

Abstract
Choroid plexus papillomas are rare, benign tumors originating from the choroid plexus. Although generally found within the ventricular system, they can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. We sought to review recent advances in our understanding of the molecular biology and oncogenic pathways associated with this disease. A comprehensive PubMed literature review was conducted to identify manuscripts discussing the clinical, molecular, and genetic features of choroid plexus papillomas. Articles concerning diagnosis, treatment, and long-term patient outcomes were also reviewed. The introduction of atypical choroid plexus papilloma as a distinct entity has increased the need for accurate histopathologic diagnosis. Advances in immunohistochemical staining have improved our ability to differentiate choroid plexus papillomas from other intracranial tumors or metastatic lesions using combinations of key markers and mitotic indices. Recent findings have implicated Notch3 signaling, the transcription factor TWIST1, platelet-derived growth factor receptor, and the tumor necrosis factor–related apoptosis-inducing ligand pathway in choroid plexus papilloma tumorigenesis. A combination of commonly occurring chromosomal duplications and deletions has also been identified. Surgical resection remains the standard of care, although chemotherapy and radiotherapy may be considered for recurrent or metastatic lesions. While generally considered benign, these tumors possess a complex biology that sheds insight into other choroid plexus tumors, particularly malignant choroid plexus carcinomas. Improving our understanding of the molecular biology, genetics, and oncogenic pathways associated with this tumor will allow for the development of targeted therapies and improved outcomes for patients with this disease.
Keywords: choroid plexus papilloma, choroid plexus tumor, diagnosis, treatment, tumorigenesis
The choroid plexus is a complex epithelial-endothelial convolute composed of an epithelium, stroma, and vascular supply. The stroma contains fibroblasts, inflammatory cells, and a rich extracellular matrix.1 There are 4 segments of the choroid plexus, each occupying either the lateral, third, or fourth ventricles. The choroid plexus is responsible for the production of cerebrospinal fluid (CSF) and has been implicated in autoimmune inflammation within the central nervous system by virtue of expression of major histocompatibility complex classes I and II on choroid plexus epithelial cells.1 Choroid plexus tumors are of neuroectodermal origin and range from benign choroid plexus papillomas (CPPs) to malignant choroid plexus carcinomas (CPCs). It is unclear whether diffuse villous hyperplasia of the choroid plexus, a rare congenital condition, represents a precursor lesion that warrants placement on this disease spectrum.2
CPPs are rare, indolent neoplasms. They have an annual incidence of 0.3 per 1 000 000 and outnumber CPCs by a factor of 5:1.3–6 They represent 0.3%–0.6% of all intracranial tumors, with a male to female ratio of 1.2:1.5 CPPs are most common within the first year of life and comprise 10%–20% of brain tumors in this demographic.3–6 This is generally a disease of childhood, with a median age at diagnosis of 3.5 years.5 There is a strong correlation with age and tumor location; over 80% of supratentorial CPPs are found in patients <20 years of age, while infratentorial CPPs are fairly evenly distributed across all ages.5 In children, CPPs are typically found in the lateral ventricles (left greater than right), while in adults they are more common in the fourth ventricle.7–9 The median ages at diagnosis for tumors in the lateral ventricle, third ventricle, fourth ventricle, and cerebellopontine angle are 1.5 years, 1.5 years, 22.5 years, and 35.5 years, respectively.5 There are reports of intraparenchymal, suprasellar, and spinal epidural CPPs, but these are rare.10–12 CPPs have also been detected in utero, suggesting a congenital origin for a subset of these tumors.13–16
Regardless of age, patients typically present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures.6,17 Since symptoms are generally attributed to overproduction of CSF or obstruction of CSF outflow, they can occur early or late in the disease course, depending on tumor location and size. Choroid plexus tumors should be included in the differential diagnosis of any intraventricular mass, particularly in the pediatric population. Treatment consists of attempted gross total resection and is associated with an excellent prognosis.5,18,19 In this review, we will summarize recent advances in histologic grading, immunostaining, and the molecular biology of CPPs, as well as provide an overview of treatment strategies and clinical outcomes.
Classification and Histologic Features
For decades, choroid plexus tumors were classified as either papillomas (World Health Organization [WHO] grade I) or carcinomas (WHO grade III); the diagnosis of atypical CPP (grade II) was introduced as an intermediate lesion in 2007. Histologically, CPPs possess an architecture that is similar to that of normal choroid plexus, although cells are generally more crowded, elongated, or stratified.6 They feature a fibrovascular stalk surrounded by a single layer of cuboidal to columnar epithelium arranged in a papillary configuration (Fig. 1).1,6 Unlike the overtly malignant choroid plexus carcinoma, CPPs feature a well-formed and continuous basement membrane and very low mitotic activity. Invasion, necrosis, blurring of the papillary pattern, and nuclear pleomorphism may occur but are unusual.6 There are also reports of CPPs with pigmentation, osseous or adipose metaplasia, psammoma bodies, oncocytic change, xanthogranulomatous reactions, and mucinous degeneration, but these features are rare.20–26 Macroscopically, CPPs are described as pink-grey, pedunculated, cauliflower-like masses and are usually well circumscribed from normal brain.17 They are typically soft but can develop foci of calcifications and contain hemorrhagic or cystic features.17 Table 1 provides a summary of the general features of CPPs that distinguish them from other choroid plexus tumors.
Fig. 1.

Representative hematoxylin/eosin and immunohistochemical staining of a CPP. These tumors feature a single layer of cuboidal or columnar epithelium in a papillary configuration (A and B) covering a fibrovascular core (C). The presence of transthyretin on immunohistochemical staining (D) can aid in confirming the diagnosis.
Table 1.
Overview of choroid plexus tumors
| General Features | Choroid Plexus Papilloma (WHO grade I) | Atypical Choroid Plexus Papilloma (WHO grade II) | Choroid Plexus Carcinoma (WHO grade III) |
|---|---|---|---|
| General histologic appearance | Fibrovascular stalk surrounded by single layer of cuboidal to epithelium arranged in papillary configuration | Intermediate histology; characterized by increased mitotic activity compared with CPP | At least 4 of the following: increased cellularity, blurring of papillary architecture, high mitotic activity, nuclear pleomorphism, and necrosis |
| Mitotic activity | <2 per 10 HPFs | >2 per 10 HPFs | >5 per 10 HPFs |
| Radiographic features | Well-circumscribed intraventricular mass with cauliflower appearance and contrast enhancement; iso- or hyperdense on CT and iso- or hypointense on T1-weighted MRI | Similar to CPP but may possess irregular or invasive margins with associated edema | Generally larger than CPPs and frequently invade adjacent parenchyma with associated edema; heterogeneous contrast enhancement with possible calcifications, hemorrhage, necrosis, or leptomeningeal enhancement (dissemination) |
Atypical CPPs (WHO grade II) are intermediate lesions formally introduced in 2007. Their most important distinguishing feature is increased mitotic activity, defined by the presence of ≥2 mitoses per 10 high power fields (HPFs), compared with <2 per 10 HPFs in CPP and >5 per 10 HPFs in CPC.27,28 The diagnosis of atypical CPP is significant because it carries a nearly 5-fold increase in recurrence risk at 5 years compared with grade I CPPs.27 Histologic features observed in a minority of atypical CPPs that may help distinguish them from other CPPs include increased cellularity, nuclear pleomorphism, blurring of the papillary growth pattern, and necrosis; however, these features are also seen in carcinomas and are not required for the diagnosis of a grade II lesion.27 CPCs, which are rarely confused with CPPs, are invasive lesions characterized by overt signs of malignancy including at least 4 of the following: increased cellularity, blurring of the papillary architecture, high mitotic activity (>5 per 10 HPFs), nuclear pleomorphism, and necrosis.1,6,29 There are reports describing these histologic features, including invasion, in both grade I and grade II lesions, but such findings are exceedingly rare.6,30–32
Despite a well-established and thoroughly validated grading scheme, choroid plexus tumors are unique because histologic appearances do not necessarily predict their behavior. While generally regarded as benign, in rare cases CPPs can possess aggressive features or behaviors generally associated with high-grade lesions. For example, CPPs with evidence of parenchymal invasion or loss of normal architecture have been found to exhibit clinically benign behavior, with good long-term outcomes after surgical resection.30 There are also reports of CPPs disseminating throughout the neuraxis and undergoing malignant transformation.31–33 Since such findings are extremely rare, the molecular events that trigger this aggressive behavior remain elusive. The phenotypic diversity, particularly among CPPs, suggests that these lesions are best described on a spectrum rather than as distinct lesions with rigidly defined criteria.
Immunohistochemical Staining
The utilization of immunohistochemical staining has improved our ability to distinguish choroid plexus tumors from other primary CNS or metastatic neoplasms. Nearly all CPPs express cytokeratins, vimentin, and podoplanin.6 Podoplanin is also expressed by ependymomas and meningiomas, making it less useful when trying to differentiate these lesions.34 While cytokeratins and vimentin are characteristic of CPPs, markers such as glial fibrillary acidic protein, transthyretin (pre-albumin), and synaptophysin exhibit variable expressivity; although generally more common in CPPs compared with CPCs, they provide little benefit in distinguishing CPPs from atypical papillomas.6,35–38 More recently, expression data generated from microarray analysis have been used to identify novel markers of CPP.38 Of the 46 genes found to be overexpressed in normal choroid plexus and CPPs by this microarray-based approach, only 11 were tested by tissue array for protein expression. Five of the 11 candidate markers (coagulation factor V, glutathione peroxidase 3, serotonin receptor 2C, lumican, and plastin-1) exhibited staining patterns weaker than expected, demonstrating a limitation of gene expression–based approaches. However, through this approach, Kir7.1 and stanniocalcin-1 were identified as novel markers of CPP. Kir7.1 is an inward rectified potassium channel found in normal choroid plexus and CPPs but not in other primary brain tumors or metastases. Stanniocalcin-1 is believed to play a role in calcium homeostasis and has been shown to confer resistance to hypoxic stress.39–41 Kir7.1 and stanniocalcin-1 are considered both sensitive and specific markers of CPPs.38
In certain cases, differentiating a choroid plexus tumor from metastatic carcinoma can prove challenging. Two particularly useful cytokeratins are CK7, a basic (type II) keratin normally found in the lung, ovary, breast, and associated adenocarcinomas arising from those tissues, and CK20, an acidic (type I) keratin found in the epithelium of the gastrointestinal and urinary tracts, as well as cancers from those tissues.42–47 A combination of CK7+/CD20− is often helpful in distinguishing primary choroid plexus tumors from metastatic carcinomas, which typically display different combinations of staining.26,42 Focal staining of these cytokeratins is also suggestive of a choroid plexus tumor, while diffuse staining is generally seen in metastatic lesions.26 Excitatory amino acid transporter 1 has been shown to stain a variety of choroid plexus tumors but not metastatic carcinomas or adenocarcinomas with papillary features.48 Conversely, monoclonal antibodies directed against epithelial cell adhesion molecules, particularly the HEA (human epithelial antigen)-125 and Ber-EP4 clones, were shown to stain metastatic carcinomas but not choroid plexus tumors.49
In addition to identifying metastatic lesions, immunohistochemical stains can help differentiate CPPs from other primary CNS neoplasms. Ependymomas and subependymomas are often included in the differential diagnosis, and distinguishing them from choroid plexus tumors is important. Subependymomas are easily identified by their histology, decreasing the need for immunostaining. In the case of distinguishing ependymomas, E-cadherin and neural cell adhesion marker (NCAM) are particularly useful. While choroid plexus tumors generally stain positive for E-cadherin and negative for NCAM, ependymomas typically display the opposite staining pattern.50 Laminin, a marker of the basement membrane, is also useful in identifying CPPs because it is rarely seen in ependymomas; this marker is less useful in identifying CPCs because their basement membranes are often fragmented.51 Additionally, epithelial membrane antigen is commonly expressed in ependymomas but not typically observed in choroid plexus tumors, thus making it useful in distinguishing these lesions.52
CPPs found in the vicinity of the third ventricle may be confused with papillary tumors of the pineal region (PTPRs). Like CPPs, these are rare neuroepithelial tumors with a papillary architecture and immunopositivity for cytokeratin, vimentin, and transthyretin.6 PTPRs can be distinguished from choroid plexus tumors by their expression of microtubule-associated protein 2 and NCAM, as well as absent staining for Kir7.1 and stanniocalcin-1.53–55 In the case of CPCs with features of concern for atypical teratoid/rhabdoid tumor (AT/RT), immunostaining for integrase interacter (INI)1 is helpful because it is retained in the majority of CPCs but lost in AT/RT.56 There are some data linking carcinoembryonic antigen to CPCs, but this relationship is inconsistent and may also suggest the presence of a metastatic carcinoma.36,57–60 Another interesting marker associated with CPCs is α1-antitrypsin, which has been found to have CSF levels that correlate with tumor recurrence and progression.61 Recent data have demonstrated the importance of p53 immunostaining, which serves as a measure of p53 dysfunction. This is significant in patients with Li-Fraumeni syndrome who are harboring CPCs, since p53 status can predict tumor phenotype and clinical outcomes.62
Ki-67/mouse intestinal bacteria (MIB)–1 indices are useful in differentiating CPPs from other choroid plexus tumors as well as for identifying aggressive lesions or those prone to recurrence, regardless of histologic grade.60 Several authors have demonstrated a direct relationship between Ki-67/MIB-1 staining and tumor grade; normal choroid plexus has an MIB-1 index of nearly zero, while mean values have been reported of 1.3%–4.5% in CPPs, 5.8%–9.1% in atypical papillomas, and 13.4%–20.3% in CPCs.63–65 Interestingly, some have observed a significant decline in MIB-1, p53, and E2F-1 expression in CPCs after chemotherapy.65 Other nuclear markers that demonstrate a direct correlation with tumor grade include proliferating cell nuclear antigen, p21, and Rb.66
Pathogenesis of Choroid Plexus Tumors
Although the majority of choroid plexus tumors are sporadic, there has been significant work to examine associations with specific genetic mutations.65,67–69 Li-Fraumeni syndrome, characterized by germline mutations of the p53 tumor suppressor, is known to increase the risk for choroid plexus tumors, particularly carcinomas.62,70,71 While most CPCs are sporadic and do not contain germline mutations of TP53, a thorough family history should be acquired, particularly in children, to rule out Li-Fraumeni syndrome. Recent analysis of 64 choroid plexus tumors found somatic TP53 mutations in 50% of CPCs but in only 5% of CPPs.62 Furthermore, 92% of wild-type TP53 CPCs featured a variant of codon 72 encoding arginine (TP53-R72), along with a polymorphism of MDM2 (SNP309), an important negative regulator of p53.62 The combination of TP53-R72 and MDM2 SNP309, which results in decreased activity of p53 in the absence of a mutation, can promote malignant transformation via deficient cell cycle arrest and highlights the role of modifiers in p53-associated phenotypes.72,73 This also suggests that either qualitative or quantitative changes in p53 are involved in CPC tumorigenesis. Interestingly, CPCs with TP53 mutations were found to contain excess chromosomal gains or losses, as well as focal amplifications or deletions, as quantified by genomic total structural variation (TSV).62 Most importantly, TP53 status can predict survival in CPC; patients with wild-type TP53 and low TSV have a favorable prognosis and do not require radiation therapy, while those with TP53-mutated tumors should be treated more aggressively given their poor prognosis.62 With respect to germline mutations of TP53, a study of 22 CPCs in southern Brazil identified a specific mutation, R337H, in 63% of patients.74 This mutation was previously described in a similar region of Brazil among children with adrenocortical cancer.75 The incidence of CPC is significantly higher in this region of Brazil, likely due to an increased frequency of the allele in this population. There was no difference in survival among CPC patients bearing this mutation, but the sample size was limited.74 Additional studies of TP53 are warranted to better understand its role in CPC and further explore its association with prognosis.
In addition to Li-Fraumeni syndrome, other genetic disorders may play a role in the development of choroid plexus tumors. CPPs are associated with Aicardi syndrome, an X-linked disorder characterized by the triad of total or partial agenesis of the corpus callosum, chorioretinal “lacunae,” and infantile spasms.76 Other genetic syndromes have shed insight into the biology of choroid plexus tumors, particularly CPCs. Rhabdoid predisposition syndrome is caused by germline mutations of hSNF5/INI1 (SMARCB1), a member of the SWI/SNF (switch/sucrose nonfermentable) ATP-dependent chromatin-remodeling complex.77–80 These patients are predisposed to malignant rhabdoid tumors—AT/RTs—when they occur in the CNS.6 However, mutations in hSNF5/INI1 have also been associated with CPCs, medulloblastomas, and central primitive neuroectodermal tumors.77,79,81 Distinguishing AT/RTs from CPCs may be challenging due to overlapping clinical and histopathologic features; however, unlike AT/RTs, CPCs demonstrate immunopositivity for INI1.56
Approximately 2 decades ago, a potential link was identified between simian virus (SV)40, a monkey polyomavirus, and CPP. In one study, 10 of the 20 choroid plexus tumors examined were found to contain DNA sequences from SV40, and 80% stained positive for the SV40 T-antigen.82 Given reports of SV40 inducing brain tumors in various animal models, as well as the viral T-antigen's ability to inactivate the tumor suppressors p53 and Rb, there was compelling evidence implicating the virus in the pathogenesis of CPP.83 However, subsequent epidemiologic studies found that the SV40 sequences were found only in populations that had received polio vaccines contaminated with the virus, suggesting that this observation was simply a result of a tumor microenvironment that favored viral replication.84,85 Additionally, the incidence of brain tumors was no different among populations exposed to the contaminated vaccines, further refuting any causal relationship.
More recently, platelet-derived growth factor receptor (PDGFR) has been implicated in the pathogenesis of choroid plexus tumors. PDGFR is a receptor tyrosine kinase with roles in CNS development, blood vessel formation, and hematopoiesis.86–89 Aberrant signaling has been implicated in tumor growth, angiogenesis, stromal recruitment, tumor invasion, and metastasis.90–93 The α and β isoforms of PDGFR are expressed in CPPs, atypical papillomas, and CPCs, but phosphorylation of the β isoform is significantly increased in CPCs compared with CPPs.94 The immortalized choroid plexus epithelial cell line Z310 was found to express only the β isoform of the receptor and demonstrated a dose-dependent proliferative response when incubated with its ligand PDGF-BB; this effect was attenuated by the tyrosine kinase inhibitor imatinib, also in a dose-dependent manner.94 This finding is significant because the authors used physiologically acceptable ranges of the drug and their findings suggest a role for imatinib in the treatment of choroid plexus tumors with abnormal PDGFR activation.
Recent genetic analysis comparing normal choroid plexus to CPP has identified differential expression of 6 genes known to play roles in tumorigenesis. These include the transcription factor TWIST1, Wnt inhibitory factor 1 (WIF1), the transmembrane protein Shrew-1 (AJAP1), the transcriptional repressor BCL (B-cell lymphoma)2-associated transcription factor 1 (BCLAF1), transient receptor potential channel (TRPM3), and interleukin-6 signal transducer (IL6ST).95 Among these targets, TWIST1 was expressed significantly higher in CPPs compared with normal choroid plexus and was also found in the immortalized choroid plexus epithelial cell line Z310.38,95 TWIST1 is notable for its role as an inhibitor of both p53 and ADP ribosylation factor (Fig. 2).96 Knockdown of TWIST1 did not affect cell migration but significantly decreased infiltrative capacity and reduced cell proliferation in a p53-dependent fashion.95 Knockdown of TWIST1 also decreased expression of vascular endothelial growth factor D (FIGF) and increased expression of cyclin-dependent kinase inhibitor 1A (CDKN1A), FLICE (Fas-associated death domain–like interleukin converting enzyme) inhibitory protein (CFLAR), and plasminogen activator inhibitor type 2 (SUPERINB2).95
Fig. 2.
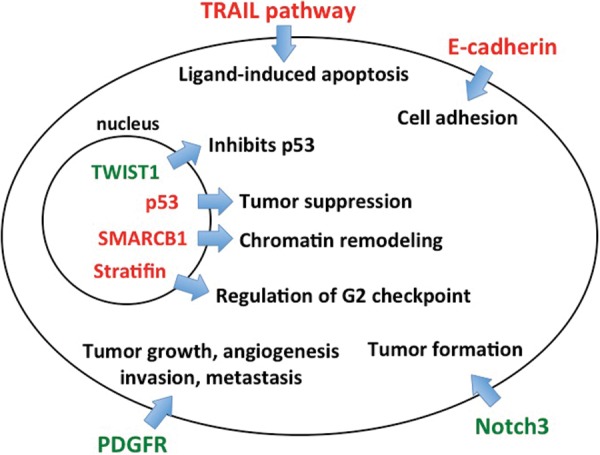
Molecular pathways associated with CPPs. Several genes in the TRAIL pathway, along with E-cadherin, are known to be methylated in CPPs, resulting in decreased tumor necrosis factor–induced apoptosis and increased cell migration, respectively. Aberrant signaling through PDGFR and Notch3 are associated with tumor formation and growth. Mutations of TP53 and hSNF5/INI1 (SMARCB1), which plays an integral role in chromatin remodeling, have been shown to promote CPP formation, while methylation of TWIST1, an inhibitor of p53, and stratifin, a regulator of the G2 checkpoint, are also associated with CPPs.
Recent evidence has implicated the Notch signaling pathway in the development of choroid plexus tumors, including papillomas. The roles of Notch1 and Notch2 in neural development have been thoroughly characterized.97,98 These pathways have also been implicated in the growth of glial and embryonal brain tumors.99,100 Less is known about the role of Notch3, in either neural development or tumorigenesis. In mice, activation of the Notch3 receptor in periventricular progenitor cells was found to induce choroid plexus tumor formation during embryonic development.101 In human CPPs, Notch receptor mRNAs are overexpressed—however, the receptor ligands Jagged1, Jagged2, and delta-like1 are expressed at levels similar to that of nonneoplastic choroid plexus.101 Current data suggest that activated Notch3 may function as an oncogene in the developing brain and drive choroid plexus tumor formation (Fig. 2). This may be of particular importance in cases of congenital choroid plexus tumors detected in utero.13,15
Epigenetics is a burgeoning field in the study of tumor biology. Methylation of important genomic sequences, particularly cytosine–phosphate–guanine islands, can induce a loss of tumor suppressor function and drive tumorigenesis. One study analyzed a group of 19 genes implicated in tumor formation and found that 71% of CPPs demonstrated methylation of ≥1 of these targets, none of which were methylated in normal cortex.102 The tumor suppressor RASSF1A was the most frequently methylated gene, followed by 3 genes of the tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) pathway: CASP8, TFRSF10C, and TFRSF10D (Fig. 2). RASSF1A stabilizes mitotic cyclins and regulates the timing and progression of mitosis.103 The TRAIL pathway is involved in ligand-induced apoptosis and has been investigated as a potential therapeutic target in the treatment of glioblastoma.104,105 After extensive analysis of the methylation patterns in CPP, the authors found no statistical difference when comparing these tumors with ependymomas, nor did they identify a relationship between methylation and clinical outcome.102
Additional genes have been identified as targets of epigenetic modification in CPP. A study of both adult and pediatric choroid plexus tumors detected a high frequency of methylation-induced silencing of E-cadherin (CDH1), retinoic acid receptor β (RARB), and stratifin (SFN) (Fig. 2).106 E-cadherin is a calcium-dependent adhesion glycoprotein; loss of function may increase cellular proliferation, migration, and invasion.107 Although present on the basolateral surface of most CPPs, it has decreased expression in atypical papillomas and CPCs, implicating loss of function as an important step in malignant progression.50,106 The retinoic acid receptor mediates signaling during embryo morphogenesis and differentiation; its exact role in the pathogenesis of CPP is unclear.108 Stratifin is involved in the G2 cell cycle checkpoint, and its expression is induced by γ-irradiation or DNA damaging agents, resulting in G2 phase arrest.109 Loss of stratifin may allow for an accumulation of genetic mutations, leading to malignant transformation, an effect that has been observed in breast cancer.110 An immunohistochemical study of primary CNS tumors found increased stratifin in a variety of lesions, including glioblastoma and CPPs.111 Methylation of CDH1 and SFN has also been detected in other malignancies, including neuroblastoma, breast, and bladder cancers.112–114 One study found decreased RARB in glial tumors, regardless of histologic grade, although methylation did not always correlate with transcriptional silencing.115 The significance and scope of epigenetic silencing, particularly with respect to tumor suppressors and regulators of the cell cycle, are biologically complex and will be active areas of future investigation.
Extensive genetic analysis of CPPs has identified specific chromosomal imbalances associated with these tumors. Duplications of chromosomes 7, 12, 15, 17, and 18 were identified in a small cohort of 9 CPPs.116 In a larger study using comparative genomic hybridization in 49 choroid plexus tumors, CPPs frequently exhibited +7q (65%), +5q (62%), +7p (59%), +5p (56%), +9p (50%), and −10q (56%), whereas CPCs commonly showed +12p, +12q, +20p (60%), +1, +4q, +20q (53%), and −22q (73%), suggesting tumor development through unique genetic pathways.117 Interestingly, the same study found that among CPPs, childhood tumors more commonly featured duplications of chromosomes 8, 12, 14, and 20, while adult tumors typically contained duplications of chromosomes 5, 6, 15, and 18 and deletions of 22. Neither the total number of mutations nor the gain or loss of a particular chromosome was found to influence overall survival in CPPs; however, gain of chromosome 9 and loss of chromosome 10 were associated with prolonged survival in patients with CPCs.117
Clinical Presentation
Choroid plexus tumors are found in patients of all ages. Some reports indicate that as many as 70%–80% occur in children, with nearly half occurring in those younger than 2 years.5,29 Furthermore, the presence of tumors in utero suggests a congenital origin for some choroid plexus tumors.13,15,29 Previous studies have failed to demonstrate differences in age, gender, symptomatology, or location when comparing CPPs with CPCs, but a more recent meta-analysis found that tumors of the cerebellopontine angle are associated with older age, benign histology, and female gender, suggesting the possibility of an X-linked tumor suppressor involved in the pathogenesis of a subset of these lesions.5,29 The most common symptoms are related to increased intracranial pressure and include headache, visual disturbances, nausea, vomiting, and generalized malaise.17 Symptoms unique to children include macrocephaly, splayed sutures, and tense fontanelles. Most of these are likely attributable to a combination of obstructive hydrocephalus, increased CSF production, and impaired CSF reabsorption at the arachnoid granulations due to scarring from hemorrhage or tumor debris. Clinical features and signs at diagnosis include hydrocephalus, papilledema, gait impairment, cranial nerve palsies, seizures, cerebellar signs, and psychomotor retardation.17 Less common presenting features include intraventricular and/or intratumoral hemorrhage, focal neurologic deficits, and, even more rarely, psychosis, or bobblehead doll syndrome.29
Radiographic Features
In children, most CPPs are found in the lateral ventricles, while in adults they are more common in the posterior fossa.118–120 Hydrocephalus is a common radiographic feature of these tumors and can be attributed to increased CSF secretion or obstruction by tumor, hemorrhage, or debris. On CT imaging, CPPs appear iso- or hyperdense with calcifications in ∼25% of cases (Figs. 3 and 4).29,121 These lesions are well vascularized, enhance with contrast, and occasionally contain cystic features.121 On MRI they are homogeneous or heterogeneous tumors with a cauliflower appearance. Papillomas are typically iso- or hypointense on T1- and T2-weighted imaging but may demonstrate a heterogeneous hyperintensity on T2-weighted imaging; they enhance after contrast injection unless the tumor is highly calcified (Figs. 3 and 4).122,123 Highly calcified tumors may exhibit corresponding areas of low signal intensity, while intratumoral hemorrhage manifests as small foci of increased signal intensity.124
Fig. 3.

Radiographic features of an adult CPP. Adult CPPs are generally located in the posterior fossa, most often in the fourth ventricle, and appear iso- or hypointense on T1-weighted MRI (A) and bright on fluid attenuated inversion recovery sequences (B and C). CPPs generally demonstrate strong enhancement on T1-weighted images with contrast (D–F).
Fig. 4.
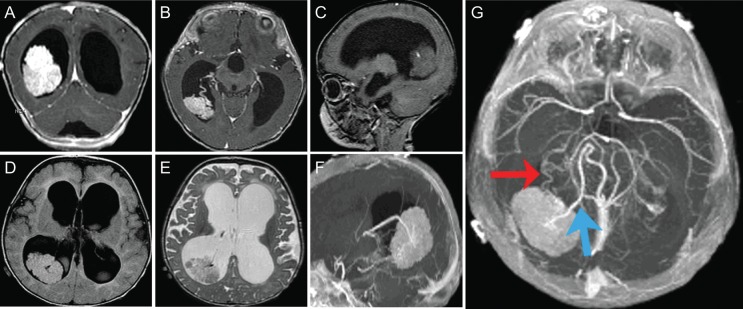
Radiographic features of a pediatric CPP. These tumors are more common in the lateral ventricles in the pediatric population. They can cause significant hydrocephalus (A–G) with transependymal flow (D) due to either increased production of CSF or obstruction by tumor debris and blood products. Intraventricular and/or intratumoral hemorrhage are not uncommon. CPPs enhance on T1-weighted MRI with contrast (A and B) due to their rich vascular supply. They are generally iso- or hypointense on T1- and T2-weighted MRI (C–E) but may demonstrate heterogeneous hyperintensity in some cases. Magnetic resonance angiography may aid in visualizing the tumor's vascular supply (F and G). These tumors are typically supplied by multiple vessels arising from the anterior and/or posterior choroidal arteries (double arrow; G), and preoperative embolization may be helpful in some cases. A large vein (bolded single arrow) arising from the tumor and draining into the right internal cerebral vein is also visible (G).
Angiograms are useful for the neurosurgeon and can aid in preoperative planning. These tumors are highly vascularized, and intraoperative hemorrhage is a serious risk, especially in pediatric patients with a low threshold for intraoperative blood loss. Historically, this was a significant source of morbidity and mortality in the surgical treatment of both adult and pediatric CPPs. It is therefore critical to visualize the tumor's vascular supply for optimal surgical planning (Fig. 4G). Certain features, such as prolonged vascular blush and intratumoral arteriovenous shunting, can mimic the appearance of a hemangioblastoma.125 The differential diagnosis of a choroid plexus tumor includes choroid plexus cyst, ependymoma, astrocytoma, germinoma, meningioma, xanthogranuloma, primitive neuroectodermal tumor, inflammatory pseudotumor, and metastatic lesion.29
Treatment
Surgical Resection
The cornerstone of treatment for CPPs is maximum safe surgical resection. Preoperative imaging helps determine the best surgical approach based on tumor location, vascular supply, and the experience and preference of the surgeon. Resection of CPPs almost always requires passing through neural structures, and large tumors may require more than one approach. Transcortical approaches provide the benefit of access to all 5 regions of the lateral ventricle, while transcallosal approaches are associated with lower risk for neuropsychological sequelae or seizure.29 Preoperative embolization has been used as an adjunct to surgical resection but is rarely successful due to the small caliber of feeding vessels.30,126–133 There is an interesting report of an infant treated with only preoperative embolization that led to total regression of the tumor with no signs of residual disease at 16 months—however, such a result is rare and should not be expected.134
Surgery remains the most important and effective treatment for patients with CPPs. Survival has improved from ∼50% in early studies to nearly 100% in more recent reports, due mainly to advances in imaging, surgical approaches, and quality of intensive care.135–140 A recent meta-analysis found 1-, 5-, and 10-year survival rates of 90%, 81%, and 77%, respectively.5 The same study found that gross total resection was associated with greater overall survival compared with partial resection in both CPPs and CPCs. There is no evidence suggesting that adjuvant therapies provide a significant increase in overall survival for patients with CPPs.5
Chemotherapy
Reports of chemotherapy use in patients with CPP are limited, mostly due to the benign nature of the disease and the fact that surgical resection is associated with excellent outcomes. There is a report on the use of vincristine in a pediatric patient whose CPP was deemed inoperable; the patient was alive recurrence free at 18 months.126 The CPT-SIOP-2000 study by the International Society of Pediatric Oncology reported on the use of etoposide, vincristine, and either carboplatin or cyclophosphamide for the treatment of various choroid plexus tumors.63 Of the 14% of CPPs treated with chemotherapy, all patients were alive at their most recent follow-up. There are additional reports of recurrent and metastatic CPPs responding to chemotherapy, although these lesions are quite rare.141–143 There are insufficient data to make recommendations on specific chemotherapeutic regimens. It is generally agreed that chemotherapy should be considered in only cases of aggressive recurrent or metastatic CPPs.
Radiotherapy
Although there are reports of irradiation used for the treatment of residual tumor after subtotal resection140,144 or as a neoadjuvant to shrink large tumors preoperatively,145 there is little evidence to support its use in such cases. Most agree that radiation should be reserved for recurrent or malignant lesions, not residual tumor after resection, particularly given the indolent nature of most CPPs.19,58,146,147 Radiosurgery has been proposed in cases of deep-seated or recurrent lesions. This has not been a particularly active area of investigation given the benign nature of the disease and the associated toxicities of radiotherapy to the brain, especially in younger pediatric patients. As a result, extensive prospective data are limited. One of the earliest reports of gamma knife radiosurgery for CPP was in a 25-year-old male with a 1.8-cm lesion of the posterior third ventricle.148 At 17 months posttreatment, the patient had left-monocular visual loss that had been present preoperatively, but an otherwise normal neurological exam. Another review,149 of 6 patients who underwent gamma knife radiosurgery for the treatment of recurrent CPPs, showed mixed results: at most recent follow-up, 4/6 patients were alive, with respective survivals of 39, 54, 57, and 120 months. Two patients died at 15 and 59 months due to tumor progression. Of particular interest, the deceased patients each had single lesions, while those alive at 57 and 120 months both had 3 separate lesions. Although these results were promising, the role of radiosurgery in the management of CPP is not yet clear. It is certainly not recommended in patients <3 years of age, but may be useful in treating recurrent or disseminated lesions or patients who are poor surgical candidates.18 More data are needed to make definitive recommendations, particularly given the cost and side effects of radiotherapy.
Conclusions
Although these are rare lesions, choroid plexus tumors pose challenges to both neuro-oncologists and neurosurgeons. Maximum safe resection remains the treatment of choice for CPPs and is associated with excellent outcomes, particularly with advances in microsurgery. In cases where gross total resection cannot be achieved, subtotal resection with watchful waiting may be the best course of action given the indolent nature of these tumors and the morbidity associated with chemotherapy and radiotherapy, particularly in children. Only in cases of tumor recurrence, metastatic spread, or malignant transformation should such aggressive strategies be considered.
There are currently no strict guidelines for long-term imaging in patients with CPP. In children, Collins' law on likelihood of recurrence is utilized, ie, MRI at 3- to 6-month intervals for a time equal to age at surgery plus 9 months.150 However, more specific follow-up criteria are needed because recurrence can occur as far as 11 years after initial diagnosis.151 As our understanding of the biology of these tumors continues to grow, the possibility of targeted therapies becomes more likely. Discoveries pertaining to epigenetic modifications, chromosomal amplifications or deletions, cell cycle regulators, and various molecular pathways, including those associated with TRAIL, PDGFR, and Notch, have greatly advanced our knowledge of all choroid plexus tumors. Future work in these areas will aid in predicting tumor behavior, expanding treatment options, and improving clinical outcomes for patients with this disease.
Future studies must focus on determining whether CPCs result from a stepwise progression in which CPPs accumulate genetic mutations and chromosomal anomalies or whether choroid plexus tumors are distinct lesions on a spectrum. From a diagnostic perspective, the greatest challenge involves distinguishing grade I CPPs from grade II atypical papillomas, or predicting which tumors will recur. Because these lesions have similar histologic appearances and lack reliable immunohistochemical stains to distinguish them or predict clinical behavior, it is important to shift our focus toward understanding the genetic susceptibilities that increase recurrence risk or malignant transformation. By incorporating genetic analysis, the current histologic grading scheme can be refined to create a more accurate, clinically relevant system for categorizing choroid plexus tumors. Microarray-based approaches are attractive due to their relative ease of use and ability to screen a large library of genes—however, the technique has limitations, particularly in cases of discordance between gene and protein data. Single nucleotide polymorphism analysis may provide insight by examining genes associated with the Notch and TRAIL pathways or those associated with epithelial-mesenchymal transition. These types of studies are challenging, particularly for choroid plexus tumor, because the disease is rare and answering these types of questions generally requires a significant number of patient samples. Genotype-to-phenotype correlations are also difficult, particularly when trying to predict clinical outcomes and treatment responses. This stresses the need for collaborative efforts among institutions with expertise in treating this disease. Multi-institutional studies that incorporate histologic, genetic, and clinical data will be essential in improving our ability to predict tumor recurrence and identify novel targets for treating the most aggressive forms of this disease.
Funding
M. S. was supported by a grant from the Doris Duke Charitable Foundation. M. C. O is funded by the Neurosurgery Research and Education Foundation from the American Association of Neurological Surgeons. G. K. and M. Z. S. are supported by the Howard Hughes Medical Institute and Ivy Foundation. A. T. P. is supported by the Reza and Georgianna Khatib Endowed Chair in Skull Base Surgery.
Acknowledgments
The authors thank the Doris Duke Charitable Foundation, Dr Peter Chin-Hong, Dr Joel Palefsky, and the Clinical and Translational Research Fellowship Program at the University of California, San Francisco.
Conflict of interest statement. None declared.
References
- 1.Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119(1):75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- 2.Cataltepe O, Liptzin D, Jolley L, Smith TW. Diffuse villous hyperplasia of the choroid plexus and its surgical management. J Neurosurg Pediatr. 2010;5(5):518–522. doi: 10.3171/2009.12.PEDS0960. [DOI] [PubMed] [Google Scholar]
- 3.Janisch W, Staneczek W. [Primary tumors of the choroid plexus. Frequency, localization and age] Zentralbl Allg Pathol. 1989;135(3):235–240. [PubMed] [Google Scholar]
- 4.Rickert CH, Paulus W. Tumors of the choroid plexus. Microsc Res Tech. 2001;52(1):104–111. doi: 10.1002/1097-0029(20010101)52:1<104::AID-JEMT12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Wolff JE, Sajedi M, Brant R, Coppes MJ, Egeler RM. Choroid plexus tumours. Br J Cancer. 2002;87(10):1086–1091. doi: 10.1038/sj.bjc.6600609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulus W, Bradner S. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC Press; 2007. [Google Scholar]
- 7.Tacconi L, Delfini R, Cantore G. Choroid plexus papillomas: consideration of a surgical series of 33 cases. Acta neurochirurgica. 1996;138(7):802–810. doi: 10.1007/BF01411257. [DOI] [PubMed] [Google Scholar]
- 8.Jooma R, Hayward RD, Grant DN. Intracranial neoplasms during the first year of life: analysis of one hundred consecutive cases. Neurosurgery. 1984;14(1):31–41. doi: 10.1227/00006123-198401000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Hopper KD, Foley LC, Nieves NL, Smirniotopoulos JG. The interventricular extension of choroid plexus papillomas. AJNR Am J Neuroradiol. 1987;8(3):469–472. [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura M, Takayasu M, Suzuki Y, et al. Primary choroid plexus papilloma located in the suprasellar region: case report. Neurosurgery. 1992;31(3):563–566. doi: 10.1227/00006123-199209000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Kurtkaya-Yapicier O, Scheithauer BW, Van Peteghem KP, Sawicki JE. Unusual case of extradural choroid plexus papilloma of the sacral canal. Case report. J Neurosurg. 2002;97(1 suppl):102–105. doi: 10.3171/spi.2002.97.1.0102. [DOI] [PubMed] [Google Scholar]
- 12.Pillai A, Rajeev K, Chandi S, Unnikrishnan M. Intrinsic brainstem choroid plexus papilloma. Case report. J Neurosurg. 2004;100(6):1076–1078. doi: 10.3171/jns.2004.100.6.1076. [DOI] [PubMed] [Google Scholar]
- 13.Krul JM, Gooskens RH, Ramos L, Veiga-Pires JA. Ultrasound detection of a choroid plexus papilloma of the third ventricle. J Neuroradiol. 1987;14(2):179–182. [PubMed] [Google Scholar]
- 14.Veyrac C, Couture A. Normal and pathologic choroid plexus ultrasound. Annales de radiologie. 1985;28(3–4):215–223. [PubMed] [Google Scholar]
- 15.Anselem O, Mezzetta L, Grange G, et al. Fetal tumors of the choroid plexus: is differential diagnosis between papilloma and carcinoma possible? Ultrasound Obstet Gynecol. 2011;38(2):229–232. doi: 10.1002/uog.8919. [DOI] [PubMed] [Google Scholar]
- 16.Hartge DR, Axt-Fliedner R, Weichert J. Prenatal diagnosis and successful postnatal therapy of an atypical choroid plexus papilloma. Case report and review of literature. J Clin Ultrasound. 2010;38(7):377–383. doi: 10.1002/jcu.20718. [DOI] [PubMed] [Google Scholar]
- 17.Gaudio RM, Tacconi L, Rossi ML. Pathology of choroid plexus papillomas: a review. Clin Neurol Neurosurg. 1998;100(3):165–186. doi: 10.1016/s0303-8467(98)00033-x. [DOI] [PubMed] [Google Scholar]
- 18.Due-Tonnessen B, Helseth E, Skullerud K, Lundar T. Choroid plexus tumors in children and young adults: report of 16 consecutive cases. Childs Nerv Syst. 2001;17(4–5):252–256. doi: 10.1007/pl00013728. [DOI] [PubMed] [Google Scholar]
- 19.Krishnan S, Brown PD, Scheithauer BW, Ebersold MJ, Hammack JE, Buckner JC. Choroid plexus papillomas: a single institutional experience. J Neurooncol. 2004;68(1):49–55. doi: 10.1023/b:neon.0000024745.06073.07. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe K, Ando Y, Iwanaga H, et al. Choroid plexus papilloma containing melanin pigment. Clin Neuropathol. 1995;14(3):159–161. [PubMed] [Google Scholar]
- 21.Corcoran GM, Frazier SR, Prayson RA. Choroid plexus papilloma with osseous and adipose metaplasia. Ann Diagn Pathol. 2001;5(1):43–47. doi: 10.1053/adpa.2001.21478. [DOI] [PubMed] [Google Scholar]
- 22.Tena-Suck ML, Lopez-Gomez M, Salinas-Lara C, Arce-Arellano RI, Biol AS, Renbao-Bojorquez D. Psammomatous choroid plexus papilloma: three cases with atypical characteristics. Surg Neurol. 2006;65(6):604–610. doi: 10.1016/j.surneu.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Aquilina K, Nanra JS, Allcutt DA, Farrell M. Choroid plexus adenoma: case report and review of the literature. Childs Nerv Syst. 2005;21(5):410–415. doi: 10.1007/s00381-004-1038-8. [DOI] [PubMed] [Google Scholar]
- 24.Buccoliero AM, Bacci S, Mennonna P, Taddei GL. Pathologic quiz case: infratentorial tumor in a middle-aged woman. Oncocytic variant of choroid plexus papilloma. Arch Pathol Lab Med. 2004;128(12):1448–1450. doi: 10.5858/2004-128-1448-PQCITI. [DOI] [PubMed] [Google Scholar]
- 25.Sarkar C, Sharma MC, Gaikwad S, Sharma C, Singh VP. Choroid plexus papilloma: a clinicopathological study of 23 cases. Surg Neurol. 1999;52(1):37–39. doi: 10.1016/s0090-3019(99)00049-x. [DOI] [PubMed] [Google Scholar]
- 26.Ikota H, Tanaka Y, Yokoo H, Nakazato Y. Clinicopathological and immunohistochemical study of 20 choroid plexus tumors: their histological diversity and the expression of markers useful for differentiation from metastatic cancer. Brain Tumor Pathol. 2011;28(3):215–221. doi: 10.1007/s10014-011-0024-6. [DOI] [PubMed] [Google Scholar]
- 27.Jeibmann A, Hasselblatt M, Gerss J, et al. Prognostic implications of atypical histologic features in choroid plexus papilloma. J Neuropathol Exp Neurol. 2006;65(11):1069–1073. doi: 10.1097/01.jnen.0000240464.26005.90. [DOI] [PubMed] [Google Scholar]
- 28.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keating RF, Goodrich JT, Packer RJ. Tumors of the Pediatric Central Nervous System. 1st ed. New York: Thieme; 2001. [Google Scholar]
- 30.Levy ML, Goldfarb A, Hyder DJ, et al. Choroid plexus tumors in children: significance of stromal invasion. Neurosurgery. 2001;48(2):303–309. doi: 10.1097/00006123-200102000-00010. [DOI] [PubMed] [Google Scholar]
- 31.Jinhu Y, Jianping D, Jun M, Hui S, Yepeng F. Metastasis of a histologically benign choroid plexus papilloma: case report and review of the literature. J Neurooncol. 2007;83(1):47–52. doi: 10.1007/s11060-006-9300-4. [DOI] [PubMed] [Google Scholar]
- 32.McCall T, Binning M, Blumenthal DT, Jensen RL. Variations of disseminated choroid plexus papilloma: 2 case reports and a review of the literature. Surg Neurol. 2006;66(1):62–67. doi: 10.1016/j.surneu.2005.09.023. discussion 67–68. [DOI] [PubMed] [Google Scholar]
- 33.Jeibmann A, Wrede B, Peters O, Wolff JE, Paulus W, Hasselblatt M. Malignant progression in choroid plexus papillomas. J Neurosurg. 2007;107(3 suppl):199–202. doi: 10.3171/PED-07/09/199. [DOI] [PubMed] [Google Scholar]
- 34.Shibahara J, Kashima T, Kikuchi Y, Kunita A, Fukayama M. Podoplanin is expressed in subsets of tumors of the central nervous system. Virchows Arch. 2006;448(4):493–499. doi: 10.1007/s00428-005-0133-x. [DOI] [PubMed] [Google Scholar]
- 35.Ang LC, Taylor AR, Bergin D, Kaufmann JC. An immunohistochemical study of papillary tumors in the central nervous system. Cancer. 1990;65(12):2712–2719. doi: 10.1002/1097-0142(19900615)65:12<2712::aid-cncr2820651219>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 36.Paulus W, Janisch W. Clinicopathologic correlations in epithelial choroid plexus neoplasms: a study of 52 cases. Acta Neuropathol. 1990;80(6):635–641. doi: 10.1007/BF00307632. [DOI] [PubMed] [Google Scholar]
- 37.Kepes JJ, Collins J. Choroid plexus epithelium (normal and neoplastic) expresses synaptophysin. A potentially useful aid in differentiating carcinoma of the choroid plexus from metastatic papillary carcinomas. J Neuropathol Exp Neurol. 1999;58(4):398–401. doi: 10.1097/00005072-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Hasselblatt M, Bohm C, Tatenhorst L, et al. Identification of novel diagnostic markers for choroid plexus tumors: a microarray-based approach. Am J Surg Pathol. 2006;30(1):66–74. doi: 10.1097/01.pas.0000176430.88702.e0. [DOI] [PubMed] [Google Scholar]
- 39.Jellinek DA, Chang AC, Larsen MR, Wang X, Robinson PJ, Reddel RR. Stanniocalcin 1 and 2 are secreted as phosphoproteins from human fibrosarcoma cells. Biochem J. 2000;350(Pt 2):453–461. [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshiko Y, Aubin JE. Stanniocalcin 1 as a pleiotropic factor in mammals. Peptides. 2004;25(10):1663–1669. doi: 10.1016/j.peptides.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 41.Zhang K, Lindsberg PJ, Tatlisumak T, Kaste M, Olsen HS, Andersson LC. Stanniocalcin: a molecular guard of neurons during cerebral ischemia. Proc Natl Acad Sci USA. 2000;97(7):3637–3642. doi: 10.1073/pnas.070045897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gyure KA, Morrison AL. Cytokeratin 7 and 20 expression in choroid plexus tumors: utility in differentiating these neoplasms from metastatic carcinomas. Mod Pathol. 2000;13(6):638–643. doi: 10.1038/modpathol.3880111. [DOI] [PubMed] [Google Scholar]
- 43.Wang NP, Zee S, Zarbo RJ, Bacchi CE, Gown AM. Coordinate expression of cytokeratins 7 and 20 defines unique subsets of carcinomas. Appl Immunohistochem. 1995;3:99–107. [Google Scholar]
- 44.Miettinen M. Keratin immunohistochemistry: update of applications and pitfalls. Pathol Annu. 1993;28(Pt 2):113–143. [PubMed] [Google Scholar]
- 45.Ramaekers F, van Niekerk C, Poels L, et al. Use of monoclonal antibodies to keratin 7 in the differential diagnosis of adenocarcinomas. Am J Pathol. 1990;136(3):641–655. [PMC free article] [PubMed] [Google Scholar]
- 46.Miettinen M. Keratin 20: immunohistochemical marker for gastrointestinal, urothelial, and Merkel cell carcinomas. Mod Pathol. 1995;8(4):384–388. [PubMed] [Google Scholar]
- 47.Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140(2):427–447. [PMC free article] [PubMed] [Google Scholar]
- 48.Beschorner R, Schittenhelm J, Schimmel H, et al. Choroid plexus tumors differ from metastatic carcinomas by expression of the excitatory amino acid transporter–1. Hum Pathol. 2006;37(7):854–860. doi: 10.1016/j.humpath.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 49.Gottschalk J, Jautzke G, Paulus W, Goebel S, Cervos-Navarro J. The use of immunomorphology to differentiate choroid plexus tumors from metastatic carcinomas. Cancer. 1993;72(4):1343–1349. doi: 10.1002/1097-0142(19930815)72:4<1343::aid-cncr2820720432>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 50.Figarella-Branger D, Lepidi H, Poncet C, et al. Differential expression of cell adhesion molecules (CAM), neural CAM and epithelial cadherin in ependymomas and choroid plexus tumors. Acta Neuropathol. 1995;89(3):248–257. doi: 10.1007/BF00309340. [DOI] [PubMed] [Google Scholar]
- 51.Furness PN, Lowe J, Tarrant GS. Subepithelial basement membrane deposition and intermediate filament expression in choroid plexus neoplasms and ependymomas. Histopathology. 1990;16(3):251–255. doi: 10.1111/j.1365-2559.1990.tb01111.x. [DOI] [PubMed] [Google Scholar]
- 52.Ishizawa K, Komori T, Shimada S, Hirose T. Podoplanin is a potential marker for the diagnosis of ependymoma: a comparative study with epithelial membrane antigen (EMA) Clin Neuropathol. 2009;28(5):373–378. [PubMed] [Google Scholar]
- 53.Hasselblatt M, Blumcke I, Jeibmann A, et al. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Neuropathol Appl Neurobiol. 2006;32(3):278–283. doi: 10.1111/j.1365-2990.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 54.Jouvet A, Fauchon F, Liberski P, et al. Papillary tumor of the pineal region. Am J Surg Pathol. 2003;27(4):505–512. doi: 10.1097/00000478-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 55.Shibahara J, Todo T, Morita A, Mori H, Aoki S, Fukayama M. Papillary neuroepithelial tumor of the pineal region. A case report. Acta Neuropathol. 2004;108(4):337–340. doi: 10.1007/s00401-004-0898-z. [DOI] [PubMed] [Google Scholar]
- 56.Judkins AR, Burger PC, Hamilton RL, et al. INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol. 2005;64(5):391–397. doi: 10.1093/jnen/64.5.391. [DOI] [PubMed] [Google Scholar]
- 57.Matsushima T, Inoue T, Takeshita I, Fukui M, Iwaki T, Kitamoto T. Choroid plexus papillomas: an immunohistochemical study with particular reference to the coexpression of prealbumin. Neurosurgery. 1988;23(3):384–389. doi: 10.1227/00006123-198809000-00021. [DOI] [PubMed] [Google Scholar]
- 58.Menon G, Nair SN, Baldawa SS, Rao RB, Krishnakumar KP, Gopalakrishnan CV. Choroid plexus tumors: an institutional series of 25 patients. Neurol India. 2010;58(3):429–435. doi: 10.4103/0028-3886.66455. [DOI] [PubMed] [Google Scholar]
- 59.Barreto AS, Vassallo J, Queiroz Lde S. Papillomas and carcinomas of the choroid plexus: histological and immunohistochemical studies and comparison with normal fetal choroid plexus. Arq Neuropsiquiatr. 2004;62(3A):600–607. doi: 10.1590/s0004-282x2004000400007. [DOI] [PubMed] [Google Scholar]
- 60.Kato T, Fujita M, Sawamura Y, et al. Clinicopathological study of choroid plexus tumors: immunohistochemical features and evaluation of proliferative potential by PCNA and Ki-67 immunostaining. Noshuyo Byori. 1996;13(2):99–105. [PubMed] [Google Scholar]
- 61.Qualman SJ, Shannon BT, Boesel CP, Jacobs D, Jinkens C, Hayes J. Ploidy analysis and cerebrospinal fluid nephelometry as measures of clinical outcome in childhood choroid plexus neoplasia. Pathol Annu. 1992;27(Pt 1):305–320. [PubMed] [Google Scholar]
- 62.Tabori U, Shlien A, Baskin B, et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28(12):1995–2001. doi: 10.1200/JCO.2009.26.8169. [DOI] [PubMed] [Google Scholar]
- 63.Wrede B, Hasselblatt M, Peters O, et al. Atypical choroid plexus papilloma: clinical experience in the CPT-SIOP-2000 study. J Neurooncol. 2009;95(3):383–392. doi: 10.1007/s11060-009-9936-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vajtai I, Varga Z, Aguzzi A. MIB-1 immunoreactivity reveals different labelling in low-grade and in malignant epithelial neoplasms of the choroid plexus. Histopathology. 1996;29(2):147–151. [PubMed] [Google Scholar]
- 65.Carlotti CG, Jr, Salhia B, Weitzman S, et al. Evaluation of proliferative index and cell cycle protein expression in choroid plexus tumors in children. Acta Neuropathol. 2002;103(1):1–10. doi: 10.1007/s004010100419. [DOI] [PubMed] [Google Scholar]
- 66.Tena-Suck ML, Salinas-Lara C, Rembao-Bojorquez D, Castillejos M. Clinicopathologic and immunohistochemical study of choroid plexus tumors: single-institution experience in Mexican population. J Neurooncol. 2010;98(3):357–365. doi: 10.1007/s11060-009-0080-5. [DOI] [PubMed] [Google Scholar]
- 67.Ohgaki H, Eibl RH, Schwab M, et al. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog. 1993;8(2):74–80. doi: 10.1002/mc.2940080203. [DOI] [PubMed] [Google Scholar]
- 68.Jay V, Ho M, Chan F, Malkin D. P53 expression in choroid plexus neoplasms: an immunohistochemical study. Arch Pathol Lab Med. 1996;120(11):1061–1065. [PubMed] [Google Scholar]
- 69.Mueller W, Eum JH, Lass U, et al. No evidence of hSNF5/INI1 point mutations in choroid plexus papilloma. Neuropathol Appl Neurobiol. 2004;30(3):304–307. doi: 10.1046/j.0305-1846.2004.00538.x. [DOI] [PubMed] [Google Scholar]
- 70.Rutherford J, Chu CE, Duddy PM, et al. Investigations on a clinically and functionally unusual and novel germline p53 mutation. Br J Cancer. 2002;86(10):1592–1596. doi: 10.1038/sj.bjc.6600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gozali AE, Britt B, Shane L, et al. Choroid plexus tumors; management, outcome, and association with the Li-Fraumeni syndrome: the Children's Hospital Los Angeles (CHLA) experience, 1991–2010. Pediatr Blood Cancer. 2012;58(6):905–909. doi: 10.1002/pbc.23349. [DOI] [PubMed] [Google Scholar]
- 72.Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9(2):95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- 73.Tabori U, Malkin D. Risk stratification in cancer predisposition syndromes: lessons learned from novel molecular developments in Li-Fraumeni syndrome. Cancer Res. 2008;68(7):2053–2057. doi: 10.1158/0008-5472.CAN-07-2091. [DOI] [PubMed] [Google Scholar]
- 74.Custodio G, Taques GR, Figueiredo BC, et al. Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PloS One. 2011;6(3):e18015. doi: 10.1371/journal.pone.0018015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci USA. 2001;98(16):9330–9335. doi: 10.1073/pnas.161479898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aicardi J. Aicardi syndrome. Brain Dev. 2005;27(3):164–171. doi: 10.1016/j.braindev.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 77.Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65(5):1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muchardt C, Sardet C, Bourachot B, Onufryk C, Yaniv M. A human protein with homology to Saccharomyces cerevisiae SNF5 interacts with the potential helicase hbrm. Nucleic Acids Res. 1995;23(7):1127–1132. doi: 10.1093/nar/23.7.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 80.Wade PA, Wolffe AP. Transcriptional regulation: SWItching circuitry. Curr Biol. 1999;9(6):R221–R224. doi: 10.1016/s0960-9822(99)80134-1. [DOI] [PubMed] [Google Scholar]
- 81.Sevenet N, Lellouch-Tubiana A, Schofield D, et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet. 1999;8(13):2359–2368. doi: 10.1093/hmg/8.13.2359. [DOI] [PubMed] [Google Scholar]
- 82.Bergsagel DJ, Finegold MJ, Butel JS, Kupsky WJ, Garcea RL. DNA sequences similar to those of simian virus 40 in ependymomas and choroid plexus tumors of childhood. N Engl J Med. 1992;326(15):988–993. doi: 10.1056/NEJM199204093261504. [DOI] [PubMed] [Google Scholar]
- 83.Marks JR, Lin J, Hinds P, Miller D, Levine AJ. Cellular gene expression in papillomas of the choroid plexus from transgenic mice that express the simian virus 40 large T antigen. J Virol. 1989;63(2):790–797. doi: 10.1128/jvi.63.2.790-797.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strickler HD, Rosenberg PS, Devesa SS, Hertel J, Fraumeni JF, Jr, Goedert JJ. Contamination of poliovirus vaccines with simian virus 40 (1955–1963) and subsequent cancer rates. JAMA. 1998;279(4):292–295. doi: 10.1001/jama.279.4.292. [DOI] [PubMed] [Google Scholar]
- 85.Ohgaki H, Huang H, Haltia M, Vainio H, Kleihues P. More about: cell and molecular biology of simian virus 40: implications for human infections and disease. J Natl Cancer Inst. 2000;92(6):495–497. doi: 10.1093/jnci/92.6.495. [DOI] [PubMed] [Google Scholar]
- 86.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Enge M, Wilhelmsson U, Abramsson A, et al. Neuron-specific ablation of PDGF-B is compatible with normal central nervous system development and astroglial response to injury. Neurochem Res. 2003;28(2):271–279. doi: 10.1023/a:1022421001288. [DOI] [PubMed] [Google Scholar]
- 88.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 89.Kaminski WE, Lindahl P, Lin NL, et al. Basis of hematopoietic defects in platelet-derived growth factor (PDGF)-B and PDGF beta-receptor null mice. Blood. 2001;97(7):1990–1998. doi: 10.1182/blood.v97.7.1990. [DOI] [PubMed] [Google Scholar]
- 90.Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58(23):5275–5279. [PubMed] [Google Scholar]
- 91.Bruna A, Darken RS, Rojo F, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11(2):147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 92.Gerhardt H, Semb H. Pericytes: gatekeepers in tumour cell metastasis? J Mol Med (Berl) 2008;86(2):135–144. doi: 10.1007/s00109-007-0258-2. [DOI] [PubMed] [Google Scholar]
- 93.Dong J, Grunstein J, Tejada M, et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23(14):2800–2810. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koos B, Paulsson J, Jarvius M, et al. Platelet-derived growth factor receptor expression and activation in choroid plexus tumors. Am J Pathol. 2009;175(4):1631–1637. doi: 10.2353/ajpath.2009.081022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hasselblatt M, Mertsch S, Koos B, et al. TWIST-1 is overexpressed in neoplastic choroid plexus epithelial cells and promotes proliferation and invasion. Cancer Res. 2009;69(6):2219–2223. doi: 10.1158/0008-5472.CAN-08-3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Puisieux A, Valsesia-Wittmann S, Ansieau S. A twist for survival and cancer progression. Br J Cancer. 2006;94(1):13–17. doi: 10.1038/sj.bjc.6602876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yoon K, Nery S, Rutlin ML, Radtke F, Fishell G, Gaiano N. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci. 2004;24(43):9497–9506. doi: 10.1523/JNEUROSCI.0993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31(4):557–568. doi: 10.1016/s0896-6273(01)00395-6. [DOI] [PubMed] [Google Scholar]
- 99.Fan X, Mikolaenko I, Elhassan I, et al. Notch1 and Notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64(21):7787–7793. doi: 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- 100.Purow BW, Haque RM, Noel MW, et al. Expression of Notch-1 and its ligands, delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65(6):2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 101.Dang L, Fan X, Chaudhry A, Wang M, Gaiano N, Eberhart CG. Notch3 signaling initiates choroid plexus tumor formation. Oncogene. 2006;25(3):487–491. doi: 10.1038/sj.onc.1209074. [DOI] [PubMed] [Google Scholar]
- 102.Michalowski MB, de Fraipont F, Michelland S, et al. Methylation of RASSF1A and TRAIL pathway-related genes is frequent in childhood intracranial ependymomas and benign choroid plexus papilloma. Cancer Genet Cytogenet. 2006;166(1):74–81. doi: 10.1016/j.cancergencyto.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 103.Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol. 2004;6(2):129–137. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 104.Pan G, O'Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 105.Badr CE, Wurdinger T, Nilsson J, et al. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro-oncology. 2011;13(11):1213–1224. doi: 10.1093/neuonc/nor067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Losi-Guembarovski R, Kuasne H, Guembarovski AL, Rainho CA, Colus IM. DNA methylation patterns of the CDH1, RARB, and SFN genes in choroid plexus tumors. Cancer Genet Cytogenet. 2007;179(2):140–145. doi: 10.1016/j.cancergencyto.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 107.Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003;94(7):575–581. doi: 10.1111/j.1349-7006.2003.tb01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soprano DR, Qin P, Soprano KJ. Retinoic acid receptors and cancers. Annu Rev Nutr. 2004;24:201–221. doi: 10.1146/annurev.nutr.24.012003.132407. [DOI] [PubMed] [Google Scholar]
- 109.Hermeking H, Lengauer C, Polyak K, et al. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1(1):3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 110.Ferguson AT, Evron E, Umbricht CB, et al. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci USA. 2000;97(11):6049–6054. doi: 10.1073/pnas.100566997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cao WD, Zhang X, Zhang JN, et al. Immunocytochemical detection of 14-3-3 in primary nervous system tumors. J Neurooncol. 2006;77(2):125–130. doi: 10.1007/s11060-005-9027-7. [DOI] [PubMed] [Google Scholar]
- 112.Nass SJ, Herman JG, Gabrielson E, et al. Aberrant methylation of the estrogen receptor and E-cadherin 5’ CpG islands increases with malignant progression in human breast cancer. Cancer Res. 2000;60(16):4346–4348. [PubMed] [Google Scholar]
- 113.Bornman DM, Mathew S, Alsruhe J, Herman JG, Gabrielson E. Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am J Pathol. 2001;159(3):831–835. doi: 10.1016/S0002-9440(10)61758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Banelli B, Gelvi I, Di Vinci A, et al. Distinct CpG methylation profiles characterize different clinical groups of neuroblastic tumors. Oncogene. 2005;24(36):5619–5628. doi: 10.1038/sj.onc.1208722. [DOI] [PubMed] [Google Scholar]
- 115.Klein O, Grignon Y, Civit T, Auque J, Marchal JC. [Methylation status of RARbeta gene promoter in low and high grade cerebral glioma. Comparison with normal tissue. Immunohistochemical study of nuclear RARbeta expression in low and high grade cerebral glioma cells. Comparison with normal cells. 48 tumors] Neuro-Chirurgie. 2005;51(3–4 Pt 1):147–154. doi: 10.1016/s0028-3770(05)83470-8. [DOI] [PubMed] [Google Scholar]
- 116.Donovan MJ, Yunis EJ, DeGirolami U, Fletcher JA, Schofield DE. Chromosome aberrations in choroid plexus papillomas. Genes Chromosomes Cancer. 1994;11(4):267–270. doi: 10.1002/gcc.2870110410. [DOI] [PubMed] [Google Scholar]
- 117.Rickert CH, Wiestler OD, Paulus W. Chromosomal imbalances in choroid plexus tumors. Am J Pathol. 2002;160(3):1105–1113. doi: 10.1016/S0002-9440(10)64931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Beskonakli E, Cayli S, Bostanci U, Kulacoglu S, Yalcinlar Y. Choroid plexus papillomas of the posterior fossa: extraventricular extension, intraventricular and primary extraventricular location. Report of four cases. J Neurosurg Sci. 1998;42(1):37–40. [PubMed] [Google Scholar]
- 119.Mitsuyama T, Ide M, Hagiwara S, Tanaka N, Kawamura H, Aiba M. [Adult choroid plexus papilloma of the posterior fossa: extraventricular location] No Shinkei Geka. 2005;33(8):825–829. [PubMed] [Google Scholar]
- 120.Bonneville F, Sarrazin JL, Marsot-Dupuch K, et al. Unusual lesions of the cerebellopontine angle: a segmental approach. Radiographics. 2001;21(2):419–438. doi: 10.1148/radiographics.21.2.g01mr13419. [DOI] [PubMed] [Google Scholar]
- 121.Li S, Savolaine ER. Imaging of atypical choroid plexus papillomas. Clin Imaging. 1996;20(2):85–90. doi: 10.1016/0899-7071(94)00078-6. [DOI] [PubMed] [Google Scholar]
- 122.Bonneville F, Savatovsky J, Chiras J. Imaging of cerebellopontine angle lesions: an update. Part 2: intra-axial lesions, skull base lesions that may invade the CPA region, and non-enhancing extra-axial lesions. Eur Radiol. 2007;17(11):2908–2920. doi: 10.1007/s00330-007-0680-4. [DOI] [PubMed] [Google Scholar]
- 123.Guermazi A, De Kerviler E, Zagdanski AM, Frija J. Diagnostic imaging of choroid plexus disease. Clin Radiol. 2000;55(7):503–516. doi: 10.1053/crad.1999.0476. [DOI] [PubMed] [Google Scholar]
- 124.Shin JH, Lee HK, Jeong AK, Park SH, Choi CG, Suh DC. Choroid plexus papilloma in the posterior cranial fossa: MR, CT, and angiographic findings. Clin Imaging. 2001;25(3):154–162. doi: 10.1016/s0899-7071(01)00284-4. [DOI] [PubMed] [Google Scholar]
- 125.Garcia-Valtuille R, Abascal F, Garcia-Valtuille AI, et al. Adult choroid plexus papilloma of the posterior fossa mimicking a hemangioblastoma. Case report. J Neurosurg. 2000;92(5):870–872. doi: 10.3171/jns.2000.92.5.0870. [DOI] [PubMed] [Google Scholar]
- 126.Addo NK, Kamaly-Asl ID, Josan VA, Kelsey AM, Estlin EJ. Preoperative vincristine for an inoperable choroid plexus papilloma: a case discussion and review of the literature. J Neurosurg Pediatr. 2011;8(2):149–153. doi: 10.3171/2011.5.PEDS1187. [DOI] [PubMed] [Google Scholar]
- 127.Pencalet P, Sainte-Rose C, Lellouch-Tubiana A, et al. Papillomas and carcinomas of the choroid plexus in children. J Neurosurg. 1998;88(3):521–528. doi: 10.3171/jns.1998.88.3.0521. [DOI] [PubMed] [Google Scholar]
- 128.McEvoy AW, Harding BN, Phipps KP, et al. Management of choroid plexus tumours in children: 20 years experience at a single neurosurgical centre. Pediatr Neurosurg. 2000;32(4):192–199. doi: 10.1159/000028933. [DOI] [PubMed] [Google Scholar]
- 129.Nagib MG, O'Fallon MT. Lateral ventricle choroid plexus papilloma in childhood: management and complications. Surg Neurol. 2000;54(5):366–372. doi: 10.1016/s0090-3019(00)00316-5. [DOI] [PubMed] [Google Scholar]
- 130.Do HM, Marx WF, Khanam H, Jensen ME. Choroid plexus papilloma of the third ventricle: angiography, preoperative embolization, and histology. Neuroradiology. 2001;43(6):503–506. doi: 10.1007/s002340000470. [DOI] [PubMed] [Google Scholar]
- 131.Di Rocco F, Caldarelli M, Sabatino G, Tamburrini G, Rocco CD. Lateral ventricle choroid plexus papilloma extending into the third ventricle. Pediatr Neurosurg. 2004;40(6):314–316. doi: 10.1159/000083747. [DOI] [PubMed] [Google Scholar]
- 132.Otten ML, Riina HA, Gobin YP, Souweidane MM. Preoperative embolization in the treatment of choroid plexus papilloma in an infant. Case report. J Neurosurg. 2006;104(6 suppl):419–421. doi: 10.3171/ped.2006.104.6.419. [DOI] [PubMed] [Google Scholar]
- 133.Picht T, Jansons J, van Baalen A, Harder A, Pietilae TA. Infant with unusually large choroid plexus papilloma undergoing emergency surgery. Case report with special emphasis on the surgical strategy. Pediatr Neurosurg. 2006;42(2):116–121. doi: 10.1159/000090467. [DOI] [PubMed] [Google Scholar]
- 134.Wind JJ, Bell RS, Bank WO, Myseros JS. Treatment of third ventricular choroid plexus papilloma in an infant with embolization alone. J Neurosurg Pediatr. 2010;6(6):579–582. doi: 10.3171/2010.9.PEDS1039. [DOI] [PubMed] [Google Scholar]
- 135.Costa JM, Ley L, Claramunt E, Lafuente J. Choroid plexus papillomas of the III ventricle in infants. Report of three cases. Childs Nerv Syst. 1997;13(5):244–249. doi: 10.1007/s003810050077. [DOI] [PubMed] [Google Scholar]
- 136.Guidetti B, Spallone A. The surgical treatment of choroid plexus papillomas: the results of 27 years experience. Neurosurg Rev. 1981;4(3):129–137. doi: 10.1007/BF01743638. [DOI] [PubMed] [Google Scholar]
- 137.Raimondi AJ, Gutierrez FA. Diagnosis and surgical treatment of choroid plexus papillomas. Childs Brain. 1975;1(2–3):81–115. doi: 10.1159/000119558. [DOI] [PubMed] [Google Scholar]
- 138.Schijman E, Monges J, Raimondi AJ, Tomita T. Choroid plexus papillomas of the III ventricle in childhood. Their diagnosis and surgical management. Childs Nerv Syst. 1990;6(6):331–334. doi: 10.1007/BF00298279. [DOI] [PubMed] [Google Scholar]
- 139.Sharma R, Rout D, Gupta AK, Radhakrishnan VV. Choroid plexus papillomas. Br J Neurosurg. 1994;8(2):169–177. doi: 10.3109/02688699409027963. [DOI] [PubMed] [Google Scholar]
- 140.Tacconi L, Delfini R, Cantore G. Choroid plexus papillomas: consideration of a surgical series of 33 cases. Acta Neurochir (Wien) 1996;138(7):802–810. doi: 10.1007/BF01411257. [DOI] [PubMed] [Google Scholar]
- 141.Chow E, Jenkins JJ, Burger PC, et al. Malignant evolution of choroid plexus papilloma. Pediatr Neurosurg. 1999;31(3):127–130. doi: 10.1159/000028847. [DOI] [PubMed] [Google Scholar]
- 142.Maria BL, Graham ML, Strauss LC, Wharam MD. Response of a recurrent choroid plexus tumor to combination chemotherapy. J Neurooncol. 1985;3(3):259–262. doi: 10.1007/BF00165187. [DOI] [PubMed] [Google Scholar]
- 143.Valencak J, Dietrich W, Raderer M, et al. Evidence of therapeutic efficacy of CCNU in recurrent choroid plexus papilloma. J Neurooncol. 2000;49(3):263–268. doi: 10.1023/a:1006405106553. [DOI] [PubMed] [Google Scholar]
- 144.Palazzi M, Di Marco A, Campostrini F, Grandinetti A, Bontempini L. The role of radiotherapy in the management of choroid plexus neoplasms. Tumori. 1989;75(5):463–469. doi: 10.1177/030089168907500512. [DOI] [PubMed] [Google Scholar]
- 145.Hawkins JC., 3rd Treatment of choroid plexus papillomas in children: a brief analysis of twenty years’ experience. Neurosurgery. 1980;6(4):380–384. [PubMed] [Google Scholar]
- 146.McGirr SJ, Ebersold MJ, Scheithauer BW, Quast LM, Shaw EG. Choroid plexus papillomas: long-term follow-up results in a surgically treated series. J Neurosurg. 1988;69(6):843–849. doi: 10.3171/jns.1988.69.6.0843. [DOI] [PubMed] [Google Scholar]
- 147.Talacchi A, De Micheli E, Lombardo C, Turazzi S, Bricolo A. Choroid plexus papilloma of the cerebellopontine angle: a twelve patient series. Surg Neurol. 1999;51(6):621–629. doi: 10.1016/s0090-3019(99)00024-5. [DOI] [PubMed] [Google Scholar]
- 148.Duke BJ, Kindt GW, Breeze RE. Pineal region choroid plexus papilloma treated with stereotactic radiosurgery: a case study. Comput Aided Surg. 1997;2(2):135–138. doi: 10.1002/(SICI)1097-0150(1997)2:2<135::AID-IGS6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 149.Kim IY, Niranjan A, Kondziolka D, Flickinger JC, Lunsford LD. Gamma knife radiosurgery for treatment resistant choroid plexus papillomas. J Neurooncol. 2008;90(1):105–110. doi: 10.1007/s11060-008-9639-9. [DOI] [PubMed] [Google Scholar]
- 150.Brown WD, Tavare CJ, Sobel EL, Gilles FH. Medulloblastoma and Collins’ law: a critical review of the concept of a period of risk for tumor recurrence and patient survival. Neurosurgery. 1995;36(4):691–697. doi: 10.1227/00006123-199504000-00008. [DOI] [PubMed] [Google Scholar]
- 151.Buetow PC, Smirniotopoulos JG, Done S. Congenital brain tumors: a review of 45 cases. AJNR Am J Neuroradiol. 1990;11(4):793–799. [PMC free article] [PubMed] [Google Scholar]