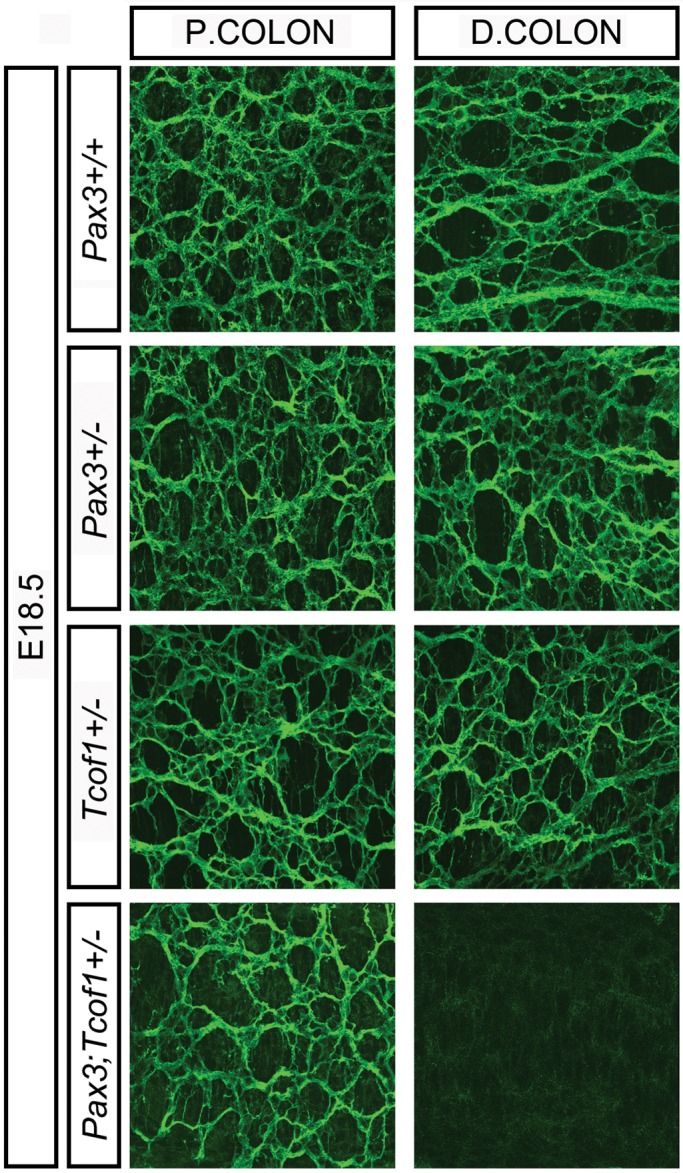
A complete ENS network is seen in Tcof1+/− guts at E18.5 while Pax3;Tcof1+/− guts show colonic aganglionosis. TuJ1 immunostaining of whole guts at E18.5 shows the ENS network within the proximal and distal colon of E18.5 Pax3+/+, Pax3+/− and Tcof1+/− guts. The distal colon is aganglionic in the majority of Pax3;Tcof1+/− guts. P, proximal; D, distal.
Altered NCC intrinsic properties result in complete colonization of the gut wall in Tcof1+/− compared with Pax;Tcof1+/− embryos
The complete colonization of the gut wall observed at E18.5 in Tcof1+/− embryos compared with the variable aganglionosis observed in Pax3;Tcof1+/− embryos could arise as a consequence of differences in NCC intrinsic properties. To determine if this was indeed the case, we analyzed NCC proliferation and neuronal differentiation at E13.5 when NCCs were advancing along the colon. The percentage of proliferating NCCs was scored by co-immunostaining whole guts with p75 and an antibody to detect BrdU that had been previously injected into pregnant females. NCCs were analyzed at the migration wavefront and along the SI. We identified a reduction in the proportion of proliferating NCCs at the migration wavefront between Pax3+/− and Pax3+/+ embryos (29.8 ± 8 versus 40.9 ± 3%, P = 0.01; Fig. 7 and Supplementary Material, Table S1). In contrast, we discovered an increase in the proportion of proliferating NCCs at the migration wavefront in Tcof1+/− embryos compared with their wild-type counterparts (49.8 ± 6.1 versus 40.9 ± 3%, P = 0.03; Fig. 7 and Supplementary Material, Table S1). Surprisingly, the proportion of proliferating NCCs at the migration wavefront in compound Pax3;Tcof1 heterozygotes was intermediate between these two results and therefore was unchanged in relation to Pax3+/+ embryos (35.9 ± 5 versus 40.9 ± 3%; Fig. 7 and Supplementary Material, Table S1). No statistical difference was observed in the percentages of BrdU+ NCC along the SI between each of these genotypes (20.9 ± 4.7%, 22 ± 4%, 27.2 ± 2%, 24.2 ± 9% for Pax3+/+, Pax3+/−, Tcof1+/− and Pax3;Tcof1+/−, respectively; Fig. 7 and Supplementary Material, Table S1). However, there was a slight increase in the percentage of BrdU+ NCCs in Tcof1+/− embryos compared with the wild-type counterparts (27.2 ± 2 versus 20.9 ± 4.7%, P = 0.05; Fig. 7 and Supplementary Material, Table S1). Of note here is that the proportion of dividing NCCs is not significantly different between Pax3;Tcof1+/− and wild-type guts despite the fact that the NCCs may have been in different positions along the length of the gut when they were analyzed. These data suggest that although there is an increase in the proportion of proliferating NCCs between the migration wavefront in the colon and along the SI in wild-type embryos, there appear to be equivalent percentages of proliferating NCCs along the SI at this stage.
Figure 7.

Increased proliferation at the migration wavefront and reduced neuronal differentiation along the gut in E13.5 Tcof1+/− guts. E13.5 whole guts were immunostained with p75 (red) and either BrdU (green in the upper panel) or the neuronal marker, Hu (green in the lower panel). Dividing cells can be identified in the upper panel by the presence of green staining in the nuclei of p75+ cells. We detected an increase in NCC proliferation specifically at the migration wavefront in Tcof1+/− guts and a small decrease at this position in Pax3+/− guts. Pax3;Tcof1+/− guts had comparable levels of proliferation to wild-type guts. Reduced neuronal differentiation was seen throughout the intestines in only Tcof1+/− embryos compared with all other genotypes by co-immunostaining for p75 and the neuronal marker Hu (lower panel). White arrowheads denote the examples of double-positive cells in each panel.
The extent of neuronal differentiation was recorded at this stage by co-immunostaining whole guts with p75 and the neuronal marker, Hu. We detected a significant decrease in the percentage of NCCs expressing Hu between the migration wavefront in the colon and along the SI within wild-type embryos (20.8 ± 4 versus 14.9 ± 1%, P = 0.01; Fig. 7 and Supplementary Material, Table S1). In addition, we observed considerably reduced neuronal differentiation in Tcof1+/− guts compared with wild types at both the NCC migration wavefront (10.9 ± 1 versus 14.9 ± 1%, P = 0.003; Fig. 7 and Supplementary Material, Table S1) and along the SI (15.9 ± 3 versus 20.8 ± 4%, P = 0.04; Fig. 7 and Supplementary Material, Table S1). No significant change in differentiation was detected in the guts in any of the other genotypes compared with wild-type embryos (Fig. 7 and Supplementary Material, Table S1). Therefore, the formation of an ENS along the entire length of the gut in Tcof1+/− embryos at E18.5 could be achieved by the combined effect of increased NCC proliferation at the migration wavefront with reduced neuronal differentiation throughout the gut. In contrast in compound Pax3;Tcof1+/− heterozygous mutants, proliferation at the wavefront is decreased, while differentiation at the wavefront is increased relative to Tcof1+/− embryos returning the levels of proliferation and differentiation to that observed in wild-type guts. Therefore, the significant loss of vagal NCCs in Pax3;Tcof1+/− embryos cannot be compensated for by changes in NCC intrinsic properties during the migration of these cells along the gut wall in these embryos. This subtle shift in the delicate balance between proliferation and differentiation at the wavefront results in the pathogenesis of colonic aganglionosis. Thus, complete NCC colonization of the gut and formation of an ENS along the entire length of the gut requires very precise regulation of progenitor cell proliferation and differentiation. The mechanisms that regulate these processes appear to be dependent upon a critical number of NCCs present within the gut wall since reducing the number of vagal NCCs to <40% of the wild-type levels as observed in Pax3;Tcof1+/− embryos results in colonic aganglionosis.
DISCUSSION
In this study, we have identified Tcof1 as a novel potential modifier of Pax3 in the etiology and pathogenesis of colonic aganglionosis. Tcof1+/− mice exhibit many of the early developmental features of aganglionosis characteristic of other animal models of HSCR. Moreover, NCCs in Tcof1+/− mice are still able to complete the formation of the ENS. Similarly, a complete ENS is observed in Pax3+/− mice. In contrast, the loss of Tcof1 sensitizes Pax3+/− mice to colonic aganglionosis. We show that neuroepithelial apoptosis reduces the progenitor cell pool in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− mice relative to controls, resulting in delayed NCC migration along the gut at E12.5 and E14.5. However, increased cell death and reduced proliferation within the NCCs that migrate toward and into the foregut in Pax3;Tcof1+/− and Pax3−/− embryos further exacerbate this phenotype.
p53-dependent cell death underlies the apoptosis observed in both Tcof1+/− and Pax3−/− mutant mice. Indeed, p53 mRNA and/or protein levels can be altered by the loss of either of these genes (19,22,32–34). In Tcof1+/− mice, apoptosis is restricted to the neuroepithelium where it diminishes the generation of NCCs thereby resulting in craniofacial and ENS defects (19; and our data). In contrast, Pax3;Tcof1+/− and Pax3−/− embryos exhibit increased cell death in the NCCs that migrate toward and into the foregut, in addition to neuroepithelial apoptosis. Together, these effects result in a cumulative reduction of the migratory progenitor cell pool in these mice which correlates with increased severity of the ENS phenotype observed in Tcof1+/−, Pax3;Tcof1+/− and Pax3−/− embryos, respectively. Therefore, the colonic aganglionosis observed in Pax3;Tcof1+/− mice appears to result from cumulative apoptotic effects within these mice. A similar mechanism has previously been described in other mouse models of HSCR and has been proposed to account for the modulation in the extent of the aganglionosis induced by ENS modifier genes (35–37). Thus, it is clear that cell death needs to be tightly regulated within the embryo during ENS development. Consistent with this observation, inhibiting apoptosis through overexpression of a dominant negative form of caspase-9 within chick embryos is capable of increasing the numbers of vagal NCCs and causing hyperganglionosis within the proximal foregut (38).
Complete colonization of the entire length of the gut has also been shown to be affected by differences in intrinsic proliferative capacities within NCCs (39,40). The size of the migratory progenitor cell pool that migrates toward and into the foregut can be influenced by regulating NCC proliferation. Therefore, the more extensive ENS defect observed in Pax3;Tcof1+/− and Pax3−/− guts compared with Tcof1+/− guts could also be due to the reduced NCC proliferation that we observed in these embryos. However, the milder phenotype observed in Tcof1+/− guts could also be attributed to the reduced proportion of progenitor cells present around the foregut that had initiated RET expression in these mice compared with Pax3;Tcof1+/− embryos. This effect contrasts to what might have been predicted from previous reports showing that RET expression is activated by Pax3 in association with Sox10 (15–17). However, heterozygous loss of Pax3 may not be sufficient to alter the levels of RET expression in these embryos and the changes in RET observed in Tcof1+/− embryos probably reflect the differentiation status of the cells rather than any direct effect on the expression of RET.
Upon entry into the foregut, a sufficient pool of progenitor cells is required to ensure that a functioning ENS is present along the entire bowel wall. This is achieved by balancing the processes of NCC proliferation and differentiation (1,3,41). These processes need to be regulated throughout the entire period of ENS formation since defects have been reported in the absence of Notch signaling and in Sox10;Zfhx1b double mutant mice despite normal initial ENS development along the intestinal wall (41,42). In contrast to these data, we see an early NCC migration delay in Tcof1+/− guts at E12.5 and E14.5 (equivalent to that described in other mouse models of HSCR. However, a normal ENS network is present along the entire length of the colon at birth. The completion of ENS formation is due to increased and sustained proliferation at the NCC migration wavefront together with a concomitant reduction in neuronal differentiation along the length of the gut. These data suggest that there may be an intrinsic mechanism that is capable of ‘sensing’ the number of NCCs within the gut wall. Thus, the degree of proliferation and differentiation can be modulated if the numbers are reduced to a threshold level of around 60% of normal numbers as shown in Tcof1+/− embryos and in more severe cases where the progenitor pool is further reduced by oxidant induced apoptosis (18).
This intrinsic regulation has also been exemplified in mice where Hand2 was deleted in nestin-expressing neural precursor cells. The number of neurons was maintained in these mice by compensatory differentiation of non-nestin-expressing neural precursors (43). In addition, compensation for the loss of specific Ednrb-iCre-expressing enteric progenitor cells has been demonstrated by increasing the proliferation and glial differentiation of the Ednrb-iCre independent population (44). Our data therefore identify reductions >60% of wild-type numbers as a critical threshold level for this intrinsic ‘sensing’ mechanism since the combined loss of one copy of Pax3 and Tcof1 results in colonic aganglionosis in the majority of guts at E18.5. Therefore, when NCC numbers are reduced to levels around 60% of wild-type, NCC cell–cell and NCC cell–environmental interactions must be sufficient to enable the intrinsic ‘sensing’ mechanism to modulate the rates of NCC proliferation and differentiation within the intestines to establish complete ENS formation along the length of the bowel. However, when NCC numbers are reduced <40% of wild-type as observed in Pax3;Tcof1+/− embryos, proliferation and differentiation of NCCs cannot be balanced to adjust for such a significant loss of cells. Hence, the intrinsic ‘sensing’ mechanism is not capable of being activated in these situations and portions of the bowel wall are unable to be colonized by the remaining NCCs.
One of the striking features noted here is that the reduced neuronal differentiation observed in Tcof1+/− embryos appears to be rescued by the additional loss of one copy of Pax3. Expression of Pax3 is restricted to the neuroepithelium and NCCs during early organogenesis (45); however, Pax3 activity diminishes as development proceeds, such that cardiac NCC for example, downregulate Pax3 prior to their arrival at their final target tissue (46,47). Therefore, the effects of loss of Pax3 in Pax3;Tcof1+/− embryos must be established prior to the arrival of NCC within the gut. Pax3 has previously been shown to be required for expansion and maintenance of neural crest stem cells and prevention of terminal differentiation of melanocytes (48–50). Hence, Pax3 may be necessary during normal development to maintain the proliferative capacity of vagal NCCs that contribute to the ENS. The loss of Pax3 would then be expected to impact the size of the progenitor cell population that migrates toward and into the gut. Whilst we did not observe any statistical difference in the numbers of NCC between wild-type and Pax3+/− embryos, there was a large variation amongst the embryos examined. We did, however, detect a dramatic loss of NCC numbers associated with reduced NCC proliferation in both Pax3;Tcof1+/− and Pax3−/− embryos compared with the other genotypes analyzed. These results suggest that Tcof1 and Pax3 play opposing roles in regulating NCC differentiation during normal development and that the effects of loss of Pax3 in the compound mutants may be dominant since the extent of neuronal differentiation in the guts of Pax3;Tcof1+/− embryos is equivalent to wild-type levels despite the extensive initial reduction in NCC number.
Thus, in conclusion we have identified Tcof1 as a novel potential modifier of HSCR since it is able to sensitize Pax3+/− mice to colonic aganglionosis. Tcof1 haploinsufficient mice recapitulate many features described in other mouse models of HSCR including a reduced pool of progenitor cells and their delayed migration along the gut wall. However, NCC proliferation and differentiation are subsequently regulated to maintain a sufficient number of cells capable of colonizing the entire length of the colon later in development. In contrast, compound Pax3;Tcof1+/− mice exhibit colonic aganlionosis, and this severe phenotype results from their synergistic loss-of-function effects on NCC formation and opposing influence on NCC differentiation. Although no common targets other than p53 are currently known for Tcof1 and Pax3, our work sets the stage for future experiments aimed at identifying common downstream partners and pathways. This will further our understanding of the pathogenesis of HSCR and hopefully shed some light on the etiology of current genetically undefined cases of HSCR. Our results therefore highlight the potential role of Tcof1 as a modifier in the etiology of HSCR, and illustrate the cellular and molecular basis of the enteric phenotype in Pax3 and Tcof1 single and combined mutants.
MATERIALS AND METHODS
Mice
The Institutional Animal Care and Use Committee of Stowers Institute for Medical Research approved all animal protocols used in this study. DBA Tcof1+/− mice were intercrossed with Pax3 heterozygous mice (The Jackson Laboratory) maintained on a C57Bl/6 background. Mice were genotyped as previously described (26,51).
Immunohistochemistry
Immunostaining of whole gut was performed following their dissection from the embryo. Guts were fixed in 4% paraformaldehyde in PBS at room temperature (RT) for 2 h. A blocking solution was then applied to the guts (10% heat-inactivated sheep serum in PBS + 0.1% Triton X-100) and they were rocked for 2 h at RT. Primary antibodies (see table) were diluted and blocking solution and incubated with the guts at 4°C overnight. Following rinsing with PBS, secondary antibodies were added to the guts for 4 h at RT (1:500 Alexa, Invitrogen see Table). Guts were mounted in Vectashield with DAPI (Vector Laboratories) prior to their analysis on an LSM5 PASCAL confocal microscope (Carl Zeiss). Adobe Photoshop software was used to compile composite images, brightness and contrast may have been modified.
Immunolabeling of 10 μm cryosections of dissected guts or whole embryos was performed by incubating the slides for 30 min in blocking solution, then immunostaining with primary antibodies for 2h at RT (see Supplementary Material, Table S1). Following PBS washing, the slides were mounted in Vectashield with DAPI (Vector Laboratories). For Sox10 labeling, amplification was performed using DSB-X™ biotin donkey anti-goat IgG (1:100, invitrogen) and Streptavidin Alexa 488 or 568 (1:300, Invitrogen). The in situ Cell Death Detection Kit Fluorescein (Roche) was utilized according to the manufacturer's instructions to detect apoptosis.
BrdU incorporation
Pregnant mice were injected intraperitoneally with BrdU (Sigma) (1 μl/g of animal weight of a 100 mg/ml stock solution). Guts were dissected from the harvested embryos 45 min post-injection. They were then immunostained as described above with p75. Following a 10 min post-fixation in 4% PFA, the slides were and treated with 2 m HCl for 30 min at 37°C before being incubated with the BrdU antibody.
NCC death and proliferation analysis of sections
The extent of NCC apoptosis was scored from embryonic day (E) 9.5–10.5 in 10 μm cryosections of embryos at the vagal neural tube level (somites 1–7). The percentage of TUNEL/p75 double-positive cells was determined in 15 sections of each embryo analyzed. The mitotic index of p75+ NCCs was counted using co-immunostaining with pHH3. Data are the mean ± standard deviation (SD). Statistical analysis was performed with ANOVA and P values of >0.05 were considered not significant.
NCC proliferation and neuronal differentiation analysis in whole guts
The extent of proliferation was determined at the migration wavefront of NCC by counting double-positive BrdU/p75 of the 50 most caudal p75+ cells within 1.5 µm optical sections captured with a 63× lens on an LSM5 PASCAL confocal microscope. Along the SI, cell proliferation was determined by scoring the double-positive cells in a minimum of four regions along the entire length. Neuronal differentiation at E13.5 was examined by immunostaining with Hu and p75+. Data are the mean ± SD. We considered P values of >0.05 not significant.
SUPPLEMENTARY MATERIAL
FUNDING
Research in the Trainor laboratory is supported by the Stowers Institute for Medical Research, March of Dimes (#6FY05-82) and National Institute of Dental and Craniofacial Research (RO1 DE 016082-01). Research in the Dixon laboratory is supported by the Medical Research Council, UK (G81/535) and the Healing Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are greatly appreciative of all members of the Trainor laboratory for their comments and suggestions during the generation of this work. We thank Dr Miles Epstein for the kind gift of the Hu antibody and Melissa Childers for her expertise with the maintenance of mutant mouse lines.
Conflicts of Interest statement. None declared.
REFERENCES
- 1.Heanue T.A., Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat. Rev. Neurosci. 2007;8:466–479. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- 2.Le Douarin N.M., Teillet M.A. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J. Embryol. Exp. Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- 3.Laranjeira C., Pachnis V. Enteric nervous system development: recent progress and future challenges. Auton. Neurosci. 2009;151:61–69. doi: 10.1016/j.autneu.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Amiel J., Sproat-Emison E., Garcia-Barcelo M., Lantieri F., Burzynski G., Borrego S., Pelet A., Arnold S., Miao X., Griseri P., et al. Hirschsprung disease, associated syndromes and genetics: a review. J. Med. Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 5.Belknap W.M. The pathogenesis of Hirschsprung disease. Curr. Opin. Gastroenterol. 2002;18:74–81. doi: 10.1097/00001574-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Parisi M.A., Kapur R.P. Genetics of Hirschsprung disease. Curr. Opin. Pediatr. 2000;12:610–617. doi: 10.1097/00008480-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 7.Gershon M.D. Genes and lineages in the formation of the enteric nervous system. Curr. Opin. Neurobiol. 1997;7:101–109. doi: 10.1016/s0959-4388(97)80127-4. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Barcelo M.M., Tang C.S., Ngan E.S., Lui V.C., Chen Y., So M.T., Leon T.Y., Miao X.P., Shum C.K., Liu F.Q., et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung's disease. Proc. Natl Acad. Sci. USA. 2009;106:2694–2699. doi: 10.1073/pnas.0809630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang C.S., Tang W.K., So M.T., Miao X.P., Leung B.M., Yip B.H., Leon T.Y., Ngan E.S., Lui V.C., Chen Y., et al. Fine mapping of the NRG1 Hirschsprung's disease locus. PloS One. 2011;6:e16181. doi: 10.1371/journal.pone.0016181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace A.S., Anderson R.B. Genetic interactions and modifier genes in Hirschsprung's disease. World J. Gastroenterol. 2011;17:4937–4944. doi: 10.3748/wjg.v17.i45.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pingault V., Ente D., Dastot-Le Moal F., Goossens M., Marlin S., Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 12.Baldwin C.T., Hoth C.F., Macina R.A., Milunsky A. Mutations in PAX3 that cause Waardenburg syndrome type I: ten new mutations and review of the literature. Am. J. Med. Genet. 1995;58:115–122. doi: 10.1002/ajmg.1320580205. [DOI] [PubMed] [Google Scholar]
- 13.Hoth C.F., Milunsky A., Lipsky N., Sheffer R., Clarren S.K., Baldwin C.T. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I) Am. J. Hum. Genet. 1993;52:455–462. [PMC free article] [PubMed] [Google Scholar]
- 14.Tassabehji M., Read A.P., Newton V.E., Harris R., Balling R., Gruss P., Strachan T. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–636. doi: 10.1038/355635a0. [DOI] [PubMed] [Google Scholar]
- 15.Lang D., Chen F., Milewski R., Li J., Lu M.M., Epstein J.A. Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret. J. Clin. Invest. 2000;106:963–971. doi: 10.1172/JCI10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang D., Epstein J.A. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum. Mol. Genet. 2003;12:937–945. doi: 10.1093/hmg/ddg107. [DOI] [PubMed] [Google Scholar]
- 17.Leon T.Y., Ngan E.S., Poon H.C., So M.T., Lui V.C., Tam P.K., Garcia-Barcelo M.M. Transcriptional regulation of RET by Nkx2–1, Phox2b, Sox10, and Pax3. J. Pediatr. Surg. 2009;44:1904–1912. doi: 10.1016/j.jpedsurg.2008.11.055. [DOI] [PubMed] [Google Scholar]
- 18.Barlow A.J., Dixon J., Dixon M., Trainor P.A. Balancing neural crest cell intrinsic processes with those of the microenvironment in Tcof1 haploinsufficient mice enables complete enteric nervous system formation. Hum. Mol. Genet. 2012;21:1782–1793. doi: 10.1093/hmg/ddr611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones N.C., Lynn M.L., Gaudenz K., Sakai D., Aoto K., Rey J.P., Glynn E.F., Ellington L., Du C., Dixon J., et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 1996;12:130–136. doi: 10.1038/ng0296-130. [DOI] [PubMed] [Google Scholar]
- 21.Dixon J., Jones N.C., Sandell L.L., Jayasinghe S.M., Crane J., Rey J.P., Dixon M.J., Trainor P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl Acad. Sci. USA. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pani L., Horal M., Loeken M.R. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuchardt A., D'Agati V., Larsson-Blomberg L., Costantini F., Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 24.Auerbach R. Analysis of the developmental effects of a lethal mutation in the house mouse. J. Exp. Zool. 1954;127:305–329. [Google Scholar]
- 25.Epstein D.J., Vekemans M., Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- 26.Epstein D.J., Vogan K.J., Trasler D.G., Gros P. A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant. Proc. Natl. Acad. Sci. USA. 1993;90:532–536. doi: 10.1073/pnas.90.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paratore C., Eichenberger C., Suter U., Sommer L. Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Hum. Mol. Genet. 2002;11:3075–3085. doi: 10.1093/hmg/11.24.3075. [DOI] [PubMed] [Google Scholar]
- 28.Kim J., Lo L., Dormand E., Anderson D.J. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 29.Bondurand N., Natarajan D., Barlow A., Thapar N., Pachnis V. Maintenance of mammalian enteric nervous system progenitors by SOX10 and endothelin 3 signalling. Development. 2006;133:2075–2086. doi: 10.1242/dev.02375. [DOI] [PubMed] [Google Scholar]
- 30.Bondurand N., Natarajan D., Thapar N., Atkins C., Pachnis V. Neuron and glia generating progenitors of the mammalian enteric nervous system isolated from foetal and postnatal gut cultures. Development. 2003;130:6387–6400. doi: 10.1242/dev.00857. [DOI] [PubMed] [Google Scholar]
- 31.Durbec P.L., Larsson-Blomberg L.B., Schuchardt A., Costantini F., Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- 32.Morgan S.C., Lee H.Y., Relaix F., Sandell L.L., Levorse J.M., Loeken M.R. Cardiac outflow tract septation failure in Pax3-deficient embryos is due to p53-dependent regulation of migrating cardiac neural crest. Mech. Dev. 2008;125:757–767. doi: 10.1016/j.mod.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phelan S.A., Ito M., Loeken M.R. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–1197. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- 34.Underwood T.J., Amin J., Lillycrop K.A., Blaydes J.P. Dissection of the functional interaction between p53 and the embryonic proto-oncoprotein PAX3. FEBS. Lett. 2007;581:5831–5835. doi: 10.1016/j.febslet.2007.11.056. [DOI] [PubMed] [Google Scholar]
- 35.Maka M., Stolt C.C., Wegner M. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev. Biol. 2005;277:155–169. doi: 10.1016/j.ydbio.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Stanchina L., Baral V., Robert F., Pingault V., Lemort N., Pachnis V., Goossens M., Bondurand N. Interactions between Sox10, Edn3 and Ednrb during enteric nervous system and melanocyte development. Dev. Biol. 2006;295:232–249. doi: 10.1016/j.ydbio.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 37.Wallace A.S., Schmidt C., Schachner M., Wegner M., Anderson R.B. L1cam acts as a modifier gene during enteric nervous system development. Neurobiol. Dis. 2010;40:622–633. doi: 10.1016/j.nbd.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Wallace A.S., Barlow A.J., Navaratne L., Delalande J.M., Tauszig-Delamasure S., Corset V., Thapar N., Burns A.J. Inhibition of cell death results in hyperganglionosis: implications for enteric nervous system development. Neurogastroenterol Motil. 2009;21:768–e49. doi: 10.1111/j.1365-2982.2009.01309.x. [DOI] [PubMed] [Google Scholar]
- 39.Barlow A.J., Wallace A.S., Thapar N., Burns A.J. Critical numbers of neural crest cells are required in the pathways from the neural tube to the foregut to ensure complete enteric nervous system formation. Development. 2008;135:1681–1691. doi: 10.1242/dev.017418. [DOI] [PubMed] [Google Scholar]
- 40.Zhang D., Brinas I.M., Binder B.J., Landman K.A., Newgreen D.F. Neural crest regionalisation for enteric nervous system formation: implications for Hirschsprung's disease and stem cell therapy. Dev. Biol. 2010;339:280–294. doi: 10.1016/j.ydbio.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 41.Stanchina L., Van de Putte T., Goossens M., Huylebroeck D., Bondurand N. Genetic interaction between Sox10 and Zfhx1b during enteric nervous system development. Dev. Biol. 2010;341:416–428. doi: 10.1016/j.ydbio.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 42.Okamura Y., Saga Y. Notch signaling is required for the maintenance of enteric neural crest progenitors. Development. 2008;135:3555–3565. doi: 10.1242/dev.022319. [DOI] [PubMed] [Google Scholar]
- 43.Lei J., Howard M.J. Targeted deletion of Hand2 in enteric neural precursor cells affects its functions in neurogenesis, neurotransmitter specification and gangliogenesis, causing functional aganglionosis. Development. 2011;138:4789–4800. doi: 10.1242/dev.060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mundell N.A., Plank J.L., LeGrone A.W., Frist A.Y., Zhu L., Shin M.K., Southard-Smith E.M., Labosky P.A. Enteric nervous system specific deletion of Foxd3 disrupts glial cell differentiation and activates compensatory enteric progenitors. Dev. Biol. 2012;363:373–387. doi: 10.1016/j.ydbio.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goulding M.D., Chalepakis G., Deutsch U., Erselius J.R., Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conway S.J., Henderson D.J., Copp A.J. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- 47.Epstein J.A., Li J., Lang D., Chen F., Brown C.B., Jin F., Lu M.M., Thomas M., Liu E., Wessels A., et al. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–1878. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- 48.Conway S.J., Bundy J., Chen J., Dickman E., Rogers R., Will B.M. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc. Res. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- 49.Lang D., Lu M.M., Huang L., Engleka K.A., Zhang M., Chu E.Y., Lipner S., Skoultchi A., Millar S.E., Epstein J.A. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature. 2005;433:884–887. doi: 10.1038/nature03292. [DOI] [PubMed] [Google Scholar]
- 50.Kubic J.D., Young K.P., Plummer R.S., Ludvik A.E., Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008;21:627–645. doi: 10.1111/j.1755-148X.2008.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon J., Brakebusch C., Fassler R., Dixon M.J. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.