

Abstract
The structure activity relationship (SAR) of 1,2,4-triazolo[1,5-a]-1,3,5-triazine derivatives related to ZM241385 as antagonists of the A2A adenosine receptor (AR) was explored through the synthesis of analogues substituted at the 5 position. The A2A AR X-ray structure was used to propose a structural basis for the activity and selectivity of the analogues and to direct the synthetic design strategy to provide access to solvent-exposed regions. Thus, we have identified a point of substitution for the attachment of solubilizing groups to enhance both aqueous solubility and physicochemical properties, maintaining potent interactions with the A2A AR and, in some cases, receptor subtype selectivity. Among the most potent and selective novel compounds were a long-chain ether-containing amine congener 20 (Ki 11.5 nM) and its urethane-protected derivative 14 (Ki 17.8 nM). Compounds 20 and 31 (Ki 11.5 and 16.9 nM, respectively) were readily water soluble up to 10 mM. The analogues were docked in the crystallographic structure of the hA2A AR and in a homology model of the hA3 AR, and the per residue electrostatic and hydrophobic contributions to the binding were assessed and stabilizing factors were proposed.
Keywords: G protein-coupled receptor, purines, molecular modeling, structure activity relationship, radioligand binding, adenylyl cyclase
Adenosine receptors (ARs) are members of the family of G protein-coupled receptors (GPCRs), and are classified as four subtypes, the A1AR, A2AAR, A2BAR, and A3AR,1 which exert their physiological functions through the activation or inhibition of various second messenger systems. In particular, the modulation of adenylyl cyclase activity could be considered to be the principal signal mediated by these receptor subtypes.2,3
Activation or blockade of ARs is responsible for a wide range of effects in numerous organ systems raising the possibility that the regulation of ARs has potential therapeutic applications. The cardioprotective2,3 and neuroprotective4,5 effects associated with AR activation have been clearly demonstrated during periods of cardiac and cerebral ischemia, respectively. In addition, the use of antagonists of distinct AR subtypes could be useful in the treatment of asthma6,7 or certain neurological diseases such as Parkinson’s disease.7,8 The patho-physiological roles of ARs and their clinical potential have been recently reviewed exhaustively.7-12
In recent years, an intensive effort performed by several groups led to the synthesis of a large variety of AR agonists and antagonists for the pharmacological characterization of this family of G protein-coupled receptors.13 Among classes of AR antagonists, diverse heterocyclic derivatives have been proposed and found to display a range of affinities and selectivities. In particular, our groups have extensively investigated the pyrazolotriazolopyrimidine nucleus for AR antagonists. Optimization of the substituents at the N5, N7, N8 and C2 positions led to potent and selective A2A and A3 AR antagonists, e.g. compounds 1 and 2 (Chart 1).14-22
Chart 1.
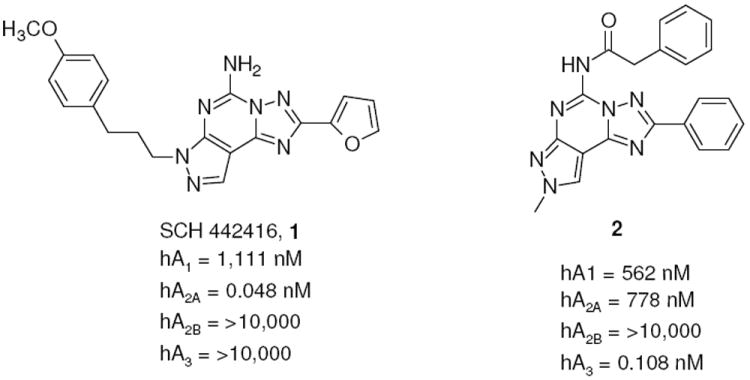
Pyrazolotriazolopyrimidines as A2A and A3 AR antagonists
Nevertheless, most of these heterocyclic derivatives, including other tricyclic structures, suffered from limited aqueous solubility and, most importantly, difficulties in their synthetic preparation.
Taking into account these problems, in recent years the synthesis of more simplified heterocyclic derivatives has been explored. In particular, bicyclic systems such as adenine,23 triazolopyrazine24-26 and triazolotriazine27-29 could be considered some of the most promising targets. The triazolotriazine nucleus was one of the most appealing bicyclic cores, which led previously to the discovery of 4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol (ZM241385, 3), i.e. one of the most potent and selective A2A AR antagonists known.30,31 This compound also binds with good affinity to the human (h) A2B AR (28 nM), and its tritiated form is a useful radioligand for this receptor subtype.32 Recently, an intensive study of the structure activity relationship (SAR) of the triazolotriazine nucleus was reported, and compound 4 proved to be one of the most potent and selective for the A2A AR as compared with the A1 AR. Nevertheless, the lack of binding data at the A2B and A3 ARs prevented a comparison with other fully characterized derivatives (Chart 2).27 Very recently, our group performed a study on this nucleus trying to optimize substitution at the C5 and N7 positions with the aim of improving affinity and selectivity versus the hA2B and hA3 AR subtypes. In particular, inclusion at the N7 position of arylcarbamoyl (for A3) or arylacetyl (for A2B) moieties, which gave good results in the pyrazolotriazolopyrimidine family, have been investigated.33 Unfortunately, none of these substitutions led to the desired selectivity; in fact introduction of bulky substituents at the N7 position (e.g., compound 5) significantly increased the potency at the hA3 AR with respect to reference compound 3 leading to low selectivity versus the A1 AR. In contrast, a free amino group at the 7 position (compound 6) provided intermediate potency at the A2B AR, but the potency at the A2A AR still predominated (Chart 3).33
Chart 2.
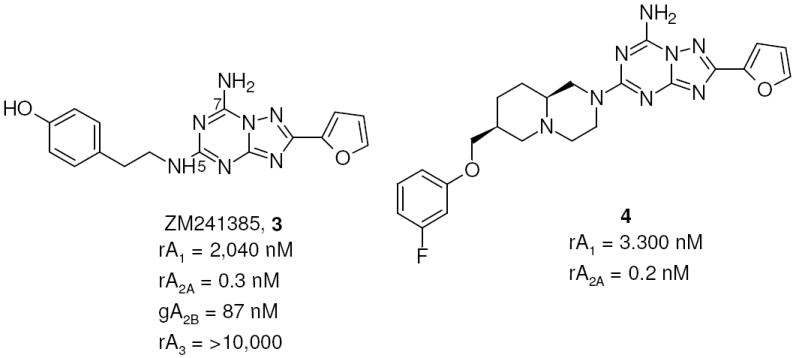
Triazolotriazines as A2A AR antagonists
Chart 3.
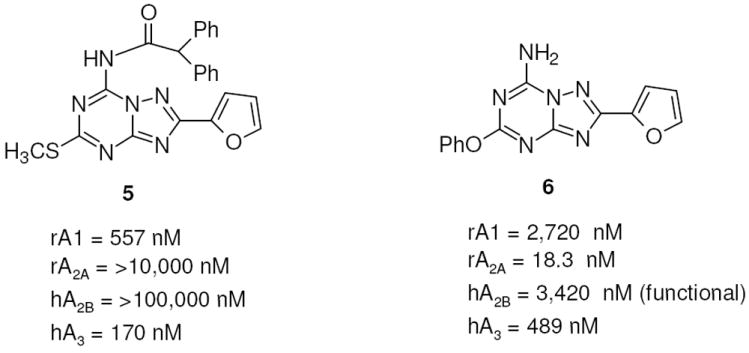
Examples of triazolotriazines as nonselective A2B and A3 AR antagonists
Considering these experimental observations, we decided to further investigate the potential of this nucleus, in particular, by exploring the C5 position through the introduction of substituted amino or diamino functions (Chart 4), with the aim to modulate the activity at the A2A and A2B ARs and importantly to improve the water solubility, which would otherwise limit their use as pharmacological tools. In contrast, this new class of derivatives mainly contained a free amino group at the 7 position, but small substituents were introduced in a few compounds with the aim of increasing the interaction in the receptor binding pocket.
Chart 4.
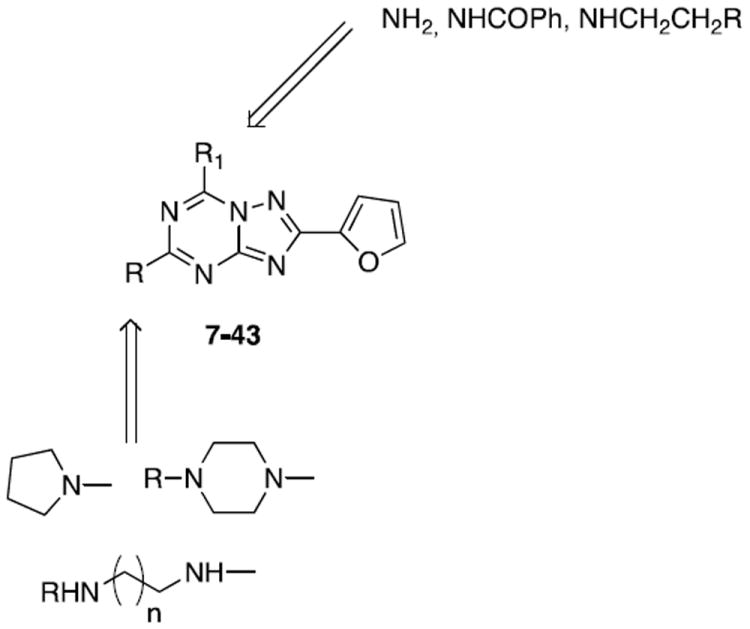
Designed and synthesized compounds 7-43
The analogues synthesized have been evaluated for potency at all four hARs, and the results obtained were interpreted with the help of computational methodologies.
Chemistry
The designed compounds (7-43) were synthesized as summarized in Schemes 1-3. Reacting the well-known key intermediate 4434 with pyrrolidine (51) or the appropriate mono-tert-butyloxy-protected diamine (45-50), prepared as previously reported,35 in ethanol solution in a sealed tube at 120 °C, the desired 5-substituted triazolotriazine derivatives (7, 10-14) were obtained. Treatment of these compounds (7, 10-14) with 10% trifluoroacetic acid in CH2Cl2 at room temperature, led to the corresponding amino derivatives (9, 20, 30-33) as trifluoroacetate salts (Scheme 1). Alternately, by reacting 44 with an excess of mono-Boc-ethylendiamine (47) in a sealed tube at 180 °C the 5,7-disubstituted derivative (37) was obtained. After N-Boc deprotection in acidic conditions, the bis-trifluoroacetate salt 38 was obtained (Scheme 1).
Scheme 1.
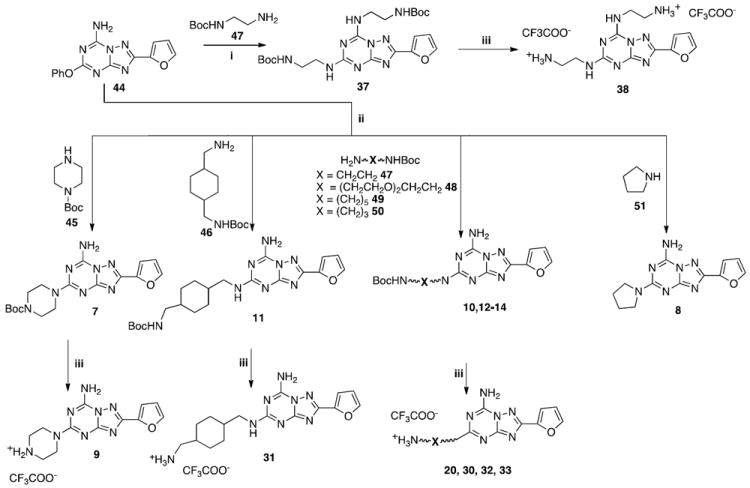
Reagents: i: EtOH, sealed tube, amine excess, 180 °C; ii: EtOH, sealed tube 120 °C; iii: CF3COOH, CH2Cl2, rt
Scheme 3.
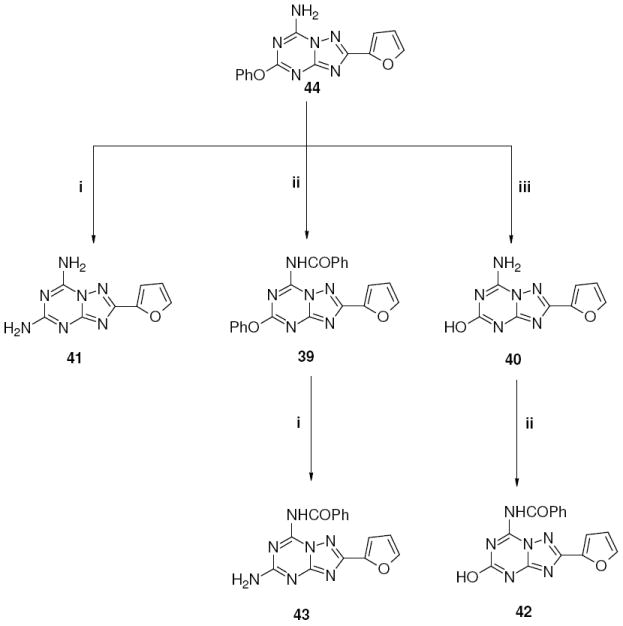
Reagents: i: NH3, EtOH, sealed tube, 120 °C; ii: PhCOCl, Et3N, dioxane reflux; iii: KOH, EtOH reflux.
By treatment of derivative 9 with the appropriate benzyl halide or acyl chloride in dry dioxane in the presence of Et3N at room temperature, the corresponding N-benzyl (15-19) or N-acyl (21-29, 34-36) derivatives were obtained (Scheme 2).
Scheme 2.
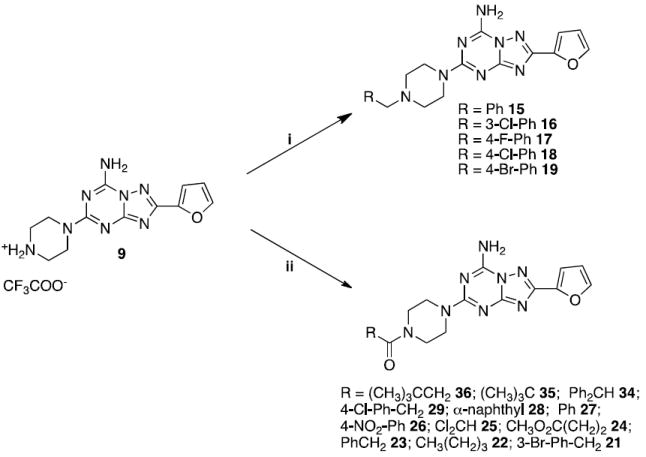
Reagents: i: RCH2Br, Et3N, dioxane, rt; ii: RCOCl, Et3N, dioxane, rt
Disubstituted derivatives at the 5 and 7 positions (39-43) were prepared starting from 44 as depicted in Scheme 3. Treating 44 with ethanolic ammonia in a sealed tube at 120 °C afforded derivative 41. Otherwise, acylation at the 7 position with benzoyl chloride in standard conditions led to compound 39, which after treatment in a sealed tube at 120 °C with ethanolic ammonia gave compound 43 (Scheme 3).
Instead, treatment of 44 with 2N KOH in ethanol at reflux led to the 5-hydroxy derivative 40, which after treatment with benzoyl chloride in the presence of Et3N afforded the desired product 42.
Molecular Modeling
To better define the SAR profile of the AR antagonists reported herein, a molecular modeling study was performed. The recently published X-ray crystallographic structure of the hA2A AR, in complex with the high affinity antagonist ZM241385 (PDB code: 3EML),36 provides useful three-dimensional structural information for performing molecular docking studies of A2A AR antagonists. Therefore, all the synthesized compounds were docked to the binding cavity of the A2A AR crystal structure, in order to evaluate their binding mode at this receptor subtype. Moreover, to explain the A2A/A3 selectivity profile, docking simulations were also performed at the hA3 AR binding site. Since no crystallographic information is available for the A3 subtype, a previously proposed homology model of the hA3 AR based on the crystal structure of the hA2A AR was used to perform the docking studies.37,38
In the initial process of selecting a reliable docking protocol to be employed in docking studies of the new derivatives, we have evaluated the ability of different docking software programs to reproduce the crystallographic pose of ZM241385 inside the binding cavity of hA2A AR. As reported in the Experimental Section, among the four different programs used to calibrate our docking protocol, the Gold program was finally chosen, because it showed the best performance with respect to the calculated RMSD values relative to the crystallographic pose of ZM241385.37,39 Consequently, based on the selected docking protocol, we performed docking simulations to identify the hypothetical binding mode of the new 1,2,4-triazolo[1,5-a]-1,3,5-triazines inside the crystallographic structure of hA2A AR and the hA3 AR model. All the newly synthesized compounds were docked into the orthosteric transmembrane (TM) binding cavities of both ARs. Additionally, in order to analyze the possible ligand-receptor recognition mechanism in a more quantitative manner, we calculated the individual electrostatic and hydrophobic contributions to the interaction energy of each receptor residue involved in the binding with ligands. Using the calculated electrostatic and hydrophobic contributions values, color maps of electrostatic and hydrophobic interactions per residue were constructed.
Results and Discussion
Table 1 reports the receptor binding affinities of the synthesized compounds (7-43). The binding properties were determined at the hA1, hA2A, and hA3 ARs expressed in human embryonic kidney (HEK)-293 cells using as radioligands: [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX, A1);40 [3H](4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-yl-amino]ethyl)-4-iodo-phenol, ([3H]ZM241385, A2A);41 or in Chinese hamster ovary (CHO) cells using [125I]N6-(4-amino-3-iodobenzyl)-5’-N-methylcarbamoyladenosine ([125I]I-AB-MECA, A3).42 Due to the lack of a suitable hA2B AR radioligand, the activity of antagonists was determined in adenylyl cyclase experiments in CHO cells expressing the hA2B AR.43,44
Table 1.
Structures and binding profile of synthesized compounds 7-43.
| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | hA1 % displ 10μM (or Ki, nM)a | hA2A (Ki, nM)b | hA2B IC50 (nM)c | hA3 % displ 10μM (or Ki, nM)d | hA1/hA2A | hA3/hA2A |
| 7 |
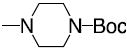
|
5400 ± 500 | 21 ± 4.5 | >10,000 | 56.6 ± 1.3% | 257 | ~400 |
| 8 |
|
1940 ± 70 | 59.1 ± 13.7 | >30,000 | 75.0 ± 4.7% | 33 | e |
| 9 |
|
28.6 ± 6.0% | 153 ± 32 | >30,000 | 29.9 ± 0.4% | >65 | >65 |
| 37 | - | 7.4 ± 4.5% | 4360 ± 1670 | >100,000 | 8490 ± 4810 | >2 | 1.9 |
| 10 | NH-(CH2)2NHBoc | 3730 ± 740 | 40.9 ± 5.7 | >10,000 | 1490 ± 480 | 91 | 36 |
| 11 |
|
3,890 ± 560 | 69.7 ± 12.7 | 7140 ± 2960 | 75.5 ± 0.3% | 56 | e |
| 12 | NH-(CH2)3NHBoc | 344 ± 16 | 38.6 ± 11.5 | 20,600 ± 1100 | 77.3 ± 0.3% | 9 | e |
| 13 | NH-(CH2)5NHBoc | 963 ± 139 | 44.7 ± 9.9 | >10,000 | 68.6 ± 0.9% | 21.5 | e |
| 14 | NH-(CH2CH2O)2-(CH2)2NHBoc | 1580 ± 100 | 17.8 ± 6.7 | >10,000 | 74.0 ± 0.8% | 89 | e |
| 15 |
|
978 ± 146 | 20.6 ± 14.5 | >30,000 | 71.9 ± 0.7% | 47.5 | e |
| 16 |
|
1510 ± 500 | 51.8 ± 23.0 | >30,000 | 52.7 ± 1.2% | 29 | ~200 |
| 17 |
|
655 ± 100 | 52.3 ± 32.3 | >10,000 | 71.3 ± 1.9% | 12.5 | e |
| 18 |
|
220 ± 88 | 102 ± 108 | >30,000 | 66.8 ± 6.0% | 2.1 | e |
| 19 |
|
86.7 ± 22.6 | 27.3 ± 18.0 | >10,000 | 49.8 ± 0.8% | 3.2 | ~300 |
| 20 | NH-(CH2CH2O)2-(CH2)2NH3+ | 769 ± 308 | 11.5 ± 2.2 | >10,000 | 59.2 ± 2.6% | 67 | e |
| 21 |
|
1,010 ± 150 | 86.2 ± 41.4 | >30,000 | 3100 ± 1100 | 11.7 | 36 |
| 22 |
|
10.8 ± 5.3% | 124 ± 50 | >100,000 | 56.2 ± 8.1% | >81 | e |
| 23 |
|
3510 ± 220 | 94.8 ± 40.7 | >100,000 | 64.5 ± 1.8% | 37 | e |
| 24 |
|
19.4 ± 2.5% | 66.2 ± 10.4 | >100,000 | 37.2 ± 6.6% | >150 | >150 |
| 25 |
|
34.6 ± 1.5% | 282 ± 164 | >100,000 | 33.6 ± 0.7% | >36 | >36 |
| 26 |
|
35.5 ± 7.4% | 212 ± 49 | >100,000 | 50.9 ± 6.9% | >47 | ~40 |
| 27 |
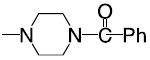
|
27.7 ± 3.3% | 348 ± 76 | >100,000 | 22.6 ± 1.7% | >29 | >29 |
| 28 |
|
4.9 ± 0.8% | 7680 ± 2190 | >100,000 | 31.1 ± 8.4% | >1.3 | >1.3 |
| 29 |
|
991 ± 226 | 39.1 ± 13.9 | >100,000 | 1260 ± 490 | 25 | 32 |
| 38 | - | 10.7 ± 1.5% | 38,200 ±14,600 | >100,000 | 17.7 ± 3.6% | 0.3 | e |
| 30 | NH-(CH2)2NH3+ | 35.8 ± 4.9% | 567 ± 43 | >100,000 | 30.8 ± 3.7% | >18 | >18 |
| 31 |
|
1350 ± 70 | 16.9 ± 2.3 | 10,700 ± 3,700 | 72.8 ± 0.4% | 79 | e |
| 32 | NH-(CH2)3NH3+ | 44.3 ± 1.6% | 270 ± 9 | >30,000 | 40.5 ± 0.2% | >37 | >37 |
| 33 | NH-(CH2)5NH3+ | 2,950 ± 260 | 90.1 ± 15.9 | >10,000 | 49.9 ± 3.5% | 33 | ~100 |
| 34 |
|
7470 ± 6820 | 92.9 ± 24.7 | >100,000 | 40.6 ± 22.2% | 80 | ~100 |
| 35 |
|
38.7 ± 4.8% | 58.4 ± 18.9 | >100,000 | 32.3 ± 3.37% | >170 | >170 |
| 36 |
|
53.1 ± 4.7% | 11.1 ± 6.3 | >100,000 | 41.9 ± 8.3% | ~900 | >900 |
| 39 | OPh | 26.2 ± 5.5% | 1000 ± 110 | >10,000 | 32.3 ± 9.7% | >10 | >10 |
| 40 | OH | 3110 ± 970 | 201 ± 61 | >10,000 | 43.9 ± 6.0% | 15.5 | ~50 |
| 41 | NH2 | 1410 ± 80 | 160 ± 42 | 11,100 ± 3,900 | 36.4 ± 0.2% | 9 | >60 |
| 42 | OH | 34.4 ± 9.2% | 1750 ± 390 | >100,000 | 75.9 ± 1.2% | >6 | e |
| 43 | NH2 | 1800 ± 150 | 44.1 ± 13.6 | 7060 ± 2935 | 67.2 ± 3.3% | 40 | e |
Displacement of specific [3H]DPCPX binding at the hA1 AR expressed in HEK-293 cells.
Displacement of specific [3H]ZM241385 binding at hA2A AR expressed in HEK-293 cells. Data are expressed as Ki ± SEM in nM (n=3-6).
Measurement of adenylyl cyclase activity in CHO cells stably transfected with recombinant hA2B AR, expressed as IC50 (nM).
Displacement of specific [125I]I-AB-MECA binding at hA3 receptors expressed in CHO cells. Data are expressed as Ki ± SEM in nM (n=3-6).
Selectivity not determined or estimated.
As clearly indicated in Table 1, all the analogues were in general nearly inactive at the hA3 AR independent of the substitution at the 5 and 7 positions, with the exception of compounds 10 and 29 having affinity in the range of 1 μM. A similar behavior, in contrast to expectations, could be observed at the hA2B AR; in fact all the analogues were inactive or poorly active (e.g. compounds 11, 31, 41, 43) in the functional assay. Nevertheless, the binding profile of this class of compounds at the hA2A AR demonstrated affinity in the nanomolar range and with different degrees of selectivity versus the hA1 AR subtype.
Insertion at the C5 position of amino or mono-Boc-diamino (compounds 7,8,10-14) functionality led to compounds with good affinity at the hA2A AR (range 18-70 nM) with variable selectivity versus the A1 AR. In particular, the presence of a pyrrolidine (8) at the 5 position favored affinity at the A2A AR (59.1 nM) but poor selectivity (33-fold) versus the A1 subtype, while introduction of Boc-piperidine (7) or Boc-ethylendiamine (10) seem to improve both affinity and selectivity at the A2A AR (e.g compound 7, hA2A=21 nM, A1/A2A=257). Interestingly, elongation of the diamino chain at the C5 position to 3 or 5 carbon atoms (compounds 12, 13) led to derivatives which still retained good affinity at the hA2A AR but less selectivity versus the A1 subtype (e.g. compound 13 hA2A=44.7 nM, A1/A2A = 21.5), while longer chains such as a Boc-triethylenoxydiamino moiety (14) gave good results both in terms of affinity and selectivity (hA2A= 17.8 nM, A1/A2A= 89).
Trifluoroacetate salt derivatives (9, 30-33) were in general less potent than the corresponding Boc derivatives (e.g. compound 7, hA2A=21 nM, A1/A2A= 257 versus compound 9, hA2A= 153 nM, A1/A2A= 65). An exception occurred when long or bulky chains were present at the C5 position. In fact, compounds 20 and 31 were more potent at the hA2A AR than the corresponding Boc derivatives (e.g. compound 31, hA2A=16.9 nM, A1/A2A= 79; compound 11: hA2A= 69.7 nM, A1/A2A= 56), but most importantly both compounds were readily water soluble up to 10 mM. In particular, compound 20, which displayed an affinity at the hA2A AR of 11.5 nM and good selectivity versus A1, was the most potent compound of this series. Despite this relevant improvement of the water solubility other pharmacokinetics properties, such as cell permeation or metabolic stability, are collecting in our laboratories before setting up any further in vivo testing.
Interestingly, double substitution with diamino functions at the C5 and N7 positions led to completely inactive compounds at all four AR subtypes, both as protected (37) or deprotected (38) forms.
An altered binding profile was observed for the piperazine derivatives alkylated or acylated at the piperazine secondary amine. In particular, in the N-benzyl series (15-19) good affinity at the hA2A AR was retained, but also an increased affinity at the hA1 was observed with a consequent reduction of selectivity, independently of the type of substitution on the phenyl ring.
A more complex profile was present when an acyl group was present on the piperazine secondary amine. In particular, when a benzoyl group was introduced on the piperazine nitrogen (26, 27) a significant reduction of affinity at the hA2A AR (range 212-348 nM) was observed with a subsequent reduction of selectivity (35-48 fold). If the phenyl ring was replaced with a more bulky substituent such as a naphthyl nucleus (compound 28) a complete loss of affinity at all four hARs was observed. If the aroyl group was replaced with an aryl acetyl moiety significant differences could be observed in binding profile. In particular, introduction of a phenylacetyl (23) or a 4-substituted-phenylacetyl group (21, 29) at the piperazine secondary amine led to compounds that still retained good affinity at the hA2A AR in the range of 40-95 nM with poor selectivity versus A1 (12-37 fold). In contrast, introduction of a bulky substituent such as a diphenylacetyl moiety (34) provided a compound with a quite good affinity at the hA2A AR (hA2A= 93 nM). Importantly, the selectivity versus the hA1 subtype (A1/A2A=80) was high, which was exactly the opposite observed for the nearly inactive bulky aroyl derivative 28.
A quite different profile could be observed when the piperazine secondary amine was acylated with alkyl groups. In particular, introduction of a small group (25) or unbranched chains (22, 24) led to compounds which proved to be quite potent (range 66-280 nM) at the hA2A AR. Branched chains such as tert-butylcarbonyl (35) and 3,3-dimethylbutanoyl (35) increased the affinity at the hA2A AR and significantly increased of selectivity versus the hA1 subtype. In particular, derivative 36 showed high affinity at the hA2A AR (11.1 nM) and 900-fold selectivity versus the hA1 subtype.
The derivatives substituted both at the C5 and N7 positions with small groups (39-43) showed in general weak affinity at the hA2A AR and low levels of selectivity. Moreover, the diamino compound 41 and the corresponding 5-hydroxy derivative 40 showed hA2A affinity in the high nanomolar range (160-200 nM), but the levels of selectivity versus the hA1 AR were very low, ranging from 2 to 12. When a benzoyl group was present at the N7 position, the presence at the C5 position of a hydroxy (42) or phenoxy (39) moiety greatly reduced activity at the four ARs, with affinity at the hA2A subtype in the micromolar range. In contrast, when an amino group was present at the C5 position, affinity at the hA2A AR (hA2A= 44.1 nM) was recovered with good levels of selectivity versus other receptor subtypes.
In order to rationalize the observed binding data, a molecular modeling investigation was performed for all the newly synthesized analogues using both the crystallographic structure of hA2A AR and the hA3 AR model. The analysis was extended to docking simulations and per residue electrostatic and hydrophobic contributions maps.
The first important consideration is that almost all the selected poses at the hA2A AR of these new analogues showed some common features, as highlighted by the calculated electrostatic and hydrophobic contributions to the interaction energy collected in Figure 1. In particular, all ligands made contacts mainly with residues belonging to TM2, TM3, TM6, TM7 and EL2.
Figure 1.
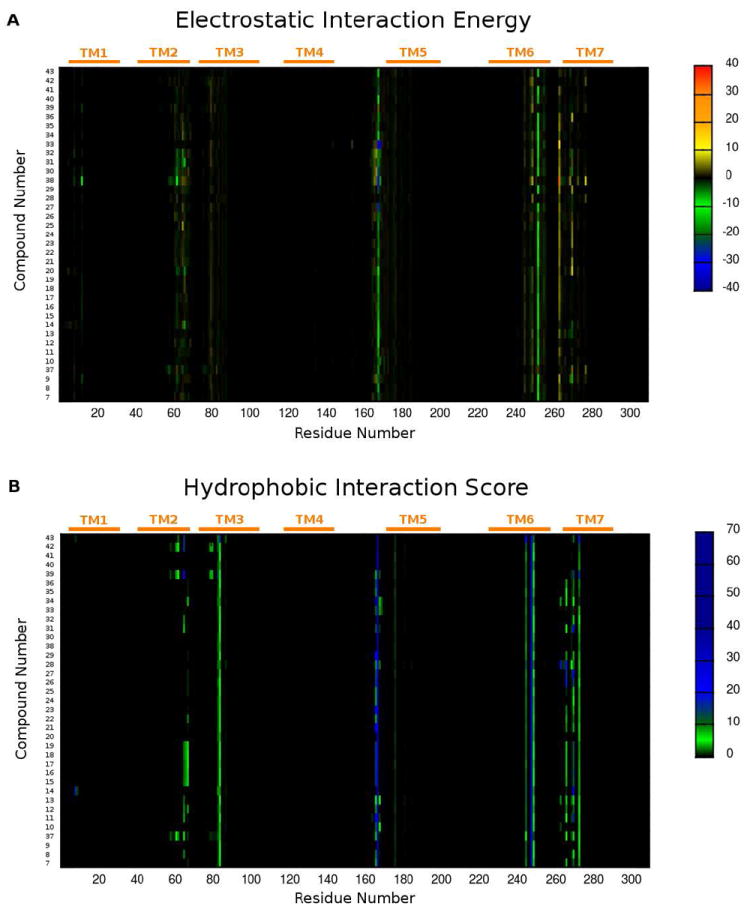
(A) per residue electrostatic interaction energy map and (B) per residue hydrophobic interaction score map. The maps are calculated for a selected pose of each compound (7-43) inside the hA2A AR binding site. Electrostatic energy values are expressed in kcal/mol while hydrophobic scores are expressed in arbitrary hydrophobic units.
The per residue electrostatic interaction energy map (Figure 1, panel A) showed two bands with negative energy (colored in green) corresponding to Glu169 in EL2 and Asn253 in TM6, indicating that these two residues were responsible for the main electrostatic interactions with all the tested analogues, with a few exceptions. On the other hand, the map of the per residue hydrophobic interaction score (Figure 1, panel B) highlighted several residues involved in hydrophobic contacts with ligands, including Leu85 in TM3, Phe168 in EL2, Trp246, Leu249, His250 in TM6 and Tyr271, Ile274 in TM7. Therefore, the analysis of these maps gave important preliminary data concerning similarity and differences in the binding modes at the hA2A AR of these new compounds; this information was then confirmed by a detailed investigation of the docking poses as reported below.
Docking of 7-amino derivatives (compounds 7-36, 40 and 41)
From the docking simulation analysis, all the new derivatives with free amino group at the 7 position, with the exception of compound 28, were seen to share a similar binding pose in the TM region of the hA2A AR. For these compounds, ligand-recognition occurred in the upper region of the TM bundle, and the triazolotriazine nucleus was surrounded by TMs 3, 5, 6, 7 with the 2-furyl ring located deep in the binding cavity.
Considering Figure 2, it is evident that the binding poses of these ligands were very similar to the crystallographic pose of ZM241385 bound to the hA2A AR.36 In fact, the triazolotriazine cores were completely superposable, while the only slight difference was in the orientation of the substituents at the 5-position. Moreover, all the crucial interactions established by ZM241385 with amino acid residues of the hA2A AR binding site were also found for all these new 7-amino derivatives.
Figure 2.
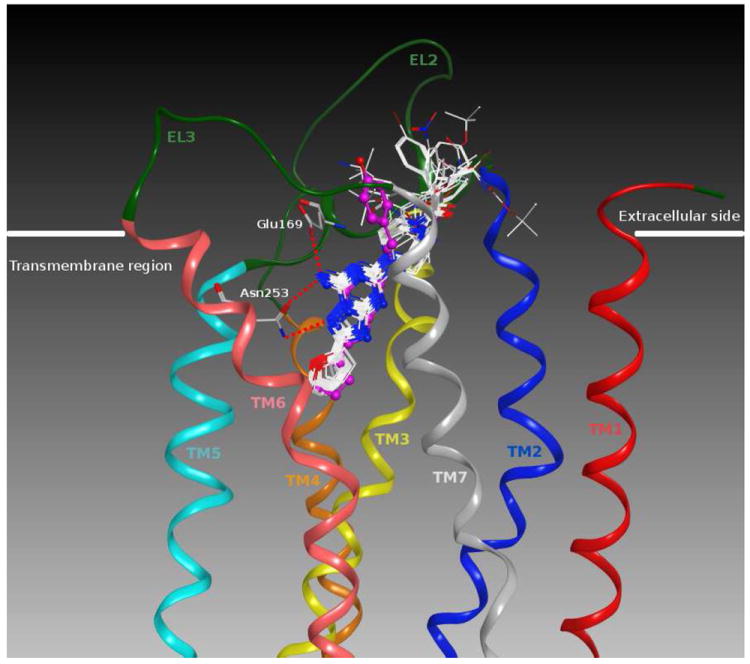
Structure superimposition of the crystallographic pose of ZM241385 (in magenta) and of the docking poses of all the 7-amino derivatives (in white) inside the hA2A AR binding site. Side chains of some amino acids important for ligand recognition and H-bonding interactions are highlighted. Hydrogen atoms are not displayed.
The analysis in Figure 3 (panel A) showing the hypothetical binding pose of compound 36 (Ki hA2A = 11.1 nM) at the hA2A AR helps to clarify this point. It appeared that the bicyclic triazolotriazine core was anchored within the binding cleft through an aromatic stacking interaction with Phe168 (EL2) and a H-bonding interaction with Asn253 (6.55). Moreover, the exocyclic amino group at the 7-position of the bicyclic core interacted with two polar residues, Asn253 (6.55) and Glu169 (EL2), forming two H-bonds. Interestingly, the important role in ligand binding of these two residues was previously revealed by site-directed mutagenesis studies.45,46 The 4-(3,3-dimethylbutanoyl)-piperazinyl chain of the ligand was directed towards the more solvent exposed extracellular region (EL2 and EL3) and interacted, through a H-bond, with Tyr271 (7.36). However, the furan ring was located deep within the ligand binding cavity and formed hydrophobic interactions with the highly conserved Trp246 (6.48), an important residue in receptor activation. Finally, compound 36 also formed hydrophobic interactions with many residues of the binding site including Val84 (3.32), Leu85 (3.33), Met177 (5.38), Leu249 (6.51) and Ile274 (7.39).
Figure 3.
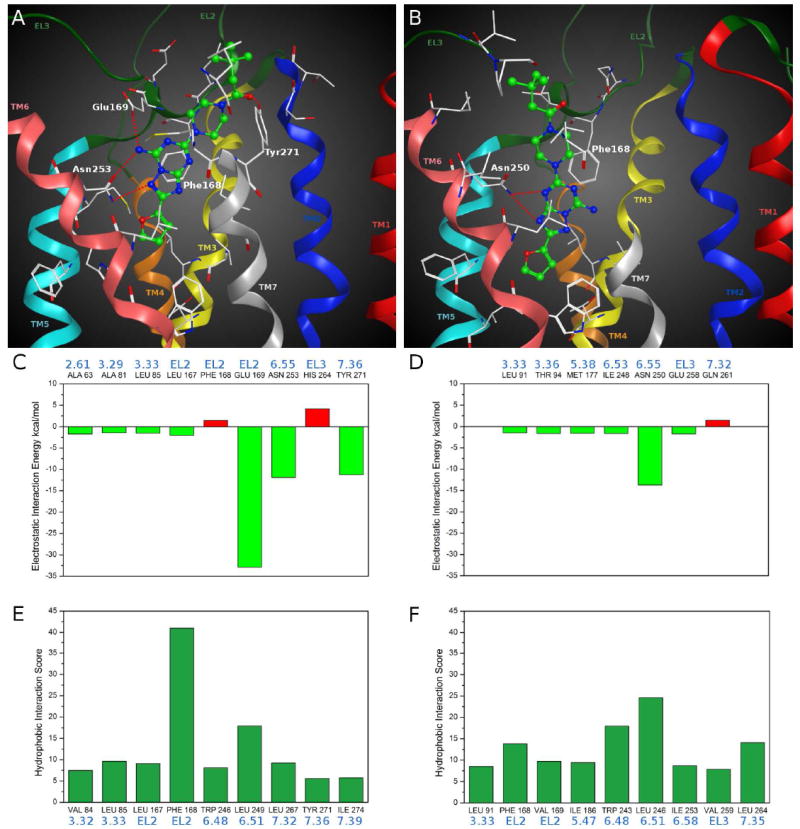
Hypothetical binding modes of compound 36 obtained after docking simulations: (A) inside the hA2A AR binding site; (B) inside the hA3 AR binding site. Poses are viewed from the membrane side facing TM6, TM7 and TM1. The view of TM7 is partially omitted. Side chains of some amino acids important for ligand recognition and H-bonding interactions are highlighted. Hydrogen atoms are not displayed. Electrostatic interaction energy (in kcal/mol) between the ligand and each single amino acid involved in ligand recognition observed from the hypothetical binding modes of compound 36 inside (C) hA2A AR and (D) hA3 AR binding sites. Hydrophobic interaction scores (in arbitrary hydrophobic units) between the ligand and each single amino acid involved in ligand recognition observed from the hypothetical binding modes of compound 36 inside (E) hA2A AR and (F) hA3 AR binding sites.
Analyzing the electrostatic contribution per residue to the whole interaction energy for the compound 36-hA2A AR complex (Figure 3, panel C), the three main stabilizing factors were found to be related to Glu169 (EL2), Asn253 (6.55) and Tyr271 (7.36), due to the H-bonding interactions with the ligand above described. Whereas, as shown in Figure 3 (panel E), the hydrophobic interaction scores pattern showed two strong stabilizing contributions corresponding to the interactions of the bicyclic core with Phe168 (EL2) and Leu249 (6.51).
Therefore, as exemplified by the binding pose of compound 36, all the newly synthesized 7-amino derivatives strongly interacted with the hA2A AR in a manner similar to the crystallographic pose of ZM241385.36 Moreover, these compounds, thanks to their different substituents at the 5 position, could variously interact with residues of the upper region of the receptor binding cavity, particularly in EL2 and EL3. Considering that the substituents at the 5 position were exposed to the highly plastic EL region and to solvent, it was difficult to define a clear SAR at the 5 position for this series.
On the other hand, the docking pose of compound 36 at the hA3 AR was located in the same region of the TM bundle as at the hA2A AR, but the orientation of the ligand was different (Figure 3, panel B). In this case the ligand formed only two H-bonds with Asn250 (6.55) and lost the aromatic interaction with Phe168 (EL2). The patterns of electrostatic and hydrophobic contribution to the energy of hA3 AR-ligand complexes (Figure 3, panels D and F) showed weaker per residue contributions compared to the ones at the hA2A AR. Moreover, the residues present at the binding pocket entrance in the two AR subtypes possess very different features, which could affect both the orientation of the ligand while approaching the binding pocket and its accommodation into the final TM binding cleft, as already proposed for other compounds.37 Therefore, both the lack of very strong interactions with the residues of the hA3 AR and the differences at the binding site entrance are consistent with a lack of hA3 affinity observed for the 7-amino derivative.
Among the 7-amino derivatives, only compound 28 (Ki hA2A = 7680 nM) showed a different docking pose at the hA2A AR (see Supporting Information). This ligand, probably due to the hindrance of the bulky and rigid α-naphthyl group, was not able to occupy the same position as the other 7-amino derivatives and consequently lost important H-bonding interactions with two critical residues of the hA2A AR binding site, such as Glu169 (EL2) and Asn253 (6.55). In fact, in this docking pose the ligand formed only one H-bond with Tyr271 (7.36). This finding explained why compound 28 showed lower hA2A AR affinity compared to the other 7-amino derivatives.
Docking of 7-(alkyl/acyl)amino derivatives (compounds 37-39, 42 and 43)
With the exception of compound 43, all the 7-(alkyl/acyl)amino derivatives showed low affinity for the hA2A AR (micromolar range). Docking studies revealed that compounds 37-39 and 42 possessed a different binding mode at the hA2A AR compared to the 7-amino derivatives (see Supporting Information). In fact, the presence of an (alkyl/acyl)amino group at the 7 position prevented these compounds from forming a H-bonding network with Asn253 (6.55) and Glu169 (EL2), already seen to be critical for the binding of ZM241385 at this receptor subtype. This fact led these compounds to assume a different orientation inside the binding cavity of the receptor, although ligand-recognition occurred in the same upper region of the TM bundle, and the triazolotriazine nucleus was surrounded by TMs 3, 5, 6, 7 with the 2-furyl ring directed towards the inner part of the binding cavity. Therefore, their binding mode showed only a weak H-bond with Glu169 (EL2) and a stacking interaction with Phe168 (EL2). These findings were in agreement with the experimental data showing micromolar Ki values for these compounds.
In contrast to the 5 position derivatives described above, compound 43 (Ki hA2A = 44.1 nM), due to the presence of a free amino group at C5, showed a characteristic mode of binding at the hA2A AR (see Supporting Information). The triazolotriazine nucleus was oriented parallel to the membrane plane, and the 2-furyl ring was directed towards TM2, while the substituent at the 7 position was located in the inner part of the binding cavity. Compound 43 formed three H-bonding interactions, two with Asn253 (6.55) and one with Glu169 (EL2), and a π-π stacking interaction with Phe168 (EL2). Therefore, the presence of a H-bonding network with the residues of the hA2A AR, similar to the one seen for the 7-amino derivatives, seemed to explain why compound 43, among the 7-(alkyl/acyl)amino derivatives, is the only one that showed affinity at the hA2A AR in the nanomolar range.
In conclusion, we have synthesized AR antagonists of the triazolotriazine class and use molecular modeling studies to explain the SAR. Among the most potent and selective novel compounds were a long-chain ether-containing amine congener 20 and its urethane-protected derivative 14. Compound 20 and a 5-(aminomethyl)cyclohexylmethyl-amino derivative 31 were readily water soluble up to 10 mM, thus overcoming a common limitation of other bicyclic and tricyclic AR antagonists. N-Alkylated and N-acylated piperidine derivatives also displayed high affinity at the human A2A AR, including an N-benzyl antagonist 19 and an N-3,3-dimethylbutanoyl derivative 36 that was roughly 900-fold selective versus both human A1 and A3 ARs. The analogues were docked in the crystallographic structure of hA2A AR and in a homology model of the hA3 AR, and the per residue electrostatic and hydrophobic contributions to the binding were assessed. The presence of a free amino group that seemed to be critical for the hA2A affinity by allowing the ligands to participate in a H-bonding network with two critical residues of the binding site, Asn253 (6.55) and Glu169 (EL2). Another interaction found to be important for the hA2A affinity of this series was the aromatic stacking between the triazole ring and Phe168 (EL2).
Conclusion
The present study has lead to the identification of a new class of promising AR antagonists, the 1,2,4-triazolo[1,5-a]-1,3,5-triazine derivatives related to ZM241385. In particular, compound 20 (Ki 11.5 nM) and a 5-(aminomethyl)cyclohexylmethyl-amino derivative 31 (Ki 16.9 nM) were readily water soluble up to 10 mM, thus overcoming a common limitation of other bicyclic and tricyclic AR antagonists. Finally, a receptor-based SAR has been proposed using a molecular modeling approach. All analogues were docked in the recently published crystallographic structure of hA2A AR and in a homology model of the hA3 AR, and the per residue electrostatic and hydrophobic contributions to the binding were assessed.
Molecular modeling results highlighted that all the newly synthesized 1,2,4-triazolo[1,5-a]-1,3,5-triazine derivatives with free amino group at the 7 position are characterized by a common binding mode very similar to the crystallographic one of ZM241385 bound to the hA2A AR. In fact, all the interactions found to be crucial for the X-ray structure, in particular the hydrogen bonding network with Asn253 (6.55), the Glu169 (EL2) and the aromatic stacking with Phe168 (EL2), were also important for anchoring these new derivatives to the hA2A AR binding site. On the contrary, substitution at the 7 position resulted to be detrimental for the affinity at the hA2A AR, as confirmed also by the orientation of the 7-(alkyl/acyl)amino derivatives inside the binding cavity that lead to the loss of the stabilizing hydrogen bonding network.
Thus, we have probed points of substitution for attachment of solubilizing groups to enhance the aqueous solubility of this class of triazolotriazines, which are characterised by poor physicochemical properties. At the same time, potent interactions with the A2A AR and, in some cases, receptor subtype selectivity have been maintained. We have used the A2A AR X-ray structure to propose a structural basis for the activity and selectivity of this class of analogues and to direct the synthetic design strategy to provide access to solvent-exposed regions.
In general, the strategy of grafting a terminal polar tail on a pharmacophore, which increases the polar surface area, can have a detrimental effect on bioavailability and ion channel activity. Therefore, it will be necessary to evaluate these molecules in further pharmacological testing to see if they will be useful for in vivo studies.
Experimental Section
Chemical synthesis
General
Reactions were routinely monitored by thin-layer chromatography (TLC) on silica gel (precoated F254 Merck plates). Infrared spectra (IR) were measured on a Jasco FT-IT instrument. 1H NMR were determined in CDCl3 or DMSO-d6 solutions with a Varian Gemini 200 spectrometer, peaks positions are given in parts per million (δ) downfield from tetramethylsilane as internal standard, and J values are given in Hz. The following abbreviations were used: s, singlet; bs, broad singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet.
Light petroleum ether refers to the fractions boiling at 40-60 °C. Melting points were determined on a Buchi-Tottoli instrument and are uncorrected. Flash chromatography was performed using Merck 60-200 mesh silica gel. Purity of compounds was detected by elemental analyses performed at the laboratory of microanalysis of the Department of Chemistry, University of Trieste (Italy), and all the compounds were confirmed to achieve 95% purity.
General procedure for nucleophilic substitution with amino compounds (7, 8, 10-14)
A mixture of compound 44 (0.29 g, 1 mmol) and the appropriate amine (45-51) (1.5 eq) in absolute ethanol (5 mL) was poured into a sealed tube and heated at 120 °C for 3 h. Then the solvent was removed under reduced pressure and the residue crystallized from EtOAc-light petroleum to afford the desired compounds (7, 8, 10-14) as solids.
7-Amino-5-(N-pyrrolidinyl)-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (8)
Yield: 88%; brown solid (EtOAc-light petroleum) mp >300 °C (dec.) 1H NMR (DMSO d6) δ: 1.9-2.1 (m, 4H); 3.4-3.7 (m, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3350-3200, 1620, 1480. Anal. (C12H13N7O) C, H, N.
7-Amino-5-[(4-tert-butyloxycarbonyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (7)
Yield 73%; white solid (EtOAc-light petroleum) mp 250 °C. 1H NMR (DMSO d6) δ: 1.4 (s, 9H); 3.4 (s, 4H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3400-3150, 1735, 1630, 1510. Anal. (C17H22N8O3) C, H, N.
7-Amino-5-[(4-tert-butyloxycarbonylaminomethyl)cyclohexylmethyl]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (11)
Yield 76%; white solid (EtOAc-light petroleum) mp 148 °C.1H NMR (CDCl3) δ: 1.2-2.0 (m, 6H); 1.4 (s, 9H); 2.9-3.2 (m, 2H); 3.3-3.5 (m, 2H); 4.6 (bs, 1H); 5.4 (bs, 1H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.3 (bs, 2H); 7.9 (bs, 2H). IR (Nujol) cm-1: 3450-3100, 1735, 1620, 1470. Anal. (C21H28N8O3) C, H, N.
7-Amino-5-[(2-tert-butyloxycarbonylamino)ethyl]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (10)
Yield 63%; white solid (EtOAc-light petroleum) mp 168 °C. 1H NMR (DMSO d6) δ: 1.4 (s, 9H); 3.1-3.4 (m, 4H); 6.7 (dd, 1H, J=2, J=4); 6.9 (bs, 1H); 7.1 (d, 1H, J=2); 7.4 (bs, 1H); 7.9 (d, 1H, J=4); 8.2 (bs, 2H). IR (Nujol) cm-1: 3450-3150, 1740, 1630, 1500. Anal. (C15H20N8O3) C, H, N.
7-Amino-5-[(3-tert-butyloxycarbonylamino)propyl]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (12)
Yield 76%; white solid (EtOAc-light petroleum) mp 155 °C. 1H NMR (CDCl3) δ: 1.5 (s, 9H); 1.80-1.85 (m, 2H); 3.20-3.25 (m, 2H); 3.5-3.6 (m, 2H); 4.9 (bs, 1H); 5.4 (bs, 1H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 7.9 (bs, 2H). IR (Nujol) cm-1: 3400-3100, 1740, 1620, 1510. Anal. (C16H22N8O3) C, H, N.
7-Amino-5-[(5-tert-butyloxycarbonylamino)n-pentyl]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (13)
Yield 71%; white solid (EtOAc-light petroleum) mp 140 °C. 1H NMR (CDCl3) δ: 1.4 (s, 9H); 1.6-1.8 (m, 6H); 3.2-3.3 (m, 2H); 3.50-3.55 (m, 2H); 4.7 (bs, 1H); 5.5 (bs, 1H); 6.40-6.43 (m, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4). IR (Nujol) cm-1: 3450-3150, 1735, 1625, 1475. Anal. (C18H26N8O3) C, H, N.
7-Amino-5-[(8-tert-butyloxycarbonylamino)triethylenoxy]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (14)
Yield 64%; brown solid (EtOAc-light petroleum) mp 130 °C. 1H NMR (CDCl3) δ: 1.5 (s, 9H); 3.4-3.8 (m, 12H); 5.5 (bs, 1H); 6.2 (bs, 1H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 9.4 (bs, 2H). IR (Nujol) cm-1: 3400-3100, 1740, 1610, 1450. Anal. (C19H28N8O5) C, H, N.
Procedure for preparation of 5,7-bis-[(2-tert-butyloxycarbonylamino) ethyl]-amino-2-(2-furyl)-1,2,4-triazolo [1,5-a] 1,3,5-triazine (37)
A mixture of compound 44 (0.29 g, 1 mmol) and the mono Boc ethylendiamine (47) (0.8g, 5 eq) in absolute ethanol (5 mL) was poured into a sealed tube and heated at 180 °C for 8 h. Then the solvent was removed under reduced pressure and the residue crystallized from EtOAc-light petroleum to afford the desired compound (37) as a white solid in a good yield (70%). mp 154 °C. 1H NMR (DMSO d6) δ: 1.4 (s, 18H); 3.0-3.4 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 6.9 (bs, 1H); 7.1 (d, 1H, J=2);7.6 (bs, 1H); 7.9 (d, 1H, J=4); 8.5 (bs, 1H); 8.7 (bs, 1H). IR (Nujol) cm-1: 3450, 1730, 1610, 1450. Anal. (C22H33N9O5) C, H, N.
General procedure for N-Boc deprotection (9, 20, 30-33, 38)
10 mmol of the appropriate N-Boc derivative (7, 8, 10-14, 37) was suspended in a 10% solution of CF3COOH in dry CH2Cl2 and stirred at room temperature for 3 h. Then the solvent was removed under reduced pressure and the residue crystallized form AcOEt-light petroleun to give the desired trifluoroacetate salts (9, 20, 30-33, 38) as solids.
5,7-bis-(Amino)ethyl-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine bis-trifluoroacetate (38)
Yield 63%; brown solid (EtOAc-light petroleum) mp 203 °C. 1H NMR (DMSO d6) δ: 3.4-3.6 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.0 (bs, 1H); 8.0 (bs, 3H). IR (Nujol) cm-1: 3500-3100, 1630, 1500. Anal. (C16H19N9O5 F6) C, H, N.
7-Amino-5-(1-piperazinyl)-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (9)
Yield 58%; brown solid (EtOAc-light petroleum) mp 243 °C. 1H NMR (DMSO d6) δ: 3.20-3.25 (m, 4H); 4.0-4.1 (m, 4H); 6.0 (bs, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 9.0 (bs, 2H). IR (Nujol) cm-1: 3500-3000, 1630, 1510. Anal. (C14H15N8O3F3) C, H, N.
7-Amino-5-(aminomethyl)cyclohexylmethyl-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (31)
Yield 66%; brown solid (EtOAc-light petroleum) mp 150 °C. 1H NMR (DMSO d6) δ: 1.2-2.0 (m, 10H); 2.4-3.0 (m, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.0 (bs, 2H). IR (Nujol) cm-1: 3500-3050, 1640, 1500. Anal. (C18H23N8O3F3) C, H, N.
7-Amino-5-(2-aminoethyl)-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (30)
Yield 54%; brown solid (EtOAc-light petroleum) mp 243 °C. 1H NMR (DMSO d6) δ: 3.4-3.6 (m, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.5 (d, 1H, J=4); 7.8 (bs, 2H); 8.4 (bs, 2H); 8.0 (bs, 3H). IR (Nujol) cm-1: 3450-3150, 1630, 1510. Anal. (C12H13N8O3F3) C, H, N.
7-Amino-5-(3-amino)propyl-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (32)
Yield 69%; brown solid (EtOAc-light petroleum) mp 235 °C. 1H NMR (DMSO d6) δ: 3.4-3.6 (m, 6H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.5 (d, 1H, J=4); 7.8 (bs, 2H); 8.4 (bs, 2H); 8.0 (bs, 3H). IR (Nujol) cm-1: 3470-3140, 1640, 1510. Anal. (C13H15N8O3F3) C, H, N.
7-Amino-5-(5-amino)pentyl-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (33)
Yield 71%; brown solid (EtOAc-light petroleum) mp 160 °C. 1H NMR (DMSO d6) δ: 1.6-1.8 (m, 6H); 3.2 (m, 2H); 3.5 (m, 2H); 5.5 (bs, 1H); 6.4 (bd, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.0 (bs, 3H). IR (Nujol) cm-1: 3480-3120, 1640, 1500. Anal. (C15H19N8O3F3) C, H, N.
7-Amino-5-[(8-amino)triethylenoxy]-amino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (20)
Yield 52%; brown solid (EtOAc-light petroleum) mp 243 °C. 1H NMR (DMSO d6) δ: 3.4-3.8 (m, 12H); 5.5 (bs, 1H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 9.4 (bs, 2H); 8.0 (bs, 3H). IR (Nujol) cm-1: 3510-3150, 1630, 1510. Anal. (C15H22N8O5F3) C, H, N.
General procedure for N-benzylation of a piperazine moiety (15-19)
A mixture of compound 9 (0.2 g, 0.05 mmol) the appropriate benzyl halide (1.2 eq), Et3N (7.2 μL, 0.06 mmol) in dry dioxane (10 mL) was stirred at room temperature for 6 h. The solvent was removed in vacuo e the residue dissolved in EtOAc (15 mL) and washed with water (3 × 5 mL). The organic phase was dried and solvent removed under reduced pressure. The crude was purified by flash chromatography (EtOAc) to give the final compounds (15-19) as solids.
7-Amino-5-[(4-benzyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (15)
Yield 63%; white solid (EtOAc-light petroleum) mp 242 °C. 1H NMR (DMSO d6) δ: 2.5 (s, 4H); 3.6 (s, 2H); 3.9 (s, 4H); 6.2 (bs, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.6 (d, 1H, J=4); 7.1-7.4 (m, 5H). IR (Nujol) cm-1: 3350-3100, 1610, 1470. Anal. (C19H20N8O) C, H, N.
7-Amino-5-[(4-(3-chloro-benzyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (16)
Yield 70%; white solid (EtOAc-light petroleum) mp 227 °C. 1H NMR (DMSO d6) δ: 2.5 (s, 4H); 3.5 (s, 2H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.3-7.5 (m, 4H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3340-3100, 1620, 1480 Anal. (C19H19N8OCl) C, H, N.
7-Amino-5-[(4-(4-fluoro-benzyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (17)
Yield 66%; white solid (EtOAc-light petroleum) mp 260 °C. 1H NMR (DMSO d6) δ: 2.5 (s, 4H); 3.5 (s, 2H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.1-7.4 (m, 4H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3340- 3110, 1630, 1480. Anal. (C19H19N8OF) C, H, N.
7-Amino-5-[(4-(4-chloro-benzyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (18)
Yield 73%; white solid (EtOAc-light petroleum) mp 274 °C. 1H NMR (DMSO d6) δ: 2.5 (s, 4H); 3.5 (s, 2H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.3-7.5 (m, 4H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3350-3110, 1620, 1470. Anal. (C19H19N8OCl) C, H, N.
7-Amino-5-[(4-(4-bromo-benzyl)-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (19)
Yield 68%; white solid (EtOAc-light petroleum) mp 284 °C. 1H NMR (DMSO d6) δ: 2.5 (s, 4H); 3.5 (s, 2H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.3 (d, 2H); 7.5 (d, 2H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3340-3110, 1630, 1480. Anal. (C19H19N8OBr) C, H, N.
General procedure for N-piperazine acylation (21-29, 34-36)
A mixture of compound 9 (0.2 g, 0.05 mmol) the appropriate acyl chloride (1.2 eq), Et3N (7.2 μL, 0.06 mmol) in dry dioxane (10 mL) was stirred at room temperature for 2 h. The solvent was removed in vacuo and the residue dissolved in EtOAc (15 mL) and washed with water (3 × 5 mL). The organic phase was dried and solvent removed under reduced pressure. The crude was purified by flash chromatography (EtOAc) to give the final compounds (21-29, 34-36) as solids.
7-Amino-5-[(4-(pentanoyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (22)
Yield 66%; yellow solid (EtOAc-light petroleum) mp 167 °C. 1H NMR (DMSO d6) δ: 0.9 (t, 3H, J=7); 3.0-3.2 (m, 10H); 3.5 (s, 2H); 3.8 (s, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3350-3100, 1685, 1620, 1500. Anal. (C17H22N8O2) C, H, N.
7-Amino-5-[(4-(benzoyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (27)
Yield 69%; white solid (EtOAc-light petroleum) mp 290 °C. 1H NMR (DMSO d6) δ: 3.4-3.9 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.5 (m, 5H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3360-3120, 1670, 1620, 1510. Anal. (C19H18N8O2) C, H, N.
7-Amino-5-[(4-(benzyl-carbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (23)
Yield 64%; pale brown solid (EtOAc-light petroleum) mp 240 °C. 1H NMR (DMSO d6) δ: 3.5-3.7 (m, 8H); 3.8 (s, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.2-7.4 (m, 5H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3320-3100, 1680, 1610, 1500. Anal. (C20H20N8O2) C, H, N.
7-Amino-5-[(4-(2-bromo-benzyl-carbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (21)
Yield 76%; white solid (EtOAc-light petroleum) mp 223 °C. 1H NMR (DMSO d6) δ: 3.5-3.7 (m, 8H); 3.8 (s, 2H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.2-7.4 (m, 4H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3315-3080, 1670, 1620, 1500. Anal. (C20H19N8O2Br) C, H, N.
7-Amino-5-[(4-(methoxycarbonylethylcarbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (24)
Yield 74%; white solid (EtOAc-light petroleum) mp 160 °C. 1H NMR (DMSO d6) δ: 2.3-2.7 (m, 4H); 3.3 (s, 3H); 3.7-3.9 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3330-3110, 1665, 1615, 1510. Anal. (C17H20N8O4) C, H, N.
7-Amino-5-[(4-(dichloromethylcarbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (25)
Yield 70%; white solid (EtOAc-light petroleum) mp 261 °C. 1H NMR (DMSO d6) δ: 3.6 (s, 4H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.0 (s, 1H); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3320-3100, 1670, 1610, 1520. Anal. (C14H14N8O2Cl2) C, H, N.
7-Amino-5-[(4-(4-nitrobenzoyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (26)
Yield 69%; pale yellow solid (EtOAc-light petroleum) mp >300 °C (dec) 1H NMR (DMSO d6) δ: 3.7-3.9 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.7 (d, 2H); 7.9 (d, 1H, J=4); 8.3 (d, 2H); 8.4 (bs, 2H). IR (Nujol) cm-1: 3300-3110, 1665, 1615, 1510. Anal. (C19H17N9O4) C, H, N.
7-Amino-5-[(4-(4-chloro-benzyl-carbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4- triazolo [1,5-a] 1,3,5-triazine (29)
Yield 74%; white solid (EtOAc-light petroleum) mp 260 °C. 1H NMR (DMSO d6) δ: 3.7-3.9 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.2 (d, 2H); 7.3 (d, 2H); 7.7 (d, 2H, J=9); 7.9 (d, 1H, J=4); 8.3 (d, 2H, J=9); 8.4 (bs, 2H). IR (Nujol) cm-1: 3300-3100, 1675, 1625, 1500. Anal. (C20H17N8O2Cl) C, H, N.
7-Amino-5-[(4-(3,3-dimethylbutanoyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (36)
Yield 77%; white solid (EtOAc-light petroleum) mp 297 °C. 1H NMR (DMSO d6) δ: 1.0 (s, 9H); 2.3 (s, 2H); 3.6 (s, 4H); 3.8 (s, 4H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3290- 3100, 1680, 1615, 1510. Anal. (C18H24N8O2) C, H, N.
7-Amino-5-[(4-(diphenylmethylcarbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (34)
Yield 63%; white solid (EtOAc-light petroleum) mp 295 °C. 1H NMR (DMSO d6) δ: 3.6 (s, 4H); 3.8 (s, 4H); 5.6 (s, 1H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.2-7.4 (m, 10H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3310-3110, 1675, 1620, 1510. Anal. (C26H24N8O2) C, H, N.
7-Amino-5-[(4-(α-naphthoyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (28)
Yield 69%; white solid (EtOAc-light petroleum) mp 305 °C. 1H NMR (DMSO d6) δ: 3.6-4.0 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.5-8.1 (m, 7H); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3305-3100, 1670, 1610, 1500. Anal. (C23H20N8O2) C, H, N.
7-Amino-5-[(4-(tert-butylcarbonyl))-1-piperazinyl]-2(2-furyl) 1,2,4-triazolo [1,5- a] 1,3,5-triazine (35)
Yield 74%; white solid (EtOAc-light petroleum) mp >300 °C. 1H NMR (DMSO d6) δ: 1.2 (s, 9H); 3.6-3.8 (m, 8H); 6.7 (dd, 1H, J=2, J=4); 7.1 (d, 1H, J=2); 7.9 (d, 1H, J=4); 8.4 (bs, 2H). IR (Nujol) cm-1: 3300-3110, 1675, 1610, 1500. Anal. (C17H22N8O2) C, H, N.
Procedure for the preparation of 5,7-diamino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (41)
A mixture of compound 44 (0.29 g, 1 mmol) in absolute ethanol saturated with gas ammonia (2 mL) was poured into a sealed tube and heated at 120 °C for 3 h. Then the solvent was removed under reduced pressure and the residue crystallized from EtOAc-light petroleum to afford the desired compound (41) as a solid in a good yield (88%). White solid (EtOAc-light petroleum) mp >300 °C. 1H NMR (DMSO d6) δ: 6.68 (dd, 1H, J=2, J=4); 6.98 (bs, 2H); 7.13 (d, 1H, J=2); 7.81 (d, 1H, J=4); 8.03 (bs, 2H). IR (Nujol) cm-1: 3300-3110, 1640, 1600, 1525. Anal. (C8H7N7O) C, H, N.
Procedure for the preparation of 7-amino 5-hydroxy-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (40)
A mixture of compound 44 (0.29 g, 1 mmol) in 2M KOH in absolute ethanol (2 mL) was poured into a sealed tube and heated at 120 °C for 3 h. Then the solvent was removed under reduced pressure and the residue crystallized from EtOAc-light petroleum to afford the desired compound (40) as a solid in a good yield (76%). White solid (EtOAc-light petroleum) mp 206 °C. 1H NMR (DMSO d6) δ: 6.65 (dd, 1H, J=2, J=4); 7.18 (d, 1H, J=2); 7.93 (d, 1H, J=4); 8.81 (bs, 2H); 12.13 (bs, 1H). IR (Nujol) cm-1: 3350-3145, 1655, 1610, 1530. Anal. (C8H6N6O2) C, H, N.
General procedure for the N7 benzoylation (39, 42)
A mixture of appropriate amino compound compound (40, 44) (0.1 mmol) benzoyl chloride (14 μL, 0.12 mmol), Et3N (14 μL, 0.12 mmol) in dry dioxane (10 mL) was stirred at reflux for 12 h. The solvent was removed in vacuo and the residue dissolved in EtOAc (15 mL) and washed with water (3 × 5 mL). The organic phase was dried and solvent removed under reduced pressure. The crude was purified by flash chromatography (EtOAc) to give the final compounds (39, 42) as solids.
5-Hydroxy-7-phenylcarbonylamino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (42)
Yield 75%; white solid (EtOAc-light petroleum) mp 201 °C. 1H NMR (DMSO d6) δ: 6.66 (dd, 1H, J=2, J=4); 7.21 (d, 1H, J=2); 7.45-7.98 (m, 7H); 10.52 (bs, 1H). IR (Nujol) cm-1: 3340-3150, 1687, 1635, 1615, 1530. Anal. (C15H10N6O3) C, H, N.
5-Phenoxy-7-phenylcarbonylamino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine (39)
Yield 87%; pale yellow solid (EtOAc-light petroleum) mp 229 °C (dec.). 1H NMR (DMSO d6) δ: 6.60 (dd, 1H, J=2, J=4); 7.12 (d, 1H, J=2); 7.25-7.65 (m, 7H); 7.80-7.99 (m, 4H). IR (Nujol) cm-1: 3325-3070, 1695, 1620, 1510. Anal. (C21H14N6O3) C, H, N.
Procedure for the preparation of 5-amino-7-phenylcarbonylamino-2(2-furyl) 1,2,4-triazolo [1,5-a] 1,3,5-triazine trifluoroacetate (43)
A mixture of compound 39 (0.39 g, 1 mmol) in absolute ethanol saturated with gas ammonia (3 mL) was poured into a sealed tube and heated at 120 °C for 3 h. Then the solvent was removed under reduced pressure and the residue crystallized from EtOAc-light petroleum to afford the desired compound (43) as a solid in a good yield (70%). Pale brown solid (EtOAc-light petroleum) mp 187 °C. 1H NMR (DMSO d6) δ: 6.62 (dd, 1H, J=2, J=4); 7.18 (d, 1H, J=2); 7.38-7.52 (m, 3H); 7.60-7.81 (m, 4H); 7.99 (d, 1H, J=4); 8.98 (bs, 1H). IR (Nujol) cm-1: 3325-3105, 1690, 1640, 1605, 1540. Anal. (C15H11N7O2) C, H, N.
Biological testing
Radioligand Binding to hA1, A2A and A3 ARs
[3H]DPCPX, [3H]ZM241385, and [125I]I-AB-MECA were utilized in radioligand binding assays to membranes prepared from HEK-293 cells expressing recombinant hA1, and hA3 ARs, and from CHO cells expressing the recombinant hA2A AR, as previously described.40-42 Adenosine deaminase (3 units/mL) was present during the preparation of the membranes, in a preincubation of 30 min at 30 °C, and during the incubation with the radioligands. All non-radioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%. Incubations were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each. At least six different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in GraphPad (Prism, San Diego, CA), were converted to Ki values as described.47 Hill coefficients of the tested compounds were in the range of 0.8-1.1.
Adenylyl cyclase activity
Due to the lack of a suitable radioligand the affinity of antagonists and the relative potency of agonists at the A2B AR was determined in adenylyl cyclase experiments. The procedure was carried out as described previously43,44 with minor modifications. Membranes were incubated with about 150,000 cpm of [α-32P]ATP for 20 min in the incubation mixture as described43,44 without EGTA and NaCl. For agonists the EC50-values for the stimulation of adenylyl cyclase were calculated with the Hill equation. Hill coefficients in all experiments were near unity. IC50 values for antagonist concentration-dependent inhibition of adenylyl cyclase stimulated by 5’-N-ethylcarboxamidoadenosine (NECA) were calculated accordingly.
Molecular modeling
All modeling studies were carried out on a 20 CPU (Intel Core2 Quad CPU 2.40 GHz) Linux cluster. Homology modeling, energy calculation, and analyses of docking poses were performed using the Molecular Operating Environment (MOE, version 2009.10) suite.48 The software package MOPAC (version 7),49 implemented in MOE suite, was utilized for all quantum mechanical calculations. Docking simulations were performed using GOLD suite (version 1.3.2).50
Three-dimensional structures of hA2A AR and hA3 AR
The recently published crystallographic structure of hA2A AR, in complex with the high affinity antagonist ZM241385 (PDB code: 3EML),36 was used to perform the molecular docking studies at this receptor subtype.
Moreover, on the basis of the assumption that GPCRs share similar TM boundaries and overall topology, a homology model of the hA3 AR was constructed with the software MOE, as previously reported,37,38 using as template the crystal structure of hA2A AR.36
The numbering of the amino acids follows the arbitrary scheme by Ballesteros and Weinstein. According to this scheme, each amino acid identifier starts with the helix number, followed by the position relative to a reference residue among the most conserved amino acid in that helix. The number 50 is arbitrarily assigned to the reference residue.51
Molecular Docking of hA2A AR Antagonists
Ligand structures were built using MOE-builder tool, part of the MOE suite,48 and were subjected to MMFF94x energy minimization until the rms conjugate gradient was <0.05 kcal mol−1 Å−1. Partial charges for the ligands were calculated using PM3/ESP methodology.
Four different programs have been used to calibrate our docking protocols: MOE-Dock, 48 GOLD,50 Glide,52 and PLANTS.53 In particular, ZM241385 was redocked into the crystal structure of the hA2A AR (PDB code: 3EML) with different docking algorithms and scoring functions, as already described.37,39 Then, rmsd values between predicted and crystallographic positions of ZM241385 were calculated for each of the docking algorithms. The results showed that docking simulations performed with GOLD gave the lowest rmsd value, the lowest mean rmsd value and the highest number of poses with rmsd value <2.5 Å.
On the basis of the best docking performance, all antagonist structures were docked into the TM binding site of the hA2A AR crystal structure and that of the hA3 AR model by using the docking tool of the GOLD suite.50 Searching was conducted within a user-specified docking sphere, using the Genetic Algorithm protocol and the GoldScore scoring function. GOLD performed a user-specified number of independent docking runs (25 in our specific case) and wrote the resulting conformations and their scores in a molecular database file. The resulting docked complexes (ligand and side chains of residues at 4.5 Å from the ligand) were subjected to MMFF94x energy minimization until the rms conjugate gradient was <1 kcal mol−1 Å−1. Partial charges for the ligands were calculated using MOPAC and PM3/ESP methodology.
Prediction of antagonist-receptor complex stability (in terms of corresponding pKi value) and the quantitative analysis for nonbonded intermolecular interactions (H-bonds, transition metal, water bridges, hydrophobic, electrostatic) were calculated and visualized using several tools implemented in MOE suite.48 Electrostatic and hydrophobic contributions to the binding energy of individual amino acids have been calculated using MOE.48 To estimate the electrostatic contributions, atomic charges for the ligands were calculated using PM3/ESP methodology. Partial charges for protein amino acids were calculated on the basis of the AMBER99 force field.
Supplementary Material
Acknowledgments
The work coordinated by S.M. was carried out with financial support from the University of Padova, Italy, and the Italian Ministry for University and Research (MIUR), Rome, Italy. This research was also supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. S.M. is very grateful to Chemical Computing Group Inc. (Montreal, Quebec, Canada) for the scientific and technical partnership.
Abbreviations
- AR
adenosine receptor
- CHO
Chinese hamster ovary
- DMSO
dimethylsulfoxide
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- EDTA
ethylenediaminetetraacetic acid
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- I-AB-MECA
1-[6-[[(4-amino-3-iodophenyl)methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-D-ribofuranuronamide; NECA, 5’-N-ethylcarboxamidoadenosine; TLC, thin layer chromatography
- ZM241385
4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol
- SAR
structure-activity relationship
- TM
transmembrane
- RMSD
root mean square deviation
- EL2
second extracellular loop
Footnotes
Supporting information available: elemental analyses and modeling figures. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Structure and function of adenosine receptors and their genes. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:364–374. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 2.Downey JM, Cohen MV, Ytrehus K, Liu Y. Cellular mechanisms in ischemic preconditioning: the role of adenosine and protein kinase Ca. Ann N Y Acad Sci. 1994;723:82–98. [PubMed] [Google Scholar]
- 3.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol. 1997;52:846–860. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson KA, von Lubitz DKJE, Daly JW, Fredholm BB. Adenosine receptor ligands: differences with acute versus chronic treatment. Trends Pharmacol Sci. 1996;17:108–113. doi: 10.1016/0165-6147(96)10002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredholm BB. Adenosine and neuroprotection. Int Rev Neurobiol. 1997;40:259–280. [PubMed] [Google Scholar]
- 6.Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Adenosine A2B receptors: a novel therapeutic target in asthma? Trends Pharmacol Sci. 1998;19:148–153. doi: 10.1016/s0165-6147(98)01179-1. [DOI] [PubMed] [Google Scholar]
- 7.Fozard JR, McCarthy C. Adenosine receptor ligands as potential therapeutics in asthma. Curr Opin Investig Drugs. 2002;3:69–77. [PubMed] [Google Scholar]
- 8.Richardson PJ, Kase H, Jenner PG. Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol Sci. 1997;18:338–344. doi: 10.1016/s0165-6147(97)01096-1. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro JA, Sebastiao AM, de Mendonca A. Adenosine receptors in the nervous system: pathophysiological implications. Prog Neurobiol. 2002;68:377–392. doi: 10.1016/s0301-0082(02)00155-7. [DOI] [PubMed] [Google Scholar]
- 10.Yan L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerg Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 11.Fredholm BB. Adenosine receptors as targets for drug development. Drug News Perspect. 2003;16:283–289. doi: 10.1358/dnp.2003.16.5.829316. [DOI] [PubMed] [Google Scholar]
- 12.Moro S, Spalluto G, Jacobson KA. Techniques: Recent developments in computer-aided engineering of GPCR ligands using the human A3 adenosine receptor as an example. Trends Pharmacol Sci. 2005;26:44–51. doi: 10.1016/j.tips.2004.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]; (b) Moro S, Gao ZG, Jacobson KA, Spalluto G. Progress in the pursuit of therapeutic adenosine receptor antagonists. Med Res Rev. 2006;26:131–159. doi: 10.1002/med.20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Monopoli A, Ongini E, Varani K, Borea PA. 7-Substituted 5-amino-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as A2A adenosine receptor antagonists: a study on the importance of modifications at the side chain on the activity and solubility. J Med Chem. 2002;45:115–126. doi: 10.1021/jm010924c. [DOI] [PubMed] [Google Scholar]
- 15.Baraldi PG, Cacciari B, Spalluto G, Bergonzoni M, Dionisotti S, Ongini E, Varani K, Borea PA. Design, synthesis, and biological evaluation of a second generation of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as potent and selective A2A adenosine receptor antagonists. J Med Chem. 1998;41:2126–2133. doi: 10.1021/jm9708689. [DOI] [PubMed] [Google Scholar]
- 16.Baraldi PG, Cacciari B, Spalluto G, Pineda de las Infantas y Villatoro MJ, Zocchi C, Dionisotti S, Ongini E. Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives: potent and selective A2A adenosine antagonists. J Med Chem. 1996;39:1164–1171. doi: 10.1021/jm950746l. [DOI] [PubMed] [Google Scholar]
- 17.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Klotz K-N, Leung E, Varani K, Gessi S, Merighi S, Borea PA. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists. J Med Chem. 1999;42:4473–4478. doi: 10.1021/jm991114s. [DOI] [PubMed] [Google Scholar]
- 18.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Moro S, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists: Influence of the chain at N8pyrazole nitrogen. J Med Chem. 2000;43:4768–4780. doi: 10.1021/jm001047y. [DOI] [PubMed] [Google Scholar]
- 19.Baraldi PG, Cacciari B, Moro S, Spalluto G, Pastorin G, Da Ros T, Klotz K-N, Varani K, Gessi S, Borea PA. Synthesis, Biological Activity, and Molecular Modeling Investigation of New Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine Derivatives as Human A3 Adenosine Receptor Antagonists. J Med Chem. 2002;45:770–780. doi: 10.1021/jm0109614. [DOI] [PubMed] [Google Scholar]
- 20.Maconi A, Pastorin G, Da Ros T, Spalluto G, Gao ZG, Jacobson KA, Baraldi PG, Cacciari B, Varani K, Borea PA. Synthesis, Biological properties and molecular modeling investigation of the first potent, selective and water soluble human A3 adenosine receptor antagonist. J Med Chem. 2002;45:3579–3582. doi: 10.1021/jm020974x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastorin G, Da Ros T, Spalluto G, Deflorian F, Moro S, Cacciari B, Baraldi PG, Gessi S, Varani K, Borea PA. Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as adenosine receptor antagonists. Influence of the N5 substituent on the affinity at the human A3 and A2B adenosine receptor subtypes: a molecular modeling investigation. J Med Chem. 2003;46:4287–96. doi: 10.1021/jm030852k. [DOI] [PubMed] [Google Scholar]
- 22.Cheong SL, Dolzhenko A, Kachler S, Paoletta S, Federico S, Cacciari B, Dolzhenko A, Klotz KN, Moro S, Spalluto G, Pastorin G. The significance of 2-furyl substitutions with a 2-(para-substituted) aryl group in a new series of pyrazolo-triazolo-pyrimidines as potent and highly selective hA3 adenosine receptor antagonists: new insight into structure activity relationship and receptor antagonist recognition. J Med Chem. 2010;53:3361–3375. doi: 10.1021/jm100049f. [DOI] [PubMed] [Google Scholar]
- 23.Pinna A, Volpini R, Cristalli G, Morelli M. New adenosine A2A receptor antagonists: actions on Parkinson’s disease models. Eur J Pharmacol. 2005;512:157–164. doi: 10.1016/j.ejphar.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 24.Vu CB, Pan D, Peng B, Kumaravel G, Phadke D, Engber T, Huamg C, Reilly J, Tam S, Petter RC. Studies on adenosine A2A receptor antagonists: comparison of three core heterocycles. Bioorg Med Chem Lett. 2004;14:4831–4834. doi: 10.1016/j.bmcl.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 25.Dowling JE, Vessels JT, Haque S, Chang HX, van Vloten K, Kumaravel G, Engber TM, Jin X, Phadke D, Wang J, Ayyub E, Petter RC. Synthesis of [1,2,4]triazolo[1,5-a]pyrazines as adenosine A2A receptor antagonists. Bioorg Med Chem Lett. 2005;15:4809–4813. doi: 10.1016/j.bmcl.2005.07.052. [DOI] [PubMed] [Google Scholar]
- 26.Gang Y, Haque S, Sha L, Kumaravel G, Wang J, Engber TM, Whalley ET, Conlon PR, Chang H, Kiesman WF, Petter RC. Synthesis of alkyne derivatives of novel triazolopyrazines as A2A adenosine receptor antagonists. Bioorg Med Chem Lett. 2005;15:511–515. doi: 10.1016/j.bmcl.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 27.Vu CB, Shields P, Peng B, Kumaravel G, Jin X, Phadke D, Wang J, Engber T, Ayyub E, Petter RC. Triamino derivatives of triazolo triazine and triazolopyrimidines as adenosine A2A receptor antagonists. Bioorg Med Chem Lett. 2004;14:4835–4838. doi: 10.1016/j.bmcl.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 28.Peng B, Kumaravel G, Yao G, Sha L, Van Vlijmen H, Bohnert T, Huang C, Vu CB, Ensinger CL, Chang H, Engber TM, Whalley E, Petter RC. Novel bicyclic piperazine derivatives of triazolotriazine and triazolopyrimidine as highly potent and selective adenosine A2A receptor antagonists. J Med Chem. 2004;47:6218–6229. doi: 10.1021/jm0494321. [DOI] [PubMed] [Google Scholar]
- 29.Vu CB, Pan D, Kumaravel G, Smits G, Jin X, Phadke D, Engber T, Huang C, Reilly J, Tam S, Grant D, Hetu G, Petter RC. Novel diamino derivatives of [1,2,4]triazolo[1,5-a] [1,3,5]triazine as potent and selective adenosine A2A receptor antagonists. J Med Chem. 2005;48:2009–2018. doi: 10.1021/jm0498396. [DOI] [PubMed] [Google Scholar]
- 30.Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, Collis MG. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br J Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeZwart M, Vollinga RC, Beukers MW, Sleegers DF, von Frijtag Drabbe Kunzel JK, De Groote M, IJzerman AP. Potent antagonists for the human adenosine A2B receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM241385 with high affinity. Drug Dev Res. 1999;48:95–103. [Google Scholar]
- 32.Ji XD, Jacobson KA. Use of triazolotriazine [3H]-ZM241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Des Discov. 1999;16:217–226. [PMC free article] [PubMed] [Google Scholar]
- 33.Pastorin G, Federico S, Paoletta S, Corradino M, Cateni F, Cacciari B, Klotz KN, Gao ZG, Jacobson KA, Spalluto G, Moro S. Synthesis and pharmacological characterization of a new series of 5,7-disubstituted [1,2,4]triazolo[1,5-a] [1,3,5]triazine derivatives as adenosine receptor antagonists: a preliminary inspection of ligand-receptor recognition process. Bioorg Med Chem. 2010;18:2524–2536. doi: 10.1016/j.bmc.2010.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caulkett PWR, Jones G, McPartlin M, Renshaw ND, Stewart SK, Wright B. Adenine isosteres with bridgehead nitrogen. Part 1. Two independent syntheses of the [1,2,4]triazolo[1,5-a][1,3,5]triazine ring system leading to a range of substituents in the 2, 5 and 7 positions. J Chem Soc Perkin Trans I. 1995:801–808. [Google Scholar]
- 35.Lee DW, Ha HJ, Lee WK. Selective Mono Boc-protection of diamines. Synth Commun. 2007;37:737–742. [Google Scholar]
- 36.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenzi O, Colotta V, Catarzi D, Varano F, Poli D, Filacchioni G, Varani K, Vincenzi F, Borea PA, Paoletta S, Morizzo E, Moro S. 2-Phenylpyrazolo[4,3-d]pyrimidin-7-one as a New Scaffold to Obtain Potent and Selective Human A3 Adenosine Receptor Antagonists: New Insights into the Receptor-Antagonist Recognition. J Med Chem. 2009;52:7640–7652. doi: 10.1021/jm900718w. [DOI] [PubMed] [Google Scholar]
- 38.Morizzo E, Federico S, Spalluto G, Moro S. Human A3 adenosine receptor as versatile G protein-coupled receptor example to validate the receptor homology modeling technology. Curr Pharm Des. 2009;15:4069–4084. doi: 10.2174/138161209789824777. [DOI] [PubMed] [Google Scholar]
- 39.Pastorin G, Federico S, Paoletta S, Corradino M, Cateni F, Cacciari B, Klotz K-N, Gao Z-G, Jacobson KA, Spalluto G, Moro S. Synthesis and pharmacological characterization of a new series of 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives as adenosine receptor antagonists: A preliminary inspection of ligand-receptor recognition process. Bioorg Med Chem. 2010;18(7):2524–2536. doi: 10.1016/j.bmc.2010.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruns RF, Fergus JH, Badger EW, Bristol JA, Santay LA, Hartman JD, Hays SJ, Huang CC. Binding of the A1-selective adenosine antagonist 8-cyclopentyl-1,3-dipropyl-xanthine to rat brain membranes. Naunyn-Schmiedeberg’s Arch Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- 41.Palmer TM, Poucher SM, Jacobson KA, Stiles GL. 125I-4-(2-[7-Amino-2-{furyl}{1,2,4}triazolo{2,3-a}{1,3,5}triazin-5-ylaminoethyl)phenol (125I-ZM241385), a high affinity antagonist radioligand selective for the A2a adenosine receptor. Mol Pharmacol. 1996;48:970–974. [PMC free article] [PubMed] [Google Scholar]
- 42.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl-5’-N-methylcarboxamidoadenosine, a highaffinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 43.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes-characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 44.Klotz KN, Cristalli G, Grifantini M, Vittori S, Lohse MJ. Photoaffinity labeling of A1-adenosine receptors. J Biol Chem. 1985;260:14 659–14664. [PubMed] [Google Scholar]
- 45.Kim J, Wess J, van Rhee M, Schoneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2A adenosine receptor. J Biol Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson KA. Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol Pharmacol. 1996;49:683–691. [PMC free article] [PubMed] [Google Scholar]
- 47.Linden J. Calculating the dissociation constant of an unlabeled compound from the concentration required to displace radiolabel binding by 50% J Cyclic Nucleotide Res. 1982;8:163–172. [PubMed] [Google Scholar]
- 48.MOE (Molecular Operating Environment), version 2009.10; software available from Chemical Computing Group Inc. (1010 Sherbrooke Street West, Suite 910, Montreal, Quebec, Canada H3A 2R7); http://www.chemcomp.com
- 49.Stewart JJP. MOPAC 7. Fujitsu Limited; Tokyo, Japan: 1993. [Google Scholar]
- 50.GOLD suite, version 1.3.2; software available from Cambridge Crystallographic Data Centre Cambridge Crystallographic Data Centre (12 Union Road Cambridge CB2 1EZ UK); http://www.ccdc.cam.ac.uk
- 51.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relationships in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 52.Halgren TA, Myrphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT. Glide: a new approach for rapid, accurate docking and scoring 1 methods and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 53.Korb O, Stützle T, Exner TE. Empirical Scoring Functions for advanced Protein-Ligand Docking with PLANTS. J Chem Inf Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.