

Abstract
MHC class I and class II are crucial for the adaptive immune system. Although regulation of MHC class II expression by CIITA (class II transactivator) has long been recognized, the mechanism of MHC class I transactivation has been largely unknown until the recent discovery of NLRC5/CITA. Here we show using Nlrc5-deficient mice that NLRC5 is required for both constitutive and inducible MHC class I expression. Loss of Nlrc5 resulted in severe reduction in the expression of MHC class I and related genes such as β2m, Tap1 or Lmp2 but did not affect MHC class II levels. IFN-γ stimulation could not overcome the impaired MHC class I expression in Nlrc5-deficient cells. Upon infection with Listeria monocyogenes, Nlrc5-deficient mice displayed impaired CD8+ T cell activation, accompanied with increased bacterial loads. These illustrate critical roles of NLRC5/CITA in MHC class I gene regulation and host defense by CD8+ T cell responses.
Keywords: NLR protein, CIITA, MHC class I
Introduction
Major histocompatibility complex (MHC) class I and class II molecules play key roles in the activation of the adaptive immune system. MHC class I molecules present peptide antigens of intracellular origin such as tumor or viral antigens to CD8+ T cells, whereas MHC class II molecules present peptide antigens of extracellular sources to CD4+ T cells (1). The expression of both constitutive and inducible MHC class II requires the master transcriptional co-activator CIITA (2). Although CIITA itself lacks a DNA binding domain, CIITA can activate the promoters of MHC class II genes by engaging in a nucleoprotein complex called the MHC-enhanceosome (3, 4), together with promoter-resident transcription factors including the trimeric RFX protein complex, CREB/ATF1 family members, and the NF-Y protein (5, 6). CIITA can also transactivate MHC class I genes at least in vitro (7, 8), although both bare lymphocyte syndrome (BLS) patients with mutations in the CIITA gene and CIITA-deficient mice retain intact MHC class I expression, indicating that there is another mechanism for the activation of MHC class I in vivo (2, 9–12).
The recent discovery of a transactivator specific for MHC class I genes, NLRC5, identified a new addition to the regulators of MHC genes (13, 14). Both NLRC5 and CIITA belong to the NLR or nucleotide binding domain (NBD), leucine-rich repeat (LRR) family of proteins and phylogenetically they are most closely related to each other (13, 15). NLRC5, or class I transactivator (CITA), is also an IFN-γ-inducible nuclear protein, but NLRC5 specifically associates with and transactivates MHC class I promoters, resulting in the expression of MHC class I and related genes such as β2-microglobulin (β2M), TAP1 and LMP2 (13, 14). The NBD is a critical domain for the function of NLRC5, as the NBD is required for both nuclear import and transactivation of MHC class I genes (16). In the nucleus, NLRC5 participates in an MHC class I specific enhanceosome, together with the RFX factor members and CREB/ATF1 family transcription factors to activate MHC class I gene promoters (17). In addition to its function as a MHC class I transactivator, NLRC5 has been reported to be involved in the regulation of Toll-like receptor and RIG-I-like receptor signaling, anti-viral responses and inflammasome activation (15, 18–21). However, those reports provide conflicting results and their conclusions are not supported by the data obtained from the initial characterization of Nlrc5-deficient mice (14, 22).
Although the identification of NLRC5 as a MHC class I gene transactivator is significant, the role of NLRC5 in MHC class I expression in vivo had not been elucidated. To address this question, we investigated the function of NLRC5 using Nlrc5-deficient mice. Here we show that Nlrc5 is required for both constitutive and inducible expression of MHC class I. Nlrc5-deficient mice were more susceptible to infection with Listeria monocytogenes, as highlighted by impaired CD8+ T cell activation and increased bacterial burden in the infected organs, indicating a critical role of Nlrc5/CITA in MHC class I-dependent CD8+ T cell responses.
Materials and Methods
Mice
Nlrc5-deficient mice were kindly provided by Drs T. Kawai and S. Akira (22). Wild-type mice (F1 mice from 129SvEv and C57BL/6 mice) were obtained from Taconic. OT-1 TCR transgenic mice were a kind gift of Dr Harvey Cantor. Mice were maintained under specific pathogen-free conditions and used in accordance with institutional and National Institutes of Health guidelines.
Cell culture and reagents
Generation and stimulation of bone marrow-derived macrophages and dendritic cells has been described previously (23). Splenocytes and thymocytes were cultured in RPMI supplemented with 10% fetal bovine serum (FBS), 55 μM 2-ME (Gibco) and penicillin/ streptomycin (Gibco). Recombinant murine IFN-γ was from BioLegend. Peritoneal cells were isolated without pre-treatment with thioglycollate.
RNA isolation and quantitative PCR
RNA isolation and quantitative PCR were performed as previously described (13). Primer sequences are listed in Supplemental Table 1.
Western blot analysis
Western blot analysis was performed as described previously (13) using following antibodies: anti-H2-Kb, anti-β2m (kind gifts from Dr T. Hansen, Washington University) and anti-Hsp90 antibody (F-8) (Santa Cruz Biotechnology).
Flow cytometric analysis
FACS analysis was performed as previously described (13) using following antibodies: FITC-anti-mouse B220, FITC- anti-mouse CD11c, APC- anti-mouse CD4, PE-/Cy5-anti-mouse CD8, APC- anti-mouse F4/80, PE- anti-mouse IAb/IAd (eBioscience), PE-anti-mouse H2 and APC-anti-mouse IFN-γ (Biolegend).
OT-1 cell co-culture assay
MACS sorted CD8+ T cells from OT-1 mice with CD45.1 background were CFSE-labeled and co-cultured with SIINFEKL (1μM) pulsed irradiated splenic B220+ cells from either Nlrc5+/− or Nlrc5−/− mice at a ratio of 1:25 (B:T) for 72 hrs. Proliferation of OT-1 T cells was determined by FACS analysis of the CFSE dilution in CD45.1 gated cells.
Listeria monocytogenes infection
L. monocytogenes (strain 10403s, kindly provided by Dr. D. Portnoy at University of California, Berkeley) was grown to mid-log phase (A600, 0.1), and injected into mice via the lateral tail vein. The bacterial burden in the spleen and liver of infected mice was determined by CFU count of serial dilution of lysates.
Statistical analysis
Data were subjected to One-way ANOVA for analysis of statistical significance using Prism (GraphPad). Results are given as the mean ± SEM. A P-value of < 0.05 was considered to be significant.
Results and Discussion
In order to clarify the role of NLRC5 in MHC class I gene expression in vivo, we analyzed Nlrc5-deficient mice. Heterozygous littermates (+/−) and F1 mice from a C57BL/6 X 129SvEv breeding (+/+) with a similar mixed genetic background to the knock-out mice (−/−), were used as controls. As can be seen in Fig. 1A, MHC class I (H2-Kb) transcript levels in the thymus, spleen, kidney and ileum were significantly reduced in Nlrc5-deficient mice compared to that of wild-type or heterozygous mice, confirming that MHC class I expression is indeed largely dependent on Nlrc5. The expression of MHC class I related genes, β2m, Tap1, Lmp2 as well as the non-classical MHC class I gene, H2-M3 was impaired in the thymus and spleen of Nlrc5-deficient mice (Fig. 1B). In contrast, Nlrc5 deficiency did not affect the expression of MHC class II (H2-Aα) and CIITA (Fig. 1C), confirming the specificity of NLRC5 for the activation of MHC class I genes. In agreement with the transcript data, protein levels of MHC class I (H2-Kb) and β2m were also reduced in the spleen and thymus of Nlrc5-deficient mice (Fig. 1D). Taken together, Nlrc5 is specifically required for the expression of MHC class I in both lymphoid and non-lymphoid organs and is dispensable for MHC class II expression.
FIGURE 1.
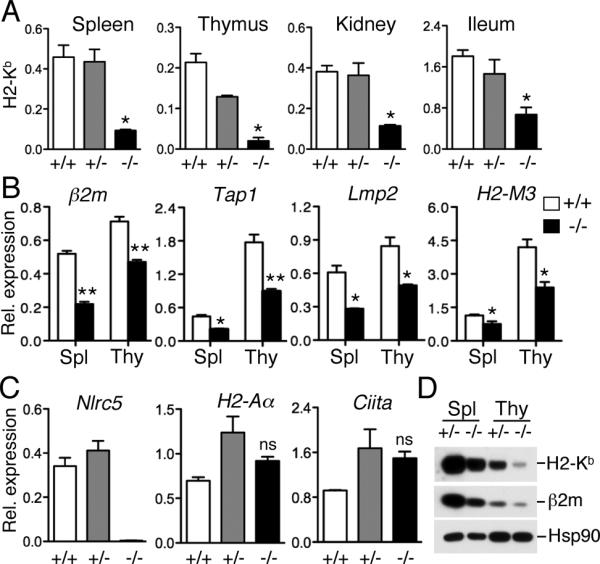
Nlrc5 is required for the constitutive expression of MHC class I in various organs. (A) Expression of MHC class I (H2-Kb) in the indicated organs of wild-type mice (+/+, n=3), or mice heterozygous (+/−, n=4) or homozygous (−/−, n=3) for the targeted disruption of the Nlrc5 gene, as determined by qPCR analysis using gene-specific primers. Error bars represent ± SEM. *p < 0.05. (B) Expression of β2m, Tap1, Lmp2 and H2-M3 in the spleen and thymus of Nlrc5+/+ (n = 3) and Nlrc5−/− mice (n = 3) were analyzed by qPCR using the indicated gene-specific primers. Error bars represent ± SEM. *p < 0.05; **p < 0.01; ns, not significant. (C) Analysis of Nlrc5, MHC class II (H2-Aa), and CIITA gene expression in splenocytes, as determined by qPCR. n = 3. Error bars represent ± SEM. ns: not significant. (D) Western blot analysis of MHC class I (H2-Kb) and β2m protein expression in the spleen and thymus from mice with the indicated genotype. The expression of Hsp90 is shown as a loading control.
As we observed a reduction of MHC class I levels in the thymus and spleen, we further investigated the cell type-specific expression of MHC class I in those organs by flow cytometry. The surface expression of MHC class I (H2-D, K, L) was severely reduced in CD4+ and CD8+ T cells in the spleen, thymus and lymph nodes from Nlrc5-deficient mice (Fig. 2ABC). Furthermore, MHC class I expression was reduced in Nlrc5-deficient B cells from the spleen, lymph nodes and peritoneal cavity, splenic F4/80+ macrophages and CD11c+ dendritic cells, although they retained significant levels of MHC class I on their surface (Fig. 2ABC and Supplemental Fig. 1A). Also, the transcript levels of MHC class I in bone marrow-derived macrophages and dendritic cells were significantly reduced (Supplemental Fig. 1B), indicating that Nlrc5 is important for the induction of MHC class I genes in macrophages and dendritic cells, whether or not they are of tissue origin or bone marrow derived. Interestingly, the surface expression of MHC class I on these bone marrow derived cells was largely unaffected (Supplemental Fig. 1A), suggesting that there appears to be a compensatory post-transcriptional mechanism to rescue the MHC class I deficiency in cultured cells. Together, these findings suggest that Nlrc5 plays a major role in the regulation of MHC class I gene expression, albeit the degree of the requirement for Nlrc5 in MHC class I expression varies between different cell types.
FIGURE 2.

Reduced MHC class I expression in various cell types of Nlrc5-deficient mice. Single cell suspension of spleen (A, left panel & D), thymus (B, left panel) and lymph node (C, left panel) from Nlrc5 +/− (grey line) and Nlrc5−/− (black line) mice were analyzed by flow cytometry for the expression of MHC class I (H2-K/D/L, in A, B & C, left panel) or MHC class II (D) molecules (IAb/IAd), gated on CD8+, CD4+, B220+, F4/80+ or CD11c+ cells. Shaded region represent the isotype control. Data are representative of two independent experiments. Bar graphs (A, B & C, right panel) present the mean and SEM of the corresponding MFI (n = 3 for each genotype). *p < 0.05; **p < 0.01.
We have previously demonstrated that NLRC5 is required for MHC class I gene expression upon IFN-γ stimulation (13). Therefore, we examined the impact of Nlrc5-deficiency on IFN-γ-inducible MHC class I expression by stimulating splenocytes from Nlrc5-deficient mice with IFN-γ. As previously shown (13, 15, 20, 21), IFN-γ treatment resulted in the upregulation of Nlrc5 transcript, which correlated with the induction of MHC class I gene (H2-Kb) expression in both wild-type and heterozygous splenocytes (Fig. 3A) (13). In Nlrc5-deficient splenocytes, however, IFN-γ stimulation could not rescue the impairment of MHC class I expression, when compared to the levels of MHC class I observed in the controls (Fig. 3A). β2m expression was also only partially rescued (Fig. 3A). In contrast, upregulation of Stat1 transcripts, which is induced by IFN-γ stimulation in an Nlrc5-independent manner (13), was comparable among the three genotypes, indicating that the JAK/STAT signaling cascade downstream of the IFN-γ receptor is intact in Nlrc5-deficient cells. Interestingly, there was a small, but distinct, induction of MHC class I expression in Nlrc5-deficient splenocytes upon IFN-γ stimulation, supporting the existence of an Nlrc5-independent mechanism(s) of MHC class I expression (Fig. 3A). In agreement with the transcript data, IFN-γ stimulation did not restore the expression of MHC class I (H2-Kb) and β2m at the protein level in Nlrc5-deficient splenocytes and thymocytes (Fig. 3B). Moreover, flow cytometric analysis showed that IFN-γ stimulation did not rescue the reduced MHC class I surface expression in CD4+, CD8+ T cells, B cells, F4/80+ macrophages and CD11c+ dendritic cells obtained from the spleen of Nlrc5-deficient mice (Fig. 3C).
FIGURE 3.
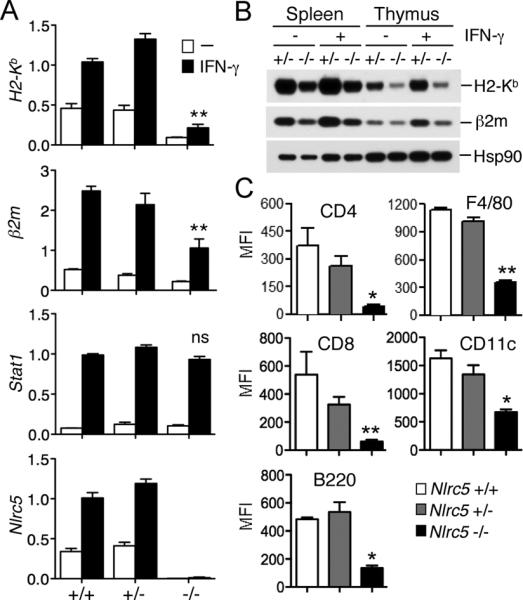
IFN-γ stimulation does not rescue the reduced MHC class I expression observed in Nlrc5-deficient mice. (A) Splenocytes from Nlrc5+/+, Nlrc5+/− and Nlrc5−/−mice (n = 3 for each genotype) were stimulated for 18 hrs with IFN-γ (100 U/ml) and transcript levels were analyzed by qPCR using the indicated gene-specific primers. Error bars represent ± SEM. **p < 0.01; ns, not significant. (B) MHC class I (H2-Kb) and β2m protein expression in splenocytes and thymocytes from mice with the indicated genotype. Cell extracts were prepared 18 hrs post stimulation with IFN-γ and analyzed by Western blotting. Hsp90 levels are depicted as a loading control. (C) Splenocytes from Nlrc5+/+, Nlrc5+/− and Nlrc5−/− mice (n = 3 for each genotype) were cultured overnight in the presence of IFN-γ and were analyzed by flow cytometry for the expression of MHC class I molecules (H2-K/D/L), gated on CD8+, CD4+, B220+, F4/80+, and CD11c+ cells. Bar graphs represent the mean and SEM of the corresponding MFI.
In order to address whether the impaired MHC class I expression caused by Nlrc5 deficiency has an impact on immune responses, OT-1 CD8+ T cells were co-cultured with peptide-loaded B cells. OT-1 T cells cultured with Nlrc5-deficient B cells displayed impaired proliferation, indicating that NLRC5 is indeed required for antigen-specific stimulation of CD8+ T cells (Fig. 4A). The role of NLRC5 in MHC class I mediated immune responses was further investigated by infection studies using an intracellular bacterium, Listeria monocytogenes, since a CD8+ T cell response is critical for the host defense against this bacterium. Although Listeria infection clearly induced antigen-specific CD8+ T cell activation in the spleen and liver of wild-type mice, as demonstrated by increased numbers of IFN-γ positive cells after ex vivo stimulation with heat-killed bacteria (Fig. 4B and Supplemental Fig. 1C), in Nlrc5-deficient mice, antigen-specific CD8+ T cell activation was impaired and the mice harbored increased numbers of the bacterium in both spleen and liver (Fig. 4BC). This Listeria-susceptible phenotype also contradicts the previously proposed function of NLRC5 as a TLR inhibitor, as in that case the Nlrc5-deficient mice should be more protected rather than being more susceptible to infection. Indeed Nlrc5-deficient macrophages expressed normal levels of IL-6, TNF-α, IL-12 p40 and IL-1β at both transcript and protein levels upon LPS, CpG oligo and poly (IC) stimulation, confirming the observations made in the previous study using Nlrc5-deficient mice (Supplemental Fig. 1DE) (22). These data collectively demonstrate a critical role of Nlrc5/CITA in MHC class I-mediated immune responses in vivo.
FIGURE 4.
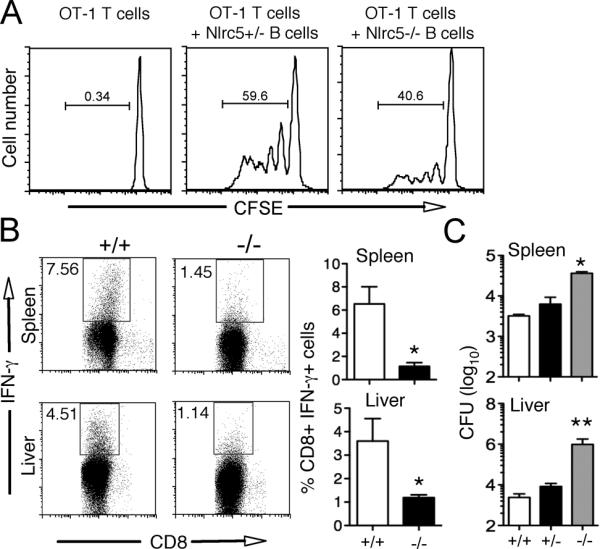
Nlrc5 is required for MHC class I mediated immune responses. (A) Nlrc5-deficient B cells display impaired antigen-specific stimulation of CD8+ T cells. CFSE-labeled OT-1 (CD45.1) CD8+ T cells were co-cultured with SIINFEKL (1μM) pulsed B220+ splenocytes from Nlrc5+/− or Nlrc5−/− mice at a ratio of 1:25 (B:T) for 72 hrs. Proliferation of CD45.1 gated cells was determined by flow cytometric analysis of CFSE dilution. Data are representative of two independent experiments (n=3). (B) Nlrc5+/+, Nlrc5+/− or Nlrc5−/− mice were infected with L. monocytogenes (LM) (1 × 104 CFU). Splenocytes and hepatic leucocytes isolated from Nlrc5+/+ and Nlrc5−/− mice at day 6 after infection were cultured with heat-killed LM for 16 hours and analyzed by flow cytometry for the expression of IFN-γ gated on CD8+ cells. Numbers indicate percentage of cells. Data are representative of two independent experiments (n=3). Bar graphs (B, right panel) present the mean and SEM of the percentage of CD8+ IFN-γ+ cells (n = 3 for each genotype). *p < 0.05. (C) Bacterial load in the spleen and liver of LM infected Nlrc5+/+, Nlrc5+/− or Nlrc5−/− mice were determined. Data are representative of two independent experiments *p < 0.05; **p < 0.01.
In addition to our previous study in which we used human lymphoid and epithelial cell lines (13), the present study compellingly demonstrates the critical role of NLRC5 in both constitutive and inducible MHC class I expression in vivo using Nlrc5-deficient mice. Moreover, we show that NLRC5 is required for CD8+ T cell responses in an antigen-specific manner. Antigen-peptide loaded Nlrc5-deficient B cells failed to activate OT-1 T cells efficiently. Strikingly, our Listeria infection study demonstrated that CD8+ T cells obtained from Nlrc5-deficient mice displayed impaired antigen-specific activation and the mice had increased bacterial loads in the spleen and liver. Therefore, we conclude that NLRC5 plays a critical role in MHC class I-mediated immune responses in vivo. This study also reveals that the requirement for NLRC5 in MHC class I gene expression varies between different cell types. The reduced MHC class I phenotype was most prominent in CD4+ and CD8+ T cells and less prominent in B cells (Fig. 2ABC). Also macrophages and dendritic cells retained residual MHC class I expression (Fig. 2A). This may suggest that an alternative, NLRC5-independent mechanism of MHC class I transactivation exists. Interestingly, similar residual expression of MHC class II genes has been reported in CIITA-deficient mice; CIITA-deficient dendritic cells retained MHC class II expression, although expression levels were significantly reduced (12). These observations indicate that although NLRC5/CITA and CIITA are critical for the expression of MHC class I and class II respectively, antigen presenting cells may possess alternative mechanisms to ensure the efficient presentation of antigens to T cells. In summary, we demonstrated the critical role of NLRC5/CITA in MHC class I expression and CD8+ T cell responses in vivo. Further analysis of Nlrc5-deficient mice will certainly extend our understanding of MHC class I biology and hence may improve therapeutic interventions in the field of infectious diseases, transplantation and cancer immunotherapy.
Supplementary Material
Acknowledgments
The authors thank Drs. Peter Cresswell and Ted Hansen for providing reagents; Yuen-Joyce Liu and Amy Li for critically proofreading the manuscript.
This work was supported by grants from the NIH and the Crohn's and Colitis Foundation of America. K.S.K. is a recipient of the Investigator Award from the Cancer Research Institute and the Claudia Adams Barr Award. T.B.M. has been a recipient of the EMBO Long Term fellowship and A.B. is a recipient of Crohn's Colitis Foundation of America fellowship.
Abbreviations used in this paper
- NLR
proteins
- (NBD)
nucleotide-binding domain
- (LRR)
leucine-rich repeat
- CIITA
containing proteins
- MHC
class II transactivator
- BLS
bare lymphocyte syndrome
- β2M
β2-microglobulin
- LMP2
large multifunctional protease 2
- RFX
regulatory factor X
- ATF1
activating transcription factor 1
- NF-Y
nuclear factor-Y
Footnotes
Disclosures The authors have no financial conflict of interest.
References
- 1.Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 2.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 3.Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat. Immunol. 2001;2:652–657. doi: 10.1038/89810. [DOI] [PubMed] [Google Scholar]
- 4.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 5.Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 6.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 7.Martin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, Ting JP. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/s1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- 8.Gobin SJ, Peijnenburg A, Keijsers V, van den Elsen PJ. Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity. 1997;6:601–611. doi: 10.1016/s1074-7613(00)80348-9. [DOI] [PubMed] [Google Scholar]
- 9.Benichou B, Strominger JL. Class II-antigen-negative patient and mutant B-cell lines represent at least three, and probably four, distinct genetic defects defined by complementation analysis. Proc. Natl. Acad. Sci. USA. 1991;88:4285–4288. doi: 10.1073/pnas.88.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4:167–178. doi: 10.1016/s1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- 11.Itoh-Lindstrom Y, Piskurich JF, Felix NJ, Wang Y, Brickey WJ, Platt JL, Koller BH, Ting JP. Reduced IL-4-, lipopolysaccharide-, and IFN-gamma-induced MHC class II expression in mice lacking class II transactivator due to targeted deletion of the GTP-binding domain. J. Immunol. 1999;163:2425–2431. [PubMed] [Google Scholar]
- 12.Williams GS, Malin M, Vremec D, Chang CH, Boyd R, Benoist C, Mathis D. Mice lacking the transcription factor CIITA--a second look. Int. Immunol. 1998;10:1957–1967. doi: 10.1093/intimm/10.12.1957. [DOI] [PubMed] [Google Scholar]
- 13.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA. 2010;107:13794–13799. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meissner TB, Li A, Kobayashi KS. NLRC5: a newly discovered MHC class I transactivator (CITA) Microbes. Infect. 2012;14:477–484. doi: 10.1016/j.micinf.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010;185:1681–1691. doi: 10.4049/jimmunol.0903900. [DOI] [PubMed] [Google Scholar]
- 16.Meissner TB, Li A, Liu YJ, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem. Biophys. Res. Commun. 2012;418:786–791. doi: 10.1016/j.bbrc.2012.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, van den Elsen PJ, Kobayashi KS. NLRC5 Cooperates with the RFX Transcription Factor Complex To Induce MHC Class I Gene Expression. J. Immunol. 2012;188:4951–4958. doi: 10.4049/jimmunol.1103160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang RF. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141:483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA, Ting JP. Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 2011;186:1333–1337. doi: 10.4049/jimmunol.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, Grotzinger J, Fickenscher H, Schreiber S, Rosenstiel P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010;184:1990–2000. doi: 10.4049/jimmunol.0900557. [DOI] [PubMed] [Google Scholar]
- 21.Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Buning H, Schwarzenbacher R, Kufer TA. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J. Biol. Chem. 2010;285:26223–26232. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, Kawai T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J. Immunol. 2011;186:994–1000. doi: 10.4049/jimmunol.1002094. [DOI] [PubMed] [Google Scholar]
- 23.Koh YS, Koo JE, Biswas A, Kobayashi KS. MyD88-dependent signaling contributes to host defense against ehrlichial infection. PLoS One. 2010;5:e11758. doi: 10.1371/journal.pone.0011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.