

Summary
PURPOSE
Increased interictal concentrations of extracellular hippocampal glutamate have been implicated in the pathophysiology of temporal lobe epilepsy (TLE) in humans. Recent studies suggest that perturbations of the glutamate metabolizing enzymes glutamine synthetase (GS) and phosphate activated glutaminase (PAG) may underlie the glutamate excess in TLE. However, the molecular mechanism of the enzyme perturbations remains unclear. A better understanding of the regulatory mechanisms of GS and PAG could facilitate the discovery of novel therapeutics for TLE.
METHODS
We used in situ hybridization on histological sections to assess the distribution and quantity of mRNA for GS and PAG in subfields of hippocampal formations from: (a) patients with TLE and concomitant hippocampal sclerosis, (b) patients with TLE and no hippocampal sclerosis, and (c) non-epilepsy autopsy subjects.
KEY FINDINGS
GS mRNA was increased by approximately 50% in the CA3 in TLE patients without hippocampal sclerosis vs. in TLE patients with sclerosis and in non-epilepsy subjects. PAG mRNA was increased by more than 100% in the subiculum in both TLE patient categories vs. in non-epilepsy subjects. PAG mRNA was also increased in the CA1, CA2, CA3 and dentate hilus in TLE without hippocampal sclerosis vs. in TLE with sclerosis. Finally, PAG mRNA was increased in the dentate gyrus in TLE with sclerosis vs. in non-epilepsy subjects, and also increased in the hilus in TLE without sclerosis vs. in TLE with sclerosis.
SIGNIFICANCE
These findings demonstrate complex changes in the expression of mRNAs for GS and PAG in the hippocampal formation in TLE, and raise the possibility that both transcriptional and post-transcriptional mechanisms may underlie the regulation of GS and PAG proteins in the epileptic brain.
Keywords: Astrocytes, brain, glutamine synthetase, glutamate-glutamine cycle, phosphate activated glutaminase
Introduction
Temporal lobe epilepsy (TLE) is one of the most common types of localization related epilepsies in humans. Many patients with TLE cannot control their seizures with current antiepileptic drugs, and the available drugs have side effects that limit their use. Improved therapies for TLE are therefore necessary. A better understanding of the cellular and molecular mechanisms underlying TLE is expected to facilitate the development of more efficacious and specific treatments for this disease.
Several studies have suggested that the brain glutamate metabolism is critically involved in the pathophysiology of TLE. For example, systemic or intracranial administration of glutamate or glutamate analogues to laboratory animals frequently causes seizures, neuronal loss and glial changes (Ben-Ari 1985, Hayashi 1954, Lucas & Newhouse 1957, Olney, et al. 1986, Olney, et al. 1972). Co-administration of glutamate antagonists prevents the behavioral seizures and many of the neuropathological features associated with these models (Berg, et al. 1993, Eid, et al. 1995, McNamara & Routtenberg 1995). Furthermore, patients with drug-resistant TLE have remarkably high concentrations of extracellular glutamate in the epileptogenic vs. the non-epileptogenic hippocampal formation between seizures (interictally) (Cavus, et al. 2005). The concentration of extracellular brain glutamate in these patients increases six-fold above the interictal level during a seizure, and the neurotransmitter remains elevated for at least 20 minutes after the cessation of the seizure activity (During & Spencer 1993). A pertinent question therefore is: What causes the glutamate excess in human TLE, and is the brain glutamate metabolism a potential therapeutic target for this disease?
Recent findings by us and others have revealed perturbations in two key glutamate metabolizing enzymes in the brain in patients with TLE – glutamine synthetase (GS) and phosphate activated glutaminase (PAG) (Eid, et al. 2007, Eid, et al. 2004). GS, which catalyzes the formation of glutamine from glutamate and ammonia (Martinez-Hernandez, et al. 1977), is severely deficient in astrocytes in the hippocampal formation in human TLE (Eid, et al. 2004, van der Hel, et al. 2005). This deficiency is most pronounced in patients with TLE and concomitant hippocampal sclerosis, which is characterized by preferential loss of neurons and proliferation of astrocytes in CA1, CA3 and the polymorphic layer of the dentate hilus (Gloor 1991, Sommer 1880). The concentration and activity of PAG, which catalyzes the formation of glutamate and ammonia from glutamine, appears to be increased per neuron in the sclerotic vs. the non-sclerotic hippocampal formation in patients with TLE (Eid, et al. 2007). The concentration of extracellular glutamate is also particularly high in the hippocampal formation in patients with TLE and concomitant hippocampal sclerosis vs. in patients without hippocampal sclerosis (Cavus, et al. 2008, Petroff, et al. 2004).
Based on the above findings, we postulated that the deficiency in hippocampal GS in TLE would slow the metabolism of glutamate to glutamine and lead to accumulation of glutamate in astrocytes and the extracellular space of the brain (Eid, et al. 2004, Eid, et al. 2008b). This postulate is partly based on the known stoichiometry of glutamate transport across the glial plasma membrane, i.e. that rapid metabolism of intracellular glutamate is a prerequisite for efficient glutamate clearance from the extracellular space (Otis & Jahr 1998). We further hypothesized that the sustained extracellular glutamate excess would result in neuronal loss and facilitate the occurrence of spontaneous recurrent seizures (Eid, et al. 2004, Eid, et al. 2008b). Recent animal studies have indeed supported the idea of GS as a causative factor in TLE. Infusion of the GS-antagonist methionine sulfoximine (MSO) into the hippocampus in rats leads to recurrent seizures and neuropathological changes very similar to human TLE (Eid, et al. 2008a, Wang, et al. 2009). Whether the increased PAG in neurons in the sclerotic hippocampus in TLE also results in glutamate excess, brain pathology and seizures, remains to be established.
If the alterations in GS and PAG in TLE are critically involved in the pathogenesis of the disease, then these enzymes may be exploited as novel therapeutic targets for TLE. An understanding of the molecular mechanisms underlying the alterations of GS and PAG in TLE could facilitate the development of such therapies. Thus, we used in situ hybridization to assess the expression of mRNA for GS and PAG in the hippocampal formation in patients with TLE (with and without hippocampal sclerosis) and in non-epilepsy autopsy control subjects. We hypothesize that the perturbed expression of GS and PAG protein in TLE is regulated at the level of gene expression, as reflected by altered expression of mRNAs for GS and PAG.
Methods
Patient Selection, Classification, and Nomenclature
Patients with medically intractable TLE underwent phased presurgical evaluation at the Yale-New Haven Hospital, and those selected for surgery had their hippocampal formation resected according to established procedures (Spencer & Spencer 1991, Spencer, et al. 1984). Informed consent from each patient and institutional approval were obtained for the use of tissue for this project.
The TLE patients were classified into two main groups based on their medical history and examination, brain imaging, EEG, and neuropathology of the resected hippocampal formation (Table 1 and Fig. 1; see also (de Lanerolle, et al. 1992, de Lanerolle, et al. 2003) for details). The first group referred to as MTLE, is characterized by hippocampal sclerosis. This pathology is recognized by an atrophic and hardened hippocampal formation with glial proliferation and extensive (>50%) neuronal loss in CA1, CA3, and the dentate hilus (Fig. 1G – I) (Sommer 1880). Loss of hilar interneurons containing neuropeptide Y, somatostatin, and substance P, is also a feature of MTLE, as well as sprouting of dynorphin-positive axons to the dentate molecular layer (de Lanerolle, et al. 1989, Houser, et al. 1990). MRI typically reveals an atrophic hippocampal formation with an increased T2 signal. Depth electrode EEG recordings indicate that the sclerotic hippocampal formation is critically involved in seizure generation (Mathern, et al. 1997). MTLE used by us corresponds to the nomenclature of mTLE with hippocampal sclerosis (mTLE-HS) used by other investigators (reviewed in (Engel, et al. 2008)). The second group referred to as non-MTLE is recognized by modest to minimal (<25%) neuronal loss throughout all hippocampal subfields and absence of axonal sprouting in the dentate gyrus (Table 1 and Fig. 1D – F). MRI is often unremarkable. Some of the non-MTLE patients may have a tumor, dysplastic lesion or vascular malformation in or near the hippocampal formation, while the rest of the non-MTLE patients have no apparent other pathology. Non-MTLE used by us corresponds to the nomenclature of mTLE without HS, used by other investigators (reviewed in (Engel, et al. 2008)). Non-MTLE patients are highly appropriate controls for MTLE patients because both groups have been: (1) Subjected to a similar regimen of antiepileptic drugs, (2) exposed to extended periods of recurrent seizures, and (3) treated with surgical resection of the hippocampal formation under similar and standardized operative conditions. It is important to control for these factors because each of them may potentially interfere with the expression of GS and PAG. Hippocampal formations obtained at autopsy from patients diagnosed with disorders other than epilepsy were also included as additional controls (Table 1 and Fig. 1A – C).
Table 1.
Characteristics of patients selected for the study
| Patient and classification | Sex | Age (years) | Years since first unprovoked seizure | AEDs at surgery | MRI findings | Pathology |
|---|---|---|---|---|---|---|
| Non-MTLE | ||||||
| 1 | M | 23 | 11 | cbz, phe | Mass in L medial temporal lobe | Anaplastic astrocytoma with oligodendroglial differentiation |
| 2 | F | 33 | 14 | cbz, pri | Mass in L anterior temporal lobe | Low grade astrocytoma |
| 3 | M | 22 | 17 | cbz, czp, vpa | Mass in L temporal lobe | Cellular astrocytoma |
| 4 | F | 7 | 6 | cbz, vpa | -- | Ganglioglioma |
| 5 | F | 30 | 25 | pri | Enlarged L hippocampus with possible tumor in L temporal lobe | Low grade astrocytoma |
| 6 | F | 25 | 14 | clo, pht | R mesial temporal lobe cystic lesion | Ganglioglioma |
| 7 | F | 35 | 20 | pb, pht | Unremarkable | Mild hippocampal gliosis and neuronal loss |
| 8 | M | 39 | 18 | pht | R lateral ventricle enlargement; atrophy and increased signal of R Hippocampus | Mild hippocampal gliosis |
| 9 | F | 43 | 34 | cbz XR, ltg, tpm | Possible tumor in L temporal lobe | Gliosis in white matter of cerebral cortex |
| 10 | F | 50 | 42 | lev | Chiari I malformation | Heterotopic neurons in molecular layer of cerebral cortex; gliosis in amygdala |
| MTLE | ||||||
| 1 | M | 17 | 1 | pht | Atrophic R hemisphere | Hippocampal sclerosis |
| 2 | M | 37 | 31 | cbz | Mucus retention cyst in R temporal lobe | Hippocampal sclerosis |
| 3 | M | 37 | -- | none | -- | Hippocampal sclerosis |
| 4 | M | 50 | -- | none | L hippocampal atrophy | Hippocampal sclerosis |
| 5 | M | 18 | <1 | cbz, czp, pht | Unremarkable | Hippocampal sclerosis |
| 6 | F | 27 | 20 | cbz | Infarct in R occipital lobe status post R occipital lobectomy; R occipital lesion, questionable cavernous hemangioma | Hippocampal sclerosis |
| 7 | M | 16 | 11 | vpa | R mesial temporal atrophy; T2 intensity changes in R anterior hippocampus and amygdala | Hippocampal sclerosis |
| 8 | M | 32 | 29 | pht, pri | L hippocampal atrophy | Hippocampal sclerosis |
| 9 | M | 25 | 16 | dpa, pht, pri | L hippocampal atrophy | Hippocampal sclerosis |
| 10 | F | 44 | 34 | cbz, czp, vpa | Hypo-intense abnormality in L anterior temporal tip; R hippocampal atrophy | Hippocampal sclerosis |
| 11 | F | 24 | 18 | ace, cbz | R hippocampal and R temporal neocortical atrophy | Hippocampal sclerosis |
| 12 | M | 26 | 23 | pht | L hippocampal atrophy | Hippocampal sclerosis |
| 13 | F | 52 | 27 | pht, vpa | R hippocampal atrophy | Hippocampal sclerosis |
| 14 | F | 18 | 7 | cbz, ltg, pht | Unremarkable | Hippocampal sclerosis |
| 15 | M | 22 | -- | none | Infarct in R inferior temporal lobe | Hippocampal sclerosis |
| 16 | F | 22 | 8 | pb, pht | Bilateral hippocampal atrophy; diffuse hemisphere atrophy; L frontal lobe encephalomalacia | Hippocampal sclerosis |
| 17 | F | 14 | 9 | cbz XR, ltg | L mesial temporal sclerosis; increased signal on FLAIR sequence in L hippocampus | Hippocampal sclerosis |
| 18 | F | 54 | 43 | ltg, pri | R mesial temporal sclerosis; increased signal on FLAIR sequence in R hippocampus | Hippocampal sclerosis |
| 19 | M | 15 | 11 | czp, pht, pri | L hippocampal atrophy; signal abnormality in R frontal lobe | Hippocampal sclerosis |
| Patient and classification | Sex | Age (years) | Postmortem interval (hrs) | Cause of death |
|---|---|---|---|---|
| Autopsy | ||||
| 1 | M | 33 | 6 | Adenocarcinoma of lung with widespread metastases |
| 2 | M | 52 | 3 | Myocardial infract, coronary atherosclerosis |
| 3 | F | 49 | 5 | Ruptured berry aneurysm, R posterior inferior cerebellar artery |
| 4 | F | 76 | 4 | Peritonitis |
Abbreviations: ace, acetazolamide; AEDs, antiepileptic drugs; cbz, carbamazepine; clo, cloracepate; czp, clonazepam; dpa, dipropylacetic acid; L, left; ltg, lamotrigine; MTLE, mesial temporal lobe epilepsy; pb, phenobarbital; pht, phenytoin; pri, primidone; R, right; tpm, topiramate; vpa, valproate; XR, extended release. (“—“ indicates data not available).
Figure 1.

Nissl-stained coronal sections of hippocampal formations obtained from representative patients diagnosed with: medical conditions other than epilepsy (autopsy, A–C), non-MTLE (D–F) and MTLE (G–I). Low-power images are shown in the left column (A, D, G), and high power fields from the hilus (polymorphic layer) of the dentate gyrus (framed areas in A, D, G) and CA1 (framed areas in A, D, G) are shown in the middle (B, E, H) and right columns (C, F, I) respectively. The cytoarchitecture of the hippocampal formation is similar in autopsy (A–C) and non-MTLE (D–F) with numerous neurons present in all subfields, including the polymorphic layer of the hilus (arrows in B and E) and pyramidal layer of CA1 (arrows in C and F). In contrast, the hippocampal formation in MTLE is shrunken (G) and there is considerable loss of neurons with proliferation of glial cells in CA1 (arrowheads in H), CA3, and the polymorphic layer of the hilus (arrowheads in I). These pathological findings are hallmarks of hippocampal sclerosis. Scale bars: 1 mm in A (same magnification of D and G), 500 μm in B and C (same magnification of E, H and F, I).
The subdivisions of the hippocampal formation are in accordance with the work of Lorente de Nó (Lorente de Nó 1934), with modifications suggested by Amaral and Insausti (Amaral & Insausti 1990). Briefly, the hippocampal formation is divided into (a) the subiculum, (b) the Ammon’s horn (hippocampus proper), which comprises fields CA1-3, and (c) the dentate gyrus, which includes the molecular, granule, and polymorphic layers of the hilus.
Tissue Preparation
Immediately after surgical resection, an approximately 5 mm thick slice was cut from the mid-anterior portion of the hippocampal formation, rapidly frozen on dry ice, and sectioned to a thickness of 50 μm using a cryostat. The tissue sections were transferred to Fisherbrand Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA) and stored at −80 °C until processed for in situ hybridization. Adjacent slices of tissue were fixed by immersion in 4% formaldehyde in 0.1 M phosphate buffer (PB), pH 7.4, for 6 hours and subsequently cut to 50 μm thick sections on a Vibratome. The fixed sections were processed for staining with cresyl violet, anti-somatostatin-, anti-neuropeptide Y- and anti-dynorphin-antibodies, as previously described (de Lanerolle, et al. 2003). These sections were used for disease classification as described above.
In Situ Hybridization
Subclones for riboprobe synthesis were prepared by amplifying unique regions of GS and PAG from a human cDNA brain library (Human Adult Brain Unamplified cDNA Library, Edge Biosystems; Gaithersburg, MD) using Polymerase Chain Reaction (PCR). Amplified cDNA segments were extracted (QIAquick Gel Extraction kit; Qiagen, Valencia, CA), subcloned (Zero Blunt TOPO PCR cloning kit; Invitrogen, Carlsbad, CA), and confirmed by nucleotide sequencing.
Riboprobes were synthesized using 100 μCi of dried [35S]UTP; 2.0 μl 5x transcription buffer; 1.0 μl of 10 mM ATP, CTP, and GTP; 2.0 μl linearized plasmid DNA; 0.5μl RNAse inhibitor; and 1.5 μl SP6 or T7 RNA polymerase; and incubated for 2 hr at 37 °C. After this incubation, 1.0 μl DNAse (RNAse free) was added and the reaction mixture was incubated again for 15 min at room temperature. The mixture was then purified with a 1 ml spin column (Micro Bio-Spin P-30 Tris Spin Columns, Bio-Rad Laboratories; Hercules, CA). Dithiothreitol (DTT) was added to each fraction for a final concentration of 0.01 M.
Two slides per subject were removed from −80 °C storage and placed in 4% (wt/vol) formaldehyde at room temperature for 1 hr. The slides were washed briefly in 2X SSC (300 mM NaCl/30 mM sodium citrate, pH 7.2), and placed in 0.1 M triethanolamine, pH 8.0/acetic anhydride, 400:1 (vol/vol) on a stir plate at room temp for 10 min. The final wash was in 2X SSC for 10 min followed by dehydration through graded alcohols and air-drying. Cover slips with 400 μl [35S] labeled riboprobe (5×106 cpm), 50% formamide buffer (50% formamide, 10% dextran sulfate, 3X SSC, 50 mM Na2HPO4, pH 7.4, 1X Denhardt’s solution, 100 μg/ml yeast tRNA), and 0.01 M DTT were placed on each slide. Slides were positioned in a covered tray with filter paper, saturated with 50% formamide buffer, and incubated at 55 °C overnight. Approximately 18 hours later, the coverslips were removed in order to wash the slides in 2X SSC at room temperature for 10 min and then incubated in RNAse A (200 mg/ml in 10 mM Tris-HCl, pH 8.0/0.5 M NaCl) at 37 °C for 30 min. Subsequently, the slides were washed twice at room temperature accordingly: 10 min in 2X SSC, 10 min in 1X SSC. The last two washes were in 0.5X SSC for 60 min at 55 °C and in 0.5 SSC for 10 min at room temperature. Finally, the slides were dehydrated in graded ethanol washes, air-dried and apposed to film.
Slides were apposed to Kodak XAR film (Eastman Kodak Company, Rochester, NY) for 15 days (GS) or 10 days (PAG). The film was developed and images analyzed (Scion Image Beta 3b for PC), as previously described (Clinton, et al. 2003, Ibrahim, et al. 2000). Images were captured, and grayscale values were obtained for the subiculum (SUB), the hippocampal subfields CA1, CA2, and CA3, the dentate molecular and granule cell layers (DG), and the polymorphic layer of the hilus (H). For each region, background readings from the tissue were subtracted from the grayscale values. Grayscale values were converted to optical density and subsequently averaged, providing one value per region per subject for each probe. Optical density values were then converted to units of bound radiation from a standard curve generated from a [14C] microscale standard (Amersham Life Sciences, Amersham UK) placed on each piece of film (Downs & Williams 1984, Williams 1982). The transcript concentration, was ultimately determined from the amount of bound radiation, the specific activity of each batch of [35S]uridine triphosphate, and the number of uracil residues contained in each probe sequence. The data were expressed as femtomoles mRNA per gram tissue (fmol/g).
We tested 12 specific a priori hypotheses (two dependent measures in 6 discrete brain regions) using ANOVA, and where indicated, ANCOVA. Using correlation analyses, we probed for associations between our dependent measures and postmortem interval, age, and tissue pH. Factors found to be associated were used, where indicated, as covariates in our primary analyses. Post-hoc analysis was performed, when appropriate, with Tukey’s HSD (honestly significant difference) test. A P-value of 0.05 was regarded as statistically significant.
Results
We first assessed the overall labeling pattern for GS mRNA on hippocampal sections from autopsy subjects (Fig. 2A) and patients classified as non-MTLE (Fig. 2C) and MTLE (Fig. 2E). Visual inspection of the scanned films from the in situ hybridization experiments revealed finely granular labeling for GS in the hippocampal formation in all patient categories (Fig. 2A, C, E). The labeling was particularly pronounced in areas of high neuronal (perikaryal) density such as the pyramidal cell layers of the hippocampal formation in autopsy and non-MTLE specimens (Fig. 2A,C). Weaker labeling was present in white matter areas, particularly in autopsy and MTLE hippocampal formations (Fig. 2A, E). In areas of neuronal loss such as the CA1 and CA3 in MTLE, the labeling was less intense than in areas of neuronal preservation, such as the subiculum in the same patients (Fig 2E). Interestingly, the labeling in neuron-dense areas appeared stronger in non-MTLE and MTLE hippocampal formations (Fig. 2C, E) than in autopsy controls (Fig. 2A). Densitometric quantitation of the labeling revealed significantly increased GS mRNA in the CA3 in non-MTLE hippocampal formations vs. autopsy and MTLE hippocampal formations (Fig. 3).
Figure 2.

In situ hybridization for GS mRNA (A, C, E) and PAG mRNA (B, D, F) in hippocampal formations (coronal sections) from representative patients diagnosed with: medical conditions other than epilepsy (autopsy, A, B), non-MTLE (C, D) and MTLE (E, F). Adjacent Nissl-stained sections were used to delineate the hippocampal subfields indicated in A and B, and densitometric measurements of the in situ hybridization signals were obtained from each subfield (see Figures 3 and 4 for details). Note the more diffuse labeling for GS mRNA vs. PAG mRNA, which is predominantly observed in areas of high neuronal density, suggesting that PAG mRNA is confined to neurons. Note the relative loss of labeling for GS mRNA in CA3 and CA1 in MTLE (arrows in E) vs. non-MTLE (C). A similar pattern is seen for PAG (arrows in F vs. D). The labeling for GS and PAG mRNAs appears to be increased in several subfields in non-MTLE and MTLE vs. autopsy (see Figures 3 and 4 for quantitative data and statistical assessments). Abbreviations: H, polymorphic layer of the hilus. Asterisks in F indicate artificially increased labeling due to a fold in the tissue section. This and other staining artifacts were excluded from the densitometric analyses.
Figure 3.
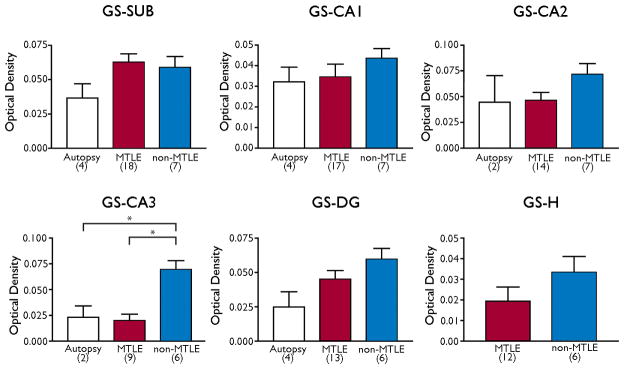
Expression of GS mRNA in different subfields of the hippocampal formation. Bars indicate the mean optical density of GS mRNA expression in the following 6 hippocampal subfields: subiculum (SUB), CA1, CA2, CA3, molecular and granular layer of the dentate gyrus (DG), polymorphic layer of the dentate hilus (H). Patient categories with number of patients included in the analysis are indicated below each bar. Statistical significant differences are indicated with asterisks. We only had anatomically intact areas of the hilus from two autopsy subjects for the analysis of GS mRNA; thus, the measurements from this area could not be used for statistical analysis. Error bars = standard error of the mean.
We next assessed the expression of PAG and observed a more defined labeling pattern than with GS (Fig. 2B, D, F). The labeling for PAG was particularly prominent over areas of high neuronal (perikaryal) density, such as the pyramidal cell layers of the subiculum and CA-fields, the polymorphic layer of the hilus, and the granule cell layer of the dentate gyrus (Fig. 2B, D, F). White matter areas had very low expression of PAG and the demarcation between labeled and minimally labeled areas was more pronounced with PAG than with GS. In areas of neuronal loss, such as the CA1 and CA3 in MTLE, the expression of PAG was clearly diminished (Fig. 2F). As with GS mRNA, the labeling for PAG mRNA appeared stronger in neuron-dense areas in non-MTLE and MTLE hippocampal formations (Fig. 2D, F) than in autopsy controls (Fig. 2B). Quantitation of the labeling revealed statistically significant increases in PAG mRNA in the CA1, CA2, CA3 and the dentate hilus in non-MTLE than in MTLE (Fig. 4). There were also significant increases in PAG mRNA expression in the subiculum of non-MTLE and MTLE vs. autopsy specimens, in the dentate gyrus in MTLE vs. autopsy specimens, and in the hilus of non-MTLE vs. autopsy specimens (Fig. 4).
Figure 4.
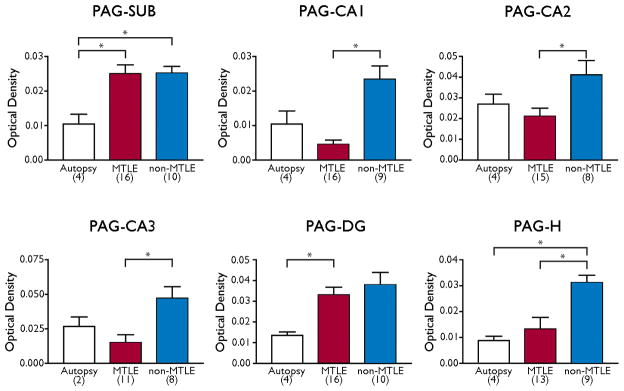
Expression of PAG mRNA in different subfields of the hippocampal formation. Bars indicate the mean optical density of PAG mRNA expression in the following 6 hippocampal subfields: subiculum (SUB), CA1, CA2, CA3, molecular and granular layer of the dentate gyrus (DG), polymorphic layer of the dentate hilus (H). Patient categories with number of patients included in the analysis are indicated below each bar. Statistical significant differences are indicated with asterisks. Error bars = standard error of the mean
Discussion
We have demonstrated for the first time that mRNA for GS and PAG are differentially expressed in the hippocampal formations among non-epilepsy (autopsy) subjects, MTLE patients and non-MTLE patients. While some of the differences in mRNA expression correlate with the previously reported patterns of GS protein expression (Eid, et al. 2004), several discrepancies are evident between the expression of mRNAs and its corresponding protein.
Prior investigations of GS in the hippocampal formation have revealed significant decreases in GS protein and enzyme activity in patients with MTLE vs. patients with non-MTLE and non-epilepsy autopsy control subjects (Eid, et al. 2004, van der Hel, et al. 2005). The decrease in GS protein is particularly pronounced in areas of significant neuronal loss, such as the CA1, CA3 and the dentate hilus in MTLE. Thus, the observation that mRNA for GS is significantly reduced in CA3 in MTLE vs. non-MTLE could, in part, explain the reduction in GS protein and enzyme activity in this region.
However, there is no significant reduction in GS mRNA in the CA1 or in the dentate hilus in MTLE vs. non-MTLE, nor is there any difference in GS mRNA between hippocampal formations from MTLE patients and non-epilepsy (autopsy) subjects. The lack of decrease in GS mRNA between MTLE and autopsy is unexpected because the hippocampal formation in autopsy subjects is strongly immunoreactive for GS protein throughout all subfields of the structure, whereas GS protein is severely deficient in CA1, CA3 and the dentate gyrus in MTLE (Eid, et al. 2004). Because the expression of GS mRNA does not consistently match that of GS protein, it is likely that mechanisms other than reduced synthesis of mRNA underlie the loss of GS protein in MTLE.
Studies have suggested that posttranscriptional modification of GS occurs in epilepsy. Protein nitration, oxidative stress, and brain deposits of amyloid β-peptide may play important roles in this process. Bidmon and colleagues recently reported that GS protein becomes nitrated and reduces its activity in the pentylentetrazole rat model of epilepsy (Bidmon, et al. 2008). The propensity for GS to undergo posttranscriptional modification with loss of enzyme activity is also evident in Alzheimer’s disease patients, who have an excess of oxidized and hence, less active GS in their brain (Castegna, et al. 2002). Injection of amyloid β-peptide into the brain of laboratory animals causes oxidation of GS (Boyd-Kimball, et al. 2005). Interestingly, amyloid β-precursor protein accumulates in the temporal lobe and hippocampal formation in patients with TLE (Sheng, et al. 1994); thus, providing a possible mechanistic link among oxidative stress, amyloid β-peptide, downregulation of GS, and the development of recurrent seizures. In addition to their effect on proteins, oxidative and nitrosative stress may prolong the half-life of some mRNAs (Gasch, et al. 2000, Ikeda, et al. 1995, Mochizuki, et al. 2012). It is possible that a similar mechanism may operate in the epileptic hippocampus, thereby resulting in increased mRNA without a corresponding increase in protein.
The observation that GS mRNA is more abundant in non-MTLE than in autopsy hippocampal formations is also unexpected because the expression of GS protein appears very similar in both subject categories (Eid, et al. 2004). It is possible that the discrepancy between GS protein and mRNA is due to the same posttranscriptional and transcriptional regulatory mechanisms postulated to occur in MTLE. After all, the hippocampal formations from non-MTLE and MTLE patients have been subjected to unique, common exposures that are not typically encountered in non-epilepsy autopsy patients, such as years of antiepileptic drugs and numerous recurrent seizures. It should also be noted that autopsy tissue is influenced by several factors such as postmortem interval, tissue pH, agonal status, manner of death, and freezer storage (McCullumsmith & Meador-Woodruff 2011). While these factors could theoretically affect the stability of tissue mRNA, a recent study did not detect a significant effect of these factors on the expression of mRNA for GS and PAG in autopsy brain tissue from human subjects (Bruneau, et al. 2005). Thus, it is unlikely that the changes in GS and PAG mRNA expression between TLE and autopsy subjects are caused by postmortem alterations.
The reduction in PAG mRNA in MTLE vs. non-MTLE is likely to result from the loss of neurons especially in CA1, CA3 and the dentate hilus in MTLE. The preferential distribution of PAG mRNA to areas of high density of neuronal perikarya, such as the pyramidal and dentate granule cell layers, suggests that PAG mRNA is predominantly found in neurons. Neuronal localization of PAG mRNA is consistent with prior immunohistochemical and immunogold electron microscopic studies of the distribution of PAG protein (Eid, et al. 2007, Laake, et al. 1999, Svenneby 1970).
We had concluded in prior studies that PAG protein is increased per neuron in the hippocampal formation in patients with MTLE vs. non-MTLE (Eid, et al. 2007) and had proposed that the increase might contribute to the glutamate excess in MTLE. However, the present study shows a significant decrease in PAG mRNA in MTLE vs. non-MTLE in CA1, CA2, CA3 and the dentate hilus. It is important to note that these findings do not rule out the possibility that PAG mRNA is increased in subpopulations of surviving neurons in the MTLE hippocampal formation. Unfortunately, the in situ hybridization method used here does not have the resolving power to properly address this issue. Future studies using high-resolution mRNA expression imaging are therefore warranted.
Similar to GS mRNA, there is also increased PAG mRNA in the hippocampal formations in non-MTLE and MTLE patients vs. autopsy controls, especially in the subiculum and the dentate gyrus/hilus. The difference in PAG mRNA expression between epileptic and non-epileptic hippocampal formations are not known, but may involve upregulation of PAG mRNA due to recurrent seizures, antiepileptic drugs or other changes associated with TLE. Further investigations are necessary to fully understand these discrepancies.
It is important to note that the glutamate excess in human TLE could be due to mechanisms other than alterations in GS and PAG. Alternative mechanisms include perturbations in high-affinity glutamate transporters (Mathern, et al. 1999, Proper, et al. 2002, Tanaka, et al. 1997, Watanabe, et al. 1999), alterations of the glutamate/cystin exchange system (Albrecht, et al. 2010), downregulation of presynaptic metabotropic glutamate receptors (Iserhot, et al. 2004, Molinari, et al. 2012), reduced currents through inwardly rectifying potassium channels (Kir 4.1) (Bordey & Sontheimer 1998, Hinterkeuser, et al. 2000), and disrupted polarization of the aquaporin-4 (AQP4) dystrophin complex (Binder, et al. 2012, Binder, et al. 2004, Eid, et al. 2005, Lee, et al. 2004). Finally, the perturbations of GS and PAG proteins in the hippocampal formation in MTLE (Eid, et al. 2007, Eid, et al. 2004) are also likely to affect the homeostasis of other metabolites, particularly ammonia and GABA, both of which can influence neuronal excitability. A more comprehensive discussion of the effects of GS and PAG perturbations in TLE can be found in recent reviews on the topic (Eid, et al. 2012, Eid & Coulter 2012, Eid, et al. 2008b).
Conclusions
We have demonstrated complex changes in the expression of mRNAs for GS and PAG in the hippocampal formation in TLE. Firstly, downregulation of mRNA can only partly explain the deficiency in GS protein in the hippocampal formation in MTLE (Eid, et al. 2004). Secondly, posttranscriptional mechanisms, such as protein oxidation or nitrosylation, are likely to contribute to the deficiency in GS protein in MTLE. Investigations of mRNA stability and posttranslational modifications of GS are justified to better understand how the protein is regulated in TLE. Thirdly, the reduction in PAG mRNA in the MTLE vs. the non-MTLE hippocampal formation probably reflects the loss of hippocampal neurons in MTLE; however, the possibility that PAG mRNA is upregulated in subpopulations of surviving neurons in MTLE cannot be entirely ruled out. High-resolution (emulsion) in situ hybridization methods may be used to address this issue by assessing the expression of PAG mRNA at the cellular level. A clear understanding of how GS and PAG are regulated in the epileptic brain is important because such information could lead to novel antiepileptic therapies targeting key enzymes in the brain glutamate homeostasis.
Acknowledgments
We thank Ms. Ilona Kovacs for excellent technical assistance. This work was supported by grants from the National Institutes of Health (NS058674 andNS070824 to T.E. and MH074016 to R.E.M.), the Swebelius Family Trust (to D.D.S.), the Doris Duke’s Charitable Foundation (to R.E.M.) and the Research Council of Norway (to F.L.).
Footnotes
Conflict of Interests
None of the authors has any conflict of interest to disclose.
References
- Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol Disord Drug Targets. 2010;9:373–382. doi: 10.2174/187152710791292567. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Insausti R. Hippocampal formation. In: Paxinos G, editor. The human nervous system. Academic Press; San Diego: 1990. pp. 711–756. [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Berg M, Bruhn T, Johansen FF, Diemer NH. Kainic acid-induced seizures and brain damage in the rat: different effects of NMDA- and AMPA receptor antagonists. Pharmacol Toxicol. 1993;73:262–268. doi: 10.1111/j.1600-0773.1993.tb00582.x. [DOI] [PubMed] [Google Scholar]
- Bidmon HJ, Gorg B, Palomero-Gallagher N, Schleicher A, Haussinger D, Speckmann EJ, Zilles K. Glutamine synthetase becomes nitrated and its activity is reduced during repetitive seizure activity in the pentylentetrazole model of epilepsy. Epilepsia. 2008;49:1733–1748. doi: 10.1111/j.1528-1167.2008.01642.x. [DOI] [PubMed] [Google Scholar]
- Binder DK, Nagelhus EA, Ottersen OP. Aquaporin-4 and epilepsy. Glia. 2012 doi: 10.1002/glia.22317. [DOI] [PubMed] [Google Scholar]
- Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport. 2004;15:259–262. doi: 10.1097/00001756-200402090-00009. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: implications for Alzheimer’s disease. Neuroscience. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, McCullumsmith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophr Res. 2005;75:27–34. doi: 10.1016/j.schres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, Abi-Saab WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Cavus I, Pan JW, Hetherington HP, Abi-Saab W, Zaveri HP, Vives KP, Krystal JH, Spencer SS, Spencer DD. Decreased hippocampal volume on MRI is associated with increased extracellular glutamate in epilepsy patients. Epilepsia. 2008;49:1358–1366. doi: 10.1111/j.1528-1167.2008.01603.x. [DOI] [PubMed] [Google Scholar]
- Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2003;160:1100–1109. doi: 10.1176/appi.ajp.160.6.1100. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Brines ML, Kim JH, Williamson A, Philips MF, Spencer DD. Neurochemical remodelling of the hippocampus in human temporal lobe epilepsy. In: Engel J Jr, Wasterlain C, Cavalheiro EA, Heinemann U, Avanzini G, editors. Molecular Neurobiology of Epilepsy. Elsevier Science Publishers, B.V; Amsterdam: 1992. pp. 205–220. [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: Evidence for distinctive patient subcategories. Epilepsia. 2003;44:677– 687. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- Downs AM, Williams MA. An improved approach to the analysis of autoradiographs containing isolated sources of simple shape: method, theoretical basis and reference data. J Microsc. 1984;136:1–22. doi: 10.1111/j.1365-2818.1984.tb02542.x. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Behar K, Dhaher R, Bumanglag AV, Lee TS. Roles of Glutamine Synthetase Inhibition in Epilepsy. Neurochem Res. 2012 doi: 10.1007/s11064-012-0766-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Coulter DA. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012 doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Du F, Schwarcz R. Differential neuronal vulnerability to amino-oxyacetate and quinolinate in the rat parahippocampal region. Neuroscience. 1995;68:645–656. doi: 10.1016/0306-4522(95)00183-j. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, de Lanerolle NC. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008a;131:2061–2070. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Hammer J, Runden-Pran E, Roberg B, Thomas MJ, Osen K, Davanger S, Laake P, Torgner IA, Lee TS, Kim JH, Spencer DD, Ottersen OP, de Lanerolle NC. Increased expression of phosphate-activated glutaminase in hippocampal neurons in human mesial temporal lobe epilepsy. Acta Neuropathol (Berl) 2007;113:137–152. doi: 10.1007/s00401-006-0158-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A. 2005;102:1193–1198. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes--key players in human mesial temporal lobe epilepsy? Epilepsia. 2008b;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- Engel J, Williamson PD, Wieser HG. Mesial temporal lobe epilepsy with hippocampal sclerosis. In: Engel J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Wolters Kluver, Lippincott Williams & Wilkins; Philadelphia: 2008. pp. 2479–2486. [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor P. Mesial temporal sclerosis: Historical background and an overview from a modern perspective. In: Luders H, editor. Epilepsy Surgery. Raven Press; New York: 1991. pp. 689–703. [Google Scholar]
- Hayashi T. Effects of sodium glutamate on the nervous system. Keiko J Med. 1954;3:183–192. [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, Steinhauser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV. Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci. 1990;10:267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim HM, Hogg AJ, Jr, Healy DJ, Haroutunian V, Davis KL, Meador-Woodruff JH. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am J Psychiatry. 2000;157:1811–1823. doi: 10.1176/appi.ajp.157.11.1811. [DOI] [PubMed] [Google Scholar]
- Ikeda E, Achen MG, Breier G, Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270:19761–19766. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- Iserhot C, Gebhardt C, Schmitz D, Heinemann U. Glutamate transporters and metabotropic receptors regulate excitatory neurotransmission in the medial entorhinal cortex of the rat. Brain Res. 2004;1027:151–160. doi: 10.1016/j.brainres.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, Ottersen OP. Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neurosci. 1999;88:1137–1151. doi: 10.1016/s0306-4522(98)00298-x. [DOI] [PubMed] [Google Scholar]
- Lee TS, Eid T, Mane S, Kim JH, Spencer DD, Ottersen OP, de Lanerolle NC. Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol (Berl) 2004 doi: 10.1007/s00401-004-0910-7. [DOI] [PubMed] [Google Scholar]
- Lorente de Nó R. Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J Psychol Neurol (Leipzig) 1934;46:113–177. [Google Scholar]
- Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58:193–201. doi: 10.1001/archopht.1957.00940010205006. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Armstrong DL. Hippocampal sclerosis. In: Engel J, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Lippincott-Raven; Philadelphia: 1997. pp. 133–155. [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE, Meador-Woodruff JH. Novel approaches to the study of postmortem brain in psychiatric illness: old limitations and new challenges. Biol Psychiatry. 2011;69:127–133. doi: 10.1016/j.biopsych.2010.09.035. [DOI] [PubMed] [Google Scholar]
- McNamara RK, Routtenberg A. NMDA receptor blockade prevents kainate induction of protein F1/GAP-43 mRNA in hippocampal granule cells and subsequent mossy fiber sprouting in the rat. Brain Res Mol Brain Res. 1995;33:22–28. doi: 10.1016/0169-328x(95)00083-5. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Murphy CJ, Brandt JD, Kiuchi Y, Russell P. Altered stability of mRNAs associated with glaucoma progression in human trabecular meshwork cells following oxidative stress. Invest Ophthalmol Vis Sci. 2012 doi: 10.1167/iovs.12-7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari F, Cattani AA, Mdzomba JB, Aniksztejn L. Glutamate transporters control metabotropic glutamate receptors activation to prevent the genesis of paroxysmal burst in the developing hippocampus. Neuroscience. 2012;207:25–36. doi: 10.1016/j.neuroscience.2012.01.036. [DOI] [PubMed] [Google Scholar]
- Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanims of epileptic brain damage. In: Delgado-Escueta AV, Ward AA Jr, Woodbury DM, Porter RJ, editors. Advances in neurology. Raven Press; New York: 1986. pp. 857–877. [PubMed] [Google Scholar]
- Olney JW, Sharpe LG, Feigin RD. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol. 1972;31:464–488. doi: 10.1097/00005072-197207000-00006. [DOI] [PubMed] [Google Scholar]
- Otis TS, Jahr CE. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroff OA, Cavus I, Kim JH, Spencer DD. Interictal extracellular glutamate concentrations are increased in hippocampal sclerosis. Ann Neurol. 2004;56:S43. [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Boop FA, Mrak RE, Griffin WS. Increased neuronal beta-amyloid precursor protein expression in human temporal lobe epilepsy: association with interleukin-1 alpha immunoreactivity. J Neurochem. 1994;63:1872–1879. doi: 10.1046/j.1471-4159.1994.63051872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer W. Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch Psychiatr Nervenkr. 1880;10:631–675. [Google Scholar]
- Spencer DD, Spencer SS. Surgery for Epilepsy. Blackwell Scientific Publishers; Boston: 1991. [Google Scholar]
- Spencer DD, Spencer SS, Mattson RH, Williamson PD, Novelly R. Access to posterior medial temporal lobe structures in the surgical treatment of temporal lobe epilepsy. Neurosurg. 1984;15:667–671. doi: 10.1227/00006123-198411000-00005. [DOI] [PubMed] [Google Scholar]
- Svenneby G. Pig brain glutaminase: purification and identification of different enzyme forms. J Neurochem. 1970;17:1591–1599. doi: 10.1111/j.1471-4159.1970.tb03729.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, de Graan PN. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology. 2005;64:326–333. doi: 10.1212/01.WNL.0000149636.44660.99. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol. 2009;220:293–302. doi: 10.1016/j.expneurol.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 1999;845:92–96. doi: 10.1016/s0006-8993(99)01945-9. [DOI] [PubMed] [Google Scholar]
- Williams MA. Autoradiography: its methodology at the present time. J Microsc. 1982;128:79–94. doi: 10.1111/j.1365-2818.1982.tb00439.x. [DOI] [PubMed] [Google Scholar]