

SUMMARY
Inflammatory bowel disease is an important risk factor for colorectal cancer. We show that sphingosine-1-phosphate (S1P) produced by upregulation of sphingosine kinase 1 (SphK1) links chronic intestinal inflammation to colitis-associated cancer (CAC) and both are exacerbated by deletion of Sphk2. S1P is essential for production of the multifunctional NF-κB-regulated cytokine IL-6, persistent activation of the transcription factor STAT3, and consequent upregulation of the S1P receptor, S1PR1. The pro-drug FTY720 decreased SphK1 and S1PR1 expression and eliminated the NF-κB/IL-6/STAT3 amplification cascade and development of CAC even in Sphk2−/− mice and may be useful in treating colon cancer in individuals with ulcerative colitis. Thus, the SphK1/S1P/S1PR1 axis is at the nexus between NF-κB and STAT3 and connects chronic inflammation and CAC.
Introduction
Chronic inflammation is now recognized to have decisive roles in the pathogenesis of cancer (Grivennikov et al., 2010). Inflammatory bowel diseases (IBD) are a salient example of the link between chronic inflammation and cancer, and one of the consequences of persistent inflammation of the colon or ulcerative colitis (UC) is an increased risk for developing colorectal cancer (Ullman and Itzkowitz, 2011). Animal models that reproduce many aspects of the human disease have provided important clues to the critical roles of inflammatory mediators and molecular events leading to development of colitis-associated cancer (CAC) (Saleh and Trinchieri, 2011) and have highlighted the key roles played by the master transcription factors NF-κB and signal transducer and activator of transcription-3 (STAT3) (Bollrath et al., 2009; Greten et al., 2004; Grivennikov et al., 2009). NF-κB activation in intestinal epithelial cells (IECs) promotes survival pathways that are required for growth of premalignant cells and subsequent formation of tumors (Greten et al., 2004). Although loss of STAT3 and NF-κB in IECs has been associated with increased inflammation (Greten et al., 2004; Grivennikov et al., 2009), activation of NF-κB in myeloid-derived inflammatory cells, on the other hand, enhances inflammation in the tumor microenvironment, mainly by increasing expression of pro-inflammatory cytokines, such as TNF-α and IL-6, that in turn stimulate proliferation of tumor and stromal cells to further fuel and promote CAC (Greten et al., 2004; Popivanova et al., 2008). It was also elegantly demonstrated that IL-6 enhances both initiation and progression of CAC and that STAT3 activation downstream of IL-6 has an essential role in intestinal mucosal regeneration after injury and in the development of CAC (Bollrath et al., 2009; Grivennikov et al., 2009). Expression of TNF-α and IL-6 and activation of NF-κB and STAT3 are also increased in patients with active UC and in those who progressed to colorectal cancer (Kim et al., 2008; Li et al., 2010; Popivanova et al., 2008).
There is growing evidence that S1P, a pleiotropic bioactive sphingolipid metabolite formed inside cells by two closely related sphingosine kinases, SphK1 and SphK2, is involved in inflammation and cancer (Pyne and Pyne, 2010; Spiegel and Milstien, 2011). S1P regulates numerous cellular processes important for cell growth, survival, invasion, lymphocyte trafficking, vascular integrity, and cytokine and chemokine production, among others. S1P and SphK1 have long been implicated in the actions of pro-inflammatory cytokines, such as TNF-α (Pettus et al., 2003; Xia et al., 1998). TNF-α activates and translocates SphK1 to the plasma membrane where it catalyzes the formation of S1P (Pitson et al., 2003). S1P in turn signals through five G protein-coupled receptors (S1PR1–5). This “inside-out signaling” by S1P is thought to promote certain TNF-α functions (De Palma et al., 2006; Scherer et al., 2010). However, it was recently demonstrated that SphK1 and intracellular S1P also play a direct role in TNF-α signaling and the canonical NF-κB activation pathway important in inflammatory, anti-apoptotic, and immune processes (Alvarez et al., 2010). Interestingly, the S1P receptor S1PR1 was recently identified as a key component for persistent activation of STAT3 in tumors (Lee et al., 2010; Liu et al., 2012). It was shown that STAT3 induced S1PR1 expression and conversely, the presence of S1PR1 in the tumor and associated immune cells was required for persistent STAT3 activation, tumor growth, and metastases (Lee et al., 2010), yet the involvement of S1P, the only known endogenous S1PR1 activator, has not been investigated.
Expression of SphK1 is elevated in the colons of patients with UC (Snider et al., 2009) and colon cancer (Kawamori et al., 2008) and deletion of Sphk1 reduced aberrant crypt formation and tumor development in a mouse CAC model (Kawamori et al., 2008). However, the molecular mechanisms by which S1P produced by SphK1 promotes early or late stages of CAC have not been elucidated. Because little is still known of the function of the other sphingosine kinase isoenzyme SphK2, in this study, we investigated its involvement in colitis and CAC. We also elucidated the roles of the SphK1/S1P/S1PR1 axis in the NF-κB/IL-6/STAT3 amplification cascade and development of CAC.
RESULTS
SphK2 Deficiency Exacerbates Colitis-Associated Tumorigenesis
To examine the involvement of SphK2 in CAC development, wild type (WT) and Sphk2−/− mice were injected with the colonotropic mutagen azoxymethane (AOM) followed by three rounds of oral administration of the luminal toxin dextran sodium sulfate (DSS) to trigger chronic inflammation (Figure 1A). In contrast to the protective effects of Sphk1 deletion (Kawamori et al., 2008), knockout of Sphk2 markedly increased tumor number (Figure 1B) and tumor size (Figure 1C), with a concomitant increase in tumor load (Figure 1D). There was also a large increase in adenomas with a high grade of dysplasia in Sphk2−/− mice (Figure 1E and 1F), while most of the tumors in WT mice were low-grade dysplasias. Consistent with the profound effect on tumor size, there was a significant increase in proliferation determined by Ki-67 staining in the Sphk2−/− mice (Figure 1G and 1H). In agreement with the causal role of cyclooxygenase-2 (COX-2) in colon carcinogenesis (Oshima et al., 1996), immunohistochemical staining revealed elevated levels of COX-2 in Sphk2−/− mice, mainly in infiltrating inflammatory cells (Figure 1I).
Figure 1. Colitis-associated tumor development is increased in Sphk2−/− mice.

(A) Schematic overview of the CAC regimen. Sphk2−/− mice and WT littermates were injected with AOM followed by three cycles of 2.5% DSS in drinking water. Intestinal tumors were analyzed on day 140.
(B–D) Tumor number (B), tumor size (C), and tumor load, sum of the diameters of all tumors in a given mouse (D), were determined.
(E,F) Representative H&E stained sections from WT and Sphk2−/− mice. (F) Higher magnification images of the boxed areas in (E).
(G,H) Proliferation was determined by Ki-67 staining (H) and percent Ki-67 positive cells within colonic crypts calculated (G).
(I) COX-2 expression was determined by immunohistochemistry.
Data are means ± SEM (n=6). *p<0.05, **p<0.005. Scale bars, 100 μm.
Increased Severity of DSS-Induced Colitis in Sphk2−/− Mice
Because inflammation plays a critical role in tumorigenesis (Grivennikov et al., 2010), we next examined whether the increased tumor multiplicity and load in Sphk2−/− mice could be due to increased intestinal inflammation. To this end, mice were subjected to a single 5-day course of DSS to induce acute colitis. Sphk2−/− mice showed more severe colitis, with significantly greater weight loss (Figure 2A), more extensive colon shortening (Figure 2B), and increased hemoccult and diarrhea scores than WT littermates (data not shown). In agreement, histopathological analysis revealed more severely damaged colonic mucosa with extensive loss of crypt structures and epithelial cell denudation, larger areas of ulceration, and more extensive infiltration of inflammatory cells (Figure 2C), which was also reflected in the pathological assessment of colitis severity scores (Figure 2D). This severe colitis was not due to dysfunction of mucosal integrity as mucosal barrier function determined with FITC-dextran during the induction phase of colitis was similar in WT and Sphk2−/− mice (Figure S1A,B). There were also no significant differences in numbers of activated Th cells or Tregs between WT and Sphk2−/− mice, even during acute colitis (Figure S1C–E).
Figure 2. Deletion of SphK2 increases S1P and the severity of colitis.

(A–D) Acute colitis was induced in Sphk2−/− mice and WT littermates with 5% DSS in drinking water for 5 days followed by recovery for 5 days on normal drinking water. Changes in body weight (A), colon shortening (B), mucosal histology examined by H&E staining (C, scale bar, 100 μm), and colitis severity scores were determined (D) in a double-blind manner. In (A), (B), and (D), data are means ± SEM (n=6). *p<0.05, **p<0.001 compared to WT treated with DSS.
(E,F) S1P in serum (E) and colon (F) was measured by LC-ESI-MS/MS.
(G,H) Expression of SphK1 and SphK2 in colons was determined by QPCR (G) and western blotting (H).
In (E–G), data are means ± SD (n=3). *p<0.05, **p<0.005.
See also Figure S1.
Deletion of SphK2 Enhances SphK1 Expression
S1P has been implicated in the development of DSS-induced colitis and deletion of SphK1 reduces colitis severity (Snider et al., 2009). Indeed, we found that circulating S1P levels in WT mice were increased during acute colitis. Surprisingly, however, basal S1P levels in the circulation and colon were greater in Sphk2−/− mice and were increased even further by DSS (Figure 2E and 2F). SphK1 expression at the message and protein level in the colon was also elevated in Sphk2−/− mice compared to littermates and markedly increased during colitis (Figure 2G and 2H), indicating that the increases in S1P and severity of colitis in Sphk2−/− mice are likely due to upregulation of SphK1, especially as there were no changes in expression or activity of the S1P metabolic enzymes, S1P phosphatase and S1P lyase (Figure S1F).
Upregulation of SphK1 was observed in Sphk2−/− MEFs (Figure S2A) or after downregulating SphK2 in cultured cells (Gao and Smith, 2011). S1P produced in the nucleus by SphK2 inhibits HDAC1/2 activity similar to classical HDAC inhibitors and conversely, downregulation of SphK2 and reduction of nuclear S1P increases HDAC activity and concomitantly decreases histone acetylations (Hait et al., 2009). In agreement, colons from Sphk2−/− mice have reduced histone acetylations (Figure 3A). As it was suggested that SphK1 expression is transcriptionally regulated by c-Jun (Paugh et al., 2009; Sobue et al., 2008), a component of AP-1 transcription factor whose expression is regulated by HDACs (Yamaguchi et al., 2005), we examined the role of c-Jun in the paradoxical increase of SphK1 expression due to Sphk2 deletion. The colons from Sphk2−/− mice have high levels of c-Jun and phospho-c-Jun (Figure 3A). Sphk2−/− MEFs also have higher expression of protein and mRNA for both c-Jun and SphK1 (Figures 3B and S2A). Moreover, the pan HDAC inhibitors, trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA), suppressed the induction of c-Jun transcription, in agreement with others (Lindstrom et al., 2008; Yamaguchi et al., 2005), and also blocked induction of Sphk1 (Figure 3B). Several putative AP-1 regulatory elements in the 8-kb-long 5′-flanking region of the Sphk1 gene have been identified (Paugh et al., 2009) (Figure S2B). Indeed, reporter activity of the Sphk1 promoter containing the AP-1 site, but not a construct without this site, was markedly induced by PMA. This Sphk1 promoter activity was blocked not only by a JNK inhibitor (SP600125) but also by SAHA (Figure S2B). Altogether, these data suggest that deletion of Sphk2 and lack of HDAC inhibition by S1P increases the induction of c-Jun and its target gene Sphk1.
Figure 3. Severity of colitis in Sphk2−/− mice correlates with upregulation of SphK1, S1PR1, and IL-6, and activation of NF-κB and STAT3.

(A) Colonic lysates or nuclear extracts from Sphk2−/− mice and WT littermates were analyzed by immunoblotting with the indicated antibodies.
(B) WT and Sphk2−/− MEFs were pretreated without or with SAHA, TSA, or SP600125, stimulated without or with PMA for 3 hr, and mRNA levels were determined by QPCR.
(C–G) Colitis was induced with 5% DSS. Equal amounts of colonic nuclear extracts were analyzed by western blotting with the indicated antibodies (C). IL-6 in serum (D) and secreted from colon (E) was measured by ELISA. Expression of IL-6 (F) and S1PR1 (G) was determined in colons by QPCR.
Data are means ± SD. *p<0.05, **p<0.005.
See also Figure S2.
Upregulation of the SphK1/S1P/S1PR1 Axis and Activation of NF-κB and STAT3 in Severity of Acute Colitis
The transcription factor NF-κB has long been known to play a critical role in acute inflammation and colitis (Karin, 2009). Because S1P produced by SphK1 has been implicated as an essential regulator of the canonical NF-κB pathway (Alvarez et al., 2010), activation of NF-κB was next examined. NF-κB was constitutively activated in Sphk2−/− colon, as shown by increased phosphorylation and nuclear translocation of its p65 subunit (Figure 3C), and was markedly stimulated even further during colitis development, correlating with the elevation of SphK1 and S1P (Figure 2F–H). Consistent with the increased NF-κB activation, serum levels of the pro-inflammatory cytokine IL-6 and its release from the colonic mucosa were elevated in Sphk2−/− mice and were further increased during DSS challenge (Figure 3D and 3E). Likewise, TNF-α, but not IL-17, IFN-γ, or IL-10, release from Sphk2−/− colonic mucosa was increased compared to littermate controls (Figure S2C). A dramatic upregulation of IL-6 mRNA expression was also evident in colonic mucosa (Figure 3F). The important functions of IL-6 are exerted via activation of STAT3 (Bollrath et al., 2009; Grivennikov et al., 2009). Indeed, nuclear accumulation of phosphorylated STAT3 was already evident in the Sphk2−/− colonic mucosa and was considerably increased by DSS compared to WT (Figure 3C). In agreement with the view that STAT3 is a direct regulator of expression of S1PR1 (Lee et al., 2010; Liu et al., 2012), S1PR1 expression was also elevated in Sphk2−/− colonic mucosa and dramatically enhanced during acute colitis (Figure 3G).
Upregulation of S1PR1 expression could result in its constitutive activation (Pyne and Pyne, 2010). However, upregulation of SphK1 in Sphk2−/− mice leads to increased levels of S1P in serum and colon (Figure 2E and 2F). Moreover, SK1-I, a SphK1 inhibitor (Paugh et al., 2008), significantly reduced the severity of DSS-induced colitis (Figure S2D-F), the sustained activation of NF-κB and STAT3 (Figure S2G), and IL-6 expression in the colonic mucosa of DSS-challenged Sphk2−/− mice (Figure S2H), concomitantly with reduction of S1P (Figure S2I). In addition, W146, a competitive S1PR1 antagonist that does not have inverse agonist activity (Sanna et al., 2006), also suppressed DSS-induced weight loss and severity of colitis in Sphk2−/− mice (Figure S2J–L), the sustained activation of STAT3 and IL-6 expression (Figure S2M–O), as well as the basal hyperactivation of STAT3 and NF-κB in naïve Sphk2−/− mice (Figure S2P). Taken together, these results suggest that although expression of S1PR1 is increased in Sphk2−/− mice, it is not constitutively activated, and highlight the importance of increased formation of S1P, the ligand for S1PR1.
SphK2 Deficiency in Hematopoietic Cells Contributes to Colitis Severity, IL-6 Production, and Activation of STAT3 and NF-κB
To determine whether the effect of SphK2 deficiency on colitis severity and activation of NF-κB and STAT3 was due to hematopoietic cells or nonhematopoietic cells (e.g. IECs), and to gain insight into the cellular source of IL-6, we generated reciprocal bone marrow chimeras by adoptively transferring bone marrow into lethally irradiated recipients. Chimerism, as determined by flow cytometric analysis of the CD45.1 (Ly5.1) and CD45.2 (Ly5.2), was approximately 90–95%. After DSS challenge, body weight loss, colitis severity with inflammatory cell infiltration (Figure 4A–D), and phosphorylation of STAT3 and p65 were greatly increased (Figure 4F) when Sphk2−/− hematopoietic cells were transplanted into wild type mice (KO→WT). Conversely, colitis severity, weight loss, and activation of STAT3 and NF-κB were markedly decreased by transplantation of WT hematopoietic cells into Sphk2−/− mice (WT→KO) (Figure 4A–F). Similarly, IL-6 expression was increased in the colons of KO→WT mice compared to WT→KO or WT→WT mice (Figure 4E). Moreover, intracellular cytokine staining and flow analysis suggest that macrophages and dendritic cells are the major producers of IL-6 in KO→WT mice (Figure 4G). These results demonstrate that Sphk2−/− hematopoietic cells, rather than IECs, orchestrate these responses.
Figure 4. SphK2 deficiency in hematopoietic cells increases colitis severity, activation of STAT3 and NF-κB, and IL-6 production.

(A–F) Acute colitis was induced in bone marrow chimeric mice with 2.5% DSS. (A) Changes in body weight. (B) Maximum percent loss of body weight. * p < 0.05. (C) Colitis severity scores. * p < 0.05. (D) Mucosal histology examined by H&E staining. Scale bar, 100 μm. (E) Immunohistochemistry staining for IL-6. Scale bar, 50 μm. (F) Colonic lysates analyzed by immunoblotting.
(G) Spleen cells were stimulated with PMA and ionomycin and intracellular IL-6 staining in T cells (CD3+), B cells (B220+), macrophages (Macs, CD11b+CD11c−), and dendritic cells (DCs, CD11b+CD11c+) was analyzed by FACS. * p < 0.05, ** p < 0.01, compared to WT→WT.
(H,I) WT and Sphk2−/− mice were subjected to the CAC regimen. (H) Immunohistochemical staining of IL-6 expression in CAC bearing mice. Scale bar, 50 μm. (I) Intracellular IL-6 staining was determined by FACS as in (G). * p < 0.05, ** p < 0.01, compared to WT.
Data are means ± SEM.
See also Figure S3.
Consistent with several studies showing that IL-6 is needed continuously for stimulation of CAC tumor growth (Becker et al., 2004; Grivennikov et al., 2009; Schiechl et al., 2011), in CAC bearing Sphk2−/− mice, strong IL-6 expression was detected in infiltrating immune cells with weaker, but still enhanced expression compared to WT in IECs (Figure 4H). Confocal fluorescence microscopy was then utilized to simultaneously visualize IL-6 and macrophages or T cells during colitis and CAC. Enhanced IL-6 expression was detected predominantly in infiltrating macrophages and much less in T cells in the colonic mucosa of DSS challenged Sphk2−/− than in WT mice (Figure S3A). Moreover, during the late phase of CAC, both macrophages and T cells were increased in the adenoma leukocyte infiltrates of Sphk2−/− mice (Figure S3B) and IL-6 was strongly expressed in macrophages, dendritic cells, and in T cells, albeit to a lesser extent (Figures 4I and S3C). Consistent with the role of IL-6 in Th17 lineage differentiation, T cells from Sphk2−/− lymph nodes during colitis and CAC secreted slightly higher levels of IL-17 (Figures S1E and S3D,E).
The S1PR1 Modulator FTY720 Suppresses Colitis in Sphk2−/− Mice
As enhanced S1PR1 levels reciprocally activate STAT3 leading to its persistent activation and upregulation of IL-6 expression (Lee et al., 2010), we therefore sought to examine the effects of interfering with S1PR1 functions and this feed-forward amplification loop. To this end, we treated mice with the immunosuppressant FTY720, known to be phosphorylated in vivo to a S1P mimetic that modulates the immune system by acting as a functional antagonist of S1PR1 and inducing its internalization and degradation (Brinkmann et al., 2010). In agreement with previous studies (Daniel et al., 2007; Deguchi et al., 2006), daily treatment with FTY720 suppressed colitis symptoms in WT mice, including weight loss, colon shortening, and colonic mucosa damage and severity (Figures 5A,B and S4A). Although it has been suggested that SphK2 is essential for FTY720-phosphate formation and function in vivo (Kharel et al., 2005; Zemann et al., 2006), unexpectedly, FTY720 also significantly reduced colitis severity in mice that do not express this enzyme (Figures 5A,B and S4A). Surprisingly, FTY720 also elicited lymphopenia in Sphk2−/− mice (Figure 5B). Consistent with these in vivo effects, significant levels of FTY720-phosphate were present in serum and colons from Sphk2−/− mice (Figure 5C), albeit less than in WT. Moreover, inhibition of SphK1 with SK1-I significantly decreased phosphorylation of FTY720 in the colons of Sphk2−/− mice (Figure S4B), suggesting that SphK1 in addition to SphK2 can also phosphorylate FTY720 in vivo.
Figure 5. FTY720 suppresses colitis, S1P signaling and persistent STAT3 activation in Sphk2−/− mice.

(A–J) Mice were given PBS or FTY720 daily during colitis regimen.
(A) Changes in body weight.
(B) Colitis severity scores and lymphocyte counts in blood were determined on day 10. In (A) and (B), data are means ± SEM (n=9). *p<0.05, **p<0.005 compared to mice treated with PBS.
(C) Serum and colon levels of FTY720 and FTY720-P in WT and Sphk2−/− mice were measured by LC-ESI-MS/MS.
(D,F) Colonic lysates were analyzed by immunoblotting with the indicated antibodies.
(E)3S1P in serum and colon was measured by LC-ESI-MS/MS.
(G,H) IL-6 in serum and secreted from colon was measured by ELISA.
(I,J) Expression of IL-6 and S1PR1 was determined in colons by QPCR.
In (C), (E), and (G-J), data are means ± SD. *p<0.05, **p<0.01.
See also Figure S4.
FTY720 Interferes with the SphK1/S1P/S1PR1 Feed-forward Loop that Leads to Persistent NF-κB and STAT3 Activation and IL-6 Production
Previous studies suggest that FTY720 is also an inhibitor of SphK1 and induces its proteasomal degradation (Lim et al., 2011; Tonelli et al., 2010). FTY720 almost completely abrogated the large increase in SphK1 protein in the inflamed colon of WT mice induced by DSS treatment, as well as dramatically decreasing the remarkable enhancement in SphK1 expression in Sphk2−/− mice (Figure 5D). Concomitantly, FTY720 administration also prevented the increased S1P levels in the circulation and the colon induced by DSS treatment (Figure 5E).
It was then of relevance to examine the effect of targeting the SphK1/S1P/S1PR1 axis with FTY720 on the NF-κB/IL-6/STAT3 cascade, a central node in the development of colitis and CAC (Grivennikov et al., 2010). FTY720 not only reduced the sustained phosphorylation of the p65 NF-κB subunit in Sphk2−/− mice with acute colitis (Figure 5F) but also dramatically decreased the elevated levels of IL-6 and S1PR1 protein, and the activation of their downstream effector STAT3 (Figures 5F–J), as well as recruitment of macrophages (Figure S4A). Similar to S1PR1, expression of other STAT3-dependent gene products that were significantly increased in these mice, such as c-Myc, was also suppressed by FTY720 (Figure 5F). To gain further insight into the positive feedback loop whereby S1PR1 elevation activates STAT3 (Lee et al., 2010), we explored the involvement of the tyrosine kinase Src, which is activated downstream of ligation of S1PR1 (Rakhit et al., 1999) and has been shown to phosphorylate and activate STAT3 (Yu et al., 1995). Consistent with activated S1PR1 signaling in the Sphk2−/− colon, higher levels of Src phosphorylation were observed in these mice (Figure 5F). Hyperactivation of Src in the colonic mucosa of mice with acute colitis was also overcome by FTY720 treatment (Figure 5F). Nevertheless, treatment of naïve Sphk2−/− mice with anti-IL-6 neutralizing antibody, but not with PP1, a Src inhibitor, decreased STAT3 hyperactivation (Figure S4C), suggesting that excess IL-6 is the major contributor to STAT3 activation in these mice.
FTY720 Suppresses Colorectal Tumorigenesis Associated with Chronic Colitis
Because our data suggest that FTY720 alleviates colitis severity by impeding the positive feedback loop initiated by SphK1 and S1P signaling, we next assessed the potential efficacy of FTY720 in prevention of the development and progression of CAC. Administration of FTY720 during CAC induction drastically reduced tumor size, multiplicity, and load (Figure 6A–D). FTY720 was also very efficacious in mice deficient in SphK2, which exacerbates colitis-associated tumorigenesis (Figure 6B–D). Moreover, the few polypoid lesions protruding into the lumen that developed in the small proportion of these FTY720 treated mice were markedly smaller (Figure 6C).
Figure 6. FTY720 represses development of CAC.

(A–D) Schematic overview of FTY720 administration during CAC induction, PBS or FTY720 (0.3 mg/kg) was administered by gavage 5 times per week from day 1 to day 140 (FTY720day 1–140) (A). Tumor number per mouse (B), tumor size (C), and tumor load (D) were analyzed on day 140.
(E–H) Schematic overview of FTY720 treatment during the late stage of CAC induction, PBS or FTY720 (0.3 mg/kg) was administered 5 times per week beginning one day after the last DSS cycle (day 51) to day 120 (FTY720day 51–120) (E). Tumor number per mouse (F), tumor size (G), and tumor load (H) were analyzed on day 140.
Data are means ± SEM, n=7–8, *p<0.05, **p<0.01
To determine whether the effects of FTY720 are mainly due to suppression of inflammation during early CAC induction or if it can also impact tumor progression, FTY720 was administered to mice only after the last DSS cycle (Figure 6E). Remarkably, even when FTY720 was administered over this shorter and delayed time period, it significantly reduced the number and size of macroscopic tumors (Figures 6F and 6G) and average tumor load (Figure 6H) compared to PBS treated WT mice. Although a similar tendency was observed in Sphk2−/− mice treated with FTY720 at this late stage of CAC, it did not reach statistical significance (Figure 6F–H).
In agreement with the profound effects of FTY720 when administered to WT mice during CAC induction or even after the last DSS cycle, nuclear Ki-67 staining was reduced and restricted to the crypt bases in these mice (Figure 7A), whereas colorectal adenomas from PBS treated WT and Sphk2−/− mice showed large increases in proliferation rates compared to normal mucosa in which only about 20% of cells were Ki-67 positive (Figures 7A and 7B). Importantly, treatment with FTY720 at the late stage of CAC also significantly reduced the proliferation rate of Sphk2−/− tumors (Figures 7A and 7B). These results suggest that FTY720 can also affect tumor growth and development.
Figure 7. Colonic epithelial cell proliferation in CAC is repressed by treatment with FTY720.
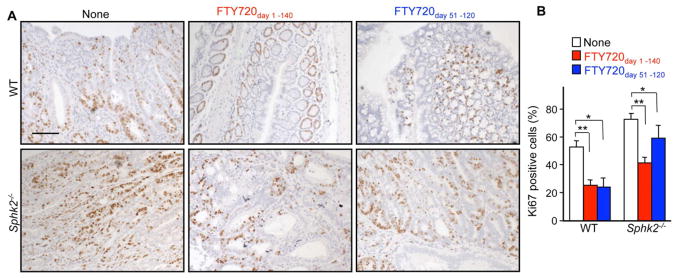
(A, B) Colons from mice treated with PBS or FTY720 from day 1–140 or from day 51–120 as indicated in Figure 6 were stained with Ki-67 antibody at the end of the CAC (A) and percent Ki-67 positive cells within colonic crypts was determined (B). Scale bar, 100 μm. Data are means ± SEM (n=5). *p<0.05, **p<0.001.
This prompted us to examine the effect of FTY720 on activation of STAT3, which is critical for proliferation and survival of IECs and development of CAC (Bollrath et al., 2009; Grivennikov et al., 2009). Immunohistochemical analysis revealed that phosphorylated STAT3 was more strongly expressed in infiltrating inflammatory cells and in nuclei of IECs of CAC adenomas from Sphk2−/− mice (Figures 8A and S5A). Administration of FTY720 even after the last DSS cycle abolished nuclear p-STAT3 staining in tumors from both WT and Sphk2−/− mice (Figure 8A), and in the much fewer infiltrating cells (Figure S5A), with marked decreases in IL-6 expression (Figure S5B). Similarly, the increased p-p65 staining in IECs of CAC adenomas from Sphk2−/− mice was reduced after this late treatment with FTY720 (Figure S5A). As with DSS-induced colitis, enhanced expression of SphK1 was clearly evident by immunohistochemistry in CAC adenomas of Sphk2−/− mice and higher levels of SphK1 were also observed in both IECs and infiltrating immune cells (Figure 8B). Late FTY720 administration also drastically reduced elevated SphK1 in CAC adenomas and in distal IECs, as well as in the few infiltrating cells (Figure 8B). Finally, late treatment with FTY720 also decreased the elevated levels of S1PR1 observed in CAC adenomas from Sphk2−/− mice (Figure 8C). Thus, FTY720 curtails the amplification loop involving SphK1, S1P, and S1PR1 leading to persistent phosphorylation of STAT3 and can suppress development of CAC even when tumors are already present.
Figure 8. Treatment with FTY720 during late stage of CAC attenuates STAT3 activation and expression of SphK1 and S1PR1.

(A–C) Immunohistochemical analysis of phospho-STAT3 (A), SphK1 (B), and S1PR1 (C) in CAC tumors from WT and Sphk2−/− mice treated with PBS or FTY720 from day 51–120. Scale bar, 100 μm.
(D) Proposed model for the role of SphK1/S1P/S1PR1 axis in a feedforward amplification loop leading to NF-κB and persistent STAT3 activation. SphK1 is upregulated in colitis and CAC, producing S1P. Extracellular S1P activates S1PR1, which induces STAT3 activation at least in part via Src. Reciprocally, STAT3 enhances transcription of its target genes, including S1PR1. Intracellular S1P is also involved in NF-κB activation, which regulates transcription of proinflammatory cytokines such as TNF-α and IL-6. IL-6 induces STAT3 activation. TNF-α also can stimulate SphK1 to amplify NF-κB activation. Treatment with FTY720 reduces upregulation of both SphK1 and S1PR1. Note: this simplified scheme is not meant to imply that all of these loops occur within a single type of cell.
See also Figure S5.
DISCUSSION
Although substantial improvements have been made in diagnosis and therapy of IBD, the risk of CAC still remains high. Significant advances have been made in recent years in understanding the link between chronic gastrointestinal inflammation and cancer, particularly in deciphering the roles of the pro-inflammatory cytokines TNF-α and IL-6 and their downstream transcription factors NF-κB and STAT3 (Ben-Neriah and Karin, 2011; Yu et al., 2009). However, negative feedback loops usually tightly limit the duration of activation of NF-κB (Ben-Neriah and Karin, 2011) and STAT3 (Yu et al., 2009), and it is not clear how their persistent activation is maintained during colitis and the progression of CAC. Our study highlights one of the previously missing links in this intriguing puzzle of the association between inflammation and cancer and suggests that strategies that disrupt this link may be of clinical benefit to IBD patients.
Here we demonstrate that SphK1, formation of S1P, and subsequent activation of its targets, NF-κB and S1PR1, play an essential role in maintaining persistent activation of NF-κB and STAT3 in a malicious feedforward amplification loop that leads to chronic inflammation and CAC development. SphK1 is upregulated in colon and gastric cancer patients and its expression correlates with cancer progression and poor prognosis (Kawamori et al., 2008; Li et al., 2009). Similar to SphK1, we found that S1PR1 is also markedly upregulated in murine models of colitis and CAC. An unanticipated observation in this study was the large basal increases in expression of SphK1 and S1PR1 in Sphk2−/− mice, which we suggest provides a priming environment that augments acute colitis and CAC. Our results suggest that myeloid cells, rather than IECs, are the initiators and orchestrators of the enhanced colitis and CAC observed in Sphk2−/− mice. During colitis, and more so in CAC, there was increased infiltration of macrophages and T cells into tumor tissues of Sphk2−/− mice that exhibited enhanced expression and secretion of IL-6. Our results support the previous conclusion that during CAC induction this tumor promoting cytokine is produced predominantly by macrophages and dendritic cells, and to a lesser extent by T cells, and possibly by IECs (Grivennikov et al., 2009; Matsumoto et al., 2010; Schiechl et al., 2011). Although recipients of Sphk2−/− CD4+ T cells in an adoptive transfer model of IBD also showed more severe colitis, it was proposed that this was due to STAT5-mediated hyperactivity of these T cells and independent of S1P (Samy et al., 2007). However, a caveat to this conclusion is that methods for measurement of low amounts of S1P in cells and tissues have only recently been developed (Shaner et al., 2009). In addition to increased IL-6, higher levels of TNF-α (but not IL-17, IFN-γ, or IL-10) were released from the colonic mucosa of Sphk2−/− mice. Our results suggest that these pro-inflammatory cytokines, TNF-α and IL-6, are responsible for the enhanced CAC in Sphk2−/− mice acting on mutant IEC cells to increase tumor incidence and growth through paracrine induction of NF-κB and STAT3 and feedforward amplification loops (Figure 8D). STAT3 then regulates expression of many genes involved in both cell survival and proliferation (Yu et al., 2009), and in addition, reciprocally upregulates transcription of S1PR1 (Yu et al., 2009). Moreover, SphK1 and intracellular S1P are also involved in activation of NF-κB by TNF-α, most likely by enhancement of the E3 ubiquitin ligase activity of TRAF2 (Alvarez et al., 2010). NF-κB and STAT3 can also act in positive feedback loops to enhance production of cytokines and chemokines that recruit additional immune cells that maintain tumor-associated inflammation (Grivennikov et al., 2010). The importance of the direct and indirect roles of NF-κB in tumorigenesis and production of tumor-promoting cytokines by inflammatory cells emerged from gene deletion studies in which inactivation of IKKβ in myeloid cells reduced tumor growth and blocked production of IL-6 and other cytokines in CAC (Greten et al., 2004). Although NF-κB does not affect proliferation of IECs, it regulates their survival by induction of anti-apoptotic genes during early tumor promotion and increases tumor incidence (Greten et al., 2004). NF-κB also enhances chemokine expression leading to further recruitment of myeloid cells that secrete pro-inflammatory cytokines, including IL-6 and TNF-α, which as mentioned are the main drivers of CAC. Thus, NF-κB can control STAT3 activation in IECs in a three-fold manner: i. by recruiting myeloid cells, most probably lamina propria macrophages, dendritic cells, and T cells, that secrete STAT3 activating cytokines; ii. by controlling the transcription of these cytokines in myeloid cells (Karin, 2009); and iii., many of these cytokines, such as TNF-α as well as IL-1, stimulate SphK1 and upregulate its expression (Spiegel and Milstien, 2011), leading to further amplification of the activation of both STAT3 (by “inside-out” signaling of S1P and ligation of S1PR1) and NF-κB (by intracellular S1P) (Figure 8D).
While NF-κB in immune cells is mainly pro-tumorigenic in CAC, the functions of STAT3 are more complex. Impaired expression of STAT3 in myeloid cells results in enhanced colitis (Takeda et al., 1999) and progression of CAC, possibly through inhibition of IL-10 signaling (Deng et al., 2010). In contrast, ablating STAT3 expression in a different colorectal cancer model decreased tumor incidence and progression by decreasing IL-17 production (Wu et al., 2009). Similarly, inhibiting STAT3 signaling in the hematopoietic system elicits multi-component antitumor immunity (Kortylewski et al., 2005). It has also been suggested that STAT3-S1PR1 upregulation in myeloid cells and persistent STAT3 activation can increase their proliferation and survival, and promote intravasation, which might facilitate colonization in future metastatic sites (Deng et al., 2012).
Like induction of NF-κB and STAT3 transcriptional programs in cancer, the most common mechanism by which SphK1 activity is increased is not through activating mutations, but rather by an excess of activating cytokines provided in an autocrine or paracrine manner. It has previously been shown that STAT3 activation and S1PR1 abundance are increased in both tumor cells and in the tumor microenvironment (Lee et al., 2010), further supporting a non-cell autonomous activation of STAT3. Likewise, SphK1 was upregulated in CAC adenomas and in inflammatory cells in the tumor microenvironment, even more so in Sphk2−/− mice (Figure 8B), and also in the mucosa and lamina propria of UC patients (Snider et al., 2009) and human colorectal cancers (Kawamori et al., 2008). Although S1P is maintained at low levels in most tissues, its abundance in the colon and circulation is congruently increased during chronic inflammation and in the tumor microenvironment (Nagahashi et al., 2012). Altered epithelial and neoplastic cells and recruited immune cells continue to produce S1P as well as cytokines (e.g. IL-6) (Bollrath et al., 2009; Grivennikov et al., 2009) that activate oncogenic STAT3 to sustain tumor-associated inflammation and to fuel tumor initiation, promotion, and progression.
Little is known about transcriptional regulation of Sphk1. We have shown that like SphK1, c-Jun, a component of the AP-1 transcription factor that regulates transcription of Sphk1, was also increased in the colons of Sphk2−/− mice. SphK2 is present in the nucleus of many types of cells and its ablation reduces nuclear levels of S1P, an endogenous inhibitor of HDAC1/2, and consequently influences expression of specific genes (Hait et al., 2009). In this regard, it has been demonstrated that inhibition of HDACs suppresses induction of c-Jun transcription by blocking the recruitment of the pre-initiation complex to the c-Jun promoter (Yamaguchi et al., 2005). In agreement, we found that HDAC inhibitors reduce expression of both c-Jun and Sphk1. Our data imply that deletion of Sphk2 and lack of HDAC inhibition by S1P increases the induction of c-Jun and its target gene Sphk1. Given the significance of c-Jun in cell transformation and carcinogenesis, our study also revealed another potential mechanism to control SphK1 expression in cancer.
The importance of the SphK1/S1P/S1PR1 amplification loop is highlighted by our findings that FTY720, which reverses the upregulation of SphK1 and S1PR1, also prevents recruitment of immune cells, excessive proinflammatory cytokine production (in particular IL-6 and TNF-α), and persistent NF-κB and STAT3 activation leading to severe colitis and CAC. Unexpectedly, however, FTY720 was also effective in Sphk2−/− mice, in contrast to previous studies indicating that SphK2 was the sole enzyme responsible for phosphorylating FTY720 in vivo to the active S1PR1 modulator (Kharel et al., 2005; Sensken et al., 2009). However, this was based on the inability of a single dose of FTY720 to induce lymphopenia in Sphk2−/− mice. As it was previously shown that FTY720 was also a weak substrate for SphK1 (Billich et al., 2003; Paugh et al., 2003), it appears that upregulation of SphK1 can compensate for the lack of SphK2.
In agreement with their immunosuppressive actions, FTY720 and other S1PR1 modulators are efficacious in several different models of colitis (Daniel et al., 2007; Deguchi et al., 2006). Although targeting the inflammatory microenvironment may be beneficial since immune cells are not subject to the same mechanisms of drug resistance as cancer cells, anti-inflammatory therapies do not kill cancer cells. Treatment with FTY720, however, blocked CAC progression even when only administered after tumors were initiated, and thus has cytostatic effects on cancer cells that are independent of immunosuppression. In agreement, FTY720 inhibits proliferation and enhances apoptosis of several types of cancer cells in vitro and suppresses xenograft tumor growth and metastasis in immunodeficient mice (Pyne and Pyne, 2010). Our work shows that FTY720 by interfering with the upregulation of both SphK1 and S1PR1 curtails the S1P-SphK1-S1PR1 feedforward amplification loop that leads to NF-κB and persistent STAT3 activation critical for CAC. These results also indicate that targeting the upstream machinery leading to STAT3 activation is a feasible alternative approach to inhibit this signaling pathway and has broad implications for pathogenesis and treatment of colitis-associated colorectal cancer in patients with IBD.
EXPERIMENTAL PROCEDURES
Mice and CAC Protocol
C57BL/6 wild type and Sphk2−/− mice were from R. Proia. Six to eight week old mice with littermate controls to assure the same genetic background were used for all experiments. Animal procedures were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University. CAC was induced as described (Grivennikov et al., 2009). Briefly, mice were injected intraperitoneally (i.p.) with 10 mg/kg azoxymethane (AOM; Sigma) and after 5 days, received drinking water containing 2.5% dextran sulfate sodium (DSS) for 5 days. Mice were then given regular drinking water for 14 days, followed by two additional DSS treatment cycles (Figure 1A). Colons were removed on day 140, flushed with PBS, and tumors counted. Macroscopic tumors were measured with calipers and software for microscopic tumors. Portions of the distal colons were either frozen in liquid nitrogen or fixed with 10% formalin and paraffin embedded for histological analyses.
Induction and Analysis of Acute Colitis
Acute colitis was induced with 5% DSS for 5 days. In some experiments, mice were given PBS or FTY720 (0.3 mg/kg) orally daily during colitis regimen. The clinical course was followed daily by measurements of body weight and monitoring signs of rectal bleeding and diarrhea and scored as described (Siegmund et al., 2001). Histological assessments of colitis and severity scores were made in a double-blinded manner after hematoxylin and eosin (H&E) staining as described (Williams et al., 2001).
Immunohistochemistry Analysis
Paraffin-embedded slides were deparaffinized and antigen unmasking was carried out by microwave heating in citrate buffer for 20 min. Slides were incubated with 3% H2O2 and then goat or horse serum (DAKO) for 30 min at room temperature. After washing with PBS, slides were incubated with primary antibodies at 4°C overnight. Biotinylated secondary anti-rat or anti-rabbit antibodies were added and incubated at room temperature for 1 hour. After 30 min with streptavidin-HRP, sections were stained with DAB substrate and counter stained with H&E. Primary antibodies used: anti-Ki-67 (DAKO); anti-COX-2, anti-p-STAT3, (Cell Signaling); anti-S1PR1 (Santa Cruz); anti-IL-6 and anti-SphK1 (Abcam).
Bone marrow chimeras
Bone marrows from femurs and tibia were collected from Sphk2+/+ congenic mice (expressing CD45.1 leukocyte antigen) and Sphk2−/− (expressing CD45.2 leukocyte antigen) donor mice and cells resupended in PBS after washing. The recipients, which were given acidified, antibiotic-treated drinking water for 2 weeks, were irradiated and then injected i.v. with 5×106 donor cells. Bone marrow reconstitution efficiency was verified after 6 weeks by staining for CD45.1 and CD45.2 in blood cells using FITC-conjugated anti-CD45.1 and PE-conjugated anti-CD45.2 (Biolegend).
Sphingolipid Analyses
Lipids were extracted from colons and serum and phosphorylated and unphosphorylated sphingoid bases were quantified by liquid chromatography, electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) as described (Hait et al., 2009).
Statistical Analysis
Experiments were repeated at least three times with consistent results. Results were analyzed for statistical significance with the Student’s t test for unpaired samples in Excel (Microsoft) and P < 0.05 was considered significant.
Supplementary Material
SIGNIFICANCE.
The pro-inflammatory cytokines TNF-α and IL-6 and their downstream master transcription factors NF-κB and STAT3 emerged as critical regulators linking inflammation to cancer. We demonstrated that upregulation of SphK1 expression, formation of the bioactive sphingolipid mediator S1P, and subsequent activation of its S1PR1 receptor play an essential role in maintaining persistent activation of these transcription factors in a malicious feedforward amplification loop that leads to chronic inflammation and CAC development. The immunosuppressive drug FTY720 interfered with the SphK1/S1P/S1PR1 axis and eliminated the NF-κB/IL-6/STAT3 amplification cascade and development of CAC. FTY720 also blocked CAC progression even after tumors were established, suggesting that FTY720 might be useful for treating cancer in individuals with chronic inflammation.
Acknowledgments
We thank R. Proia for providing Sphk2−/− mice and Dr. Michael Maceyka for helpful discussions. This work was supported by grants from the National Institutes of Health (R37GM043880, R01CA61774, U19AI077435 to S.S.). M.N. is a Japan Society for the Promotion of Science Postdoctoral Fellow. E.Y.K. and J.C.A. were supported by NIH Training Grant T32HL094290. The Flow Cytometry and Confocal Microscopy Shared Resource Cores were supported in part by NIH Grant P30CA16059 to the Massey Cancer Center and NINDS Center core Grant 5P30NS047463, respectively.
Footnotes
The Supplemental Data include Supplemental Experimental Procedures and 5 figures and can be found with this article online at http://www.cancercell.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the imunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al. gp130-mediated STAT3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Daniel C, Sartory NA, Zahn N, Schmidt R, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Mol Immunol. 2007;44:3305–3316. doi: 10.1016/j.molimm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- De Palma C, Meacci E, Perrotta C, Bruni P, Clementi E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: a novel pathway relevant to the pathophysiology of endothelium. Arterioscler Thromb Vasc Biol. 2006;26:99–105. doi: 10.1161/01.ATV.0000194074.59584.42. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Andoh A, Yagi Y, Bamba S, Inatomi O, Tsujikawa T, Fujiyama Y. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16:699–703. [PubMed] [Google Scholar]
- Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21:642–654. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Zhou JF, Sellers RS, Li JF, Nguyen AV, Wang Y, Orlofsky A, Liu Q, Hume DA, Pollard JW, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. 2010;176:952–967. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Smith CD. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol Cancer Res. 2011;9:1509–1519. doi: 10.1158/1541-7786.MCR-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. NF-κB as a Critical Link Between Inflammation and Cancer. In: Staudt LM, Karin M, editors. Cold Spring Harbor Perspectives in Biology. Cold Spring Harbor: 2009. pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, Hannun YA, Obeid LM. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2008;23:405–414. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- Kim S, Keku TO, Martin C, Galanko J, Woosley JT, Schroeder JC, Satia JA, Halabi S, Sandler RS. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008;68:323–328. doi: 10.1158/0008-5472.CAN-07-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, et al. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, Kong QL, Hu LJ, Zeng MS, Zeng YX, et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, Xia B, Kuipers EJ, van der Woude CJ. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59:227–235. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- Lim KG, Tonelli F, Li Z, Lu X, Bittman R, Pyne S, Pyne NJ. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangment in MCF-7 breast cancer cells. J Biol Chem. 2011;286:18633–18640. doi: 10.1074/jbc.M111.220756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom TM, Mohan AR, Johnson MR, Bennett PR. Histone deacetylase inhibitors exert time-dependent effects on nuclear factor-kappaB but consistently suppress the expression of proinflammatory genes in human myometrial cells. Mol Pharmacol. 2008;74:109–121. doi: 10.1124/mol.107.042838. [DOI] [PubMed] [Google Scholar]
- Liu Y, Deng J, Wang L, Lee H, Armstrong B, Scuto A, Kowolik C, Weiss LM, Forman S, Yu H. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood. 2012;120:1458–1465. doi: 10.1182/blood-2011-12-399030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M, Scheller J, Rose-John S, Kado S, Takada T. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543–1551. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Milstien S, Spiegel S, Takabe K. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by tumor-induced angiogenesis and lymphangiogenesis. Cancer Res. 2012;72:726–735. doi: 10.1158/0008-5472.CAN-11-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM, Singh SK, Kapitonov D, Rokita H, Wright S, Griswold-Prenner I, et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem. 2009;284:3408–3417. doi: 10.1074/jbc.M807170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554:189–193. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 2003;17:1411–1421. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22:5491–5500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Rakhit S, Conway AM, Tate R, Bower T, Pyne NJ, Pyne S. Sphingosine 1-phosphate stimulation of the p42/p44 mitogen-activated protein kinase pathway in airway smooth muscle. Role of endothelial differentiation gene 1, c-src tyrosine kinase and phosphoinositide 3- kinase. Biochem J. 1999;338:643–649. [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- Samy ET, Meyer CA, Caplazi P, Langrish CL, Lora JM, Bluethmann H, Peng SL. Cutting Edge: Modulation of Intestinal Autoimmunity and IL-2 Signaling by Sphingosine Kinase 2 Independent of Sphingosine 1-Phosphate. J Immunol. 2007;179:5644–5648. doi: 10.4049/jimmunol.179.9.5644. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P(1) antagonist in vivo. Nat Chem Biol. 2006;2:434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- Scherer EQ, Yang J, Canis M, Reimann K, Ivanov K, Diehl CD, Backx PH, Wier WG, Strieth S, Wangemann P, et al. Tumor necrosis factor-alpha enhances microvascular tone and reduces blood flow in the cochlea via enhanced sphingosine-1-phosphate signaling. Stroke. 2010;41:2618–2624. doi: 10.1161/STROKEAHA.110.593327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiechl G, Bauer B, Fuss I, Lang SA, Moser C, Ruemmele P, Rose-John S, Neurath MF, Geissler EK, Schlitt HJ, et al. Tumor development in murine ulcerative colitis depends on MyD88 signaling of colonic F4/80+CD11b(high)Gr1(low) macrophages. J Clin Invest. 2011;121:1692–1708. doi: 10.1172/JCI42540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensken SC, Bode C, Graler MH. Accumulation of fingolimod (FTY720) in lymphoid tissues contributes to prolonged efficacy. J Pharmacol Exp Ther. 2009;328:963–969. doi: 10.1124/jpet.108.148163. [DOI] [PubMed] [Google Scholar]
- Shaner RL, Allegood JC, Park H, Wang E, Kelly S, Haynes CA, Sullards MC, Merrill AH., Jr Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009;50:1692–1707. doi: 10.1194/jlr.D800051-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23:143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue S, Murakami M, Banno Y, Ito H, Kimura A, Gao S, Furuhata A, Takagi A, Kojima T, Suzuki M, et al. v-Src oncogene product increases sphingosine kinase 1 expression through mRNA stabilization: alteration of AU-rich element-binding proteins. Oncogene. 2008;27:6023–6033. doi: 10.1038/onc.2008.198. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of STAT3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, Bittman R, Pyne S, Pyne NJ. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal. 2010;22:1536–1542. doi: 10.1016/j.cellsig.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- Williams KL, Fuller CR, Dieleman LA, DaCosta CM, Haldeman KM, Sartor RB, Lund PK. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–937. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Gamble JR, Rye KA, Wang L, Hii CST, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA. Tumor necrosis factor-a induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci USA. 1998;95:14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Lantowski A, Dannenberg AJ, Subbaramaiah K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including CO. J Biol Chem. 2005;280:32569–32577. doi: 10.1074/jbc.M503201200. [DOI] [PubMed] [Google Scholar]
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a STAT3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemann B, Kinzel B, Muller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006;107:1454–1458. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.