

Abstract
Alpers-Huttenlocher syndrome is an uncommon mitochondrial disease most often associated with mutations in the mitochondrial DNA replicase, polymerase gamma. Alterations in enzyme activity result in reduced levels and/or deletions within the mitochondrial DNA with phenotypic manifestations occurring when the functional content of mitochondrial DNA reaches a critical nadir. The tempo of disease progression and onset varies among patients, even those with identical genotypes. The classical clinical triad of seizures, liver degeneration, and progressive developmental regression helps define the disorder, but there is a wide range of clinical expression. The majority of patients are healthy before the disease onset with seizures heralding the disorder in most patients. Seizures can rapidly progress to medical intractability with frequent episodes of epilepsia partialis continua or status epilepticus. Liver involvement may precede or occur after onset of seizures. Regardless, eventual liver failure is common. Both the tempo of disease progression and range of organ involvement vary from patient to patient and are only partially explained by the pathogenic effects of a given genetic mutation(s). Diagnosis is made by the constellation of organ involvement, not the sequence of symptoms. This disorder is relentlessly progressive and ultimately fatal.
Keywords: Alpers-Huttenlocher syndrome, Alpers syndrome, DNA polymerase gamma, mitochondrial disease, mitochondrial DNA depletion, cerebrohepatopathy
INTRODUCTION
Bernard Alpers is credited with the influential description of a 4-month-old girl with normal development who developed intractable seizures in the context of a 1-month illness [1]. Although the first description of this disease likely occurred much earlier [2], Alpers description lead to the recognition and fostered further reports of this disorder. Forty-five years later, Huttenlocher et al. [3] described the associated hepatic features, cerebral spinal fluid findings, and confirmed earlier suggestions of autosomal recessive inheritance [4]. Some 25 years passed until Harding’s [5] seminal symptomatic description of 32 patients with a distinctive liver and brain pathology, and defined the typical clinical course of the disease. Harding described normal early development; an insidious onset of developmental delay, failure to gain weight (thrive), bouts of vomiting, and pronounced hypotonia. Seizures onset occurred during these early manifestations, but in some children seizures heralded the disease. Once the seizure onset occurred, the clinical course became rapidly progressive. Liver involvement was variable in onset, in some patients it preceded seizure onset and in others only occurred at the terminal stages of the disease [6]. Post-mortem liver examination demonstrated a characteristic combination of findings [Table 1; 7]. Examination of the cerebral cortex revealed variability but a constant involvement of the calcarine cortex with microscopic changes was found including spongiosis, neuronal loss, and astrocytosis; which progressed down the cortical layers.
Table 1.
Specific liver histological findings in Alpers-Huttenlocher syndrome.
At least three of the following must be present: (Wilson disease has been ruled out)
|
Adapted from [9]
The cloning and characterization of polymerase gamma by Ropp and Copeland [8] ushered in the molecular era of polymerase gamma related disorders; and in particular Alpers-Huttenlocher syndrome. Eventually, polymerase gamma-induced disorders helped defined a category of mitochondria-nuclear communication disorders, diseases of mitochondrial DNA maintenance. The biochemical and enzymatic relevance of polymerase gamma to Alpers-Huttenlocher syndrome was made by Naviaux and colleagues with the description of mitochondrial DNA depletion and reduced polymerase gamma enzyme activity [9]. In 2001, the first patients were reported with pathogenic mutations in polymerase gamma, presenting with progressive external ophthalmoplegia and “progressive external ophthalmoplegia plus” syndromes [10]. The Alpers-Huttenlocher syndrome genotype to phenotype link was established in 2004, when Naviaux and Nguyen [11] described mutations in polymerase gamma responsible for the clinical entity of Alpers-Huttenlocher syndrome. These data completed the full pathophysiology of clinical phenotype, identification of genetic etiology, and physiological changes of reduced mitochondrial DNA content inducing the mitochondrial DNA depletion syndrome of Alpers-Huttenlocher syndrome. Within the next three years a number of publications outlining the full spectrum and clinical descriptions of the polymerase gamma disorders were nearly completed, with both dominant and recessive mutations now known to cause a wide spectrum of clinical disease [12].
CLINICAL FEATURES OF ALPERS-HUTTENLOCHER SYNDROME
Range of polymerase gamma diseases
There are over 180 mutations and counting in polymerase gamma producing mitochondrial diseases that vary in age of onset, mode of inheritance, and expression of clinical symptoms [12 - 22]. Most of the dominant mutations cause illnesses with onset in the later adult years, and these mutations reside in the catalytic residues of the polymerase domain of polymerase gamma [23]. Illness caused by recessive mutations is seen throughout the life span and represent the predominant disease inheritance pattern in polymerase gamma diseases [14, 15].
Disease-causing mutations occur throughout all exons in polymerase gamma and produce many clinical syndromes as well as multiple organ system diseases that do not cluster within distinct phenotypic syndromes [12 - 14, 24: http://niehs.niehs.nih.gov/polg/]. Although there are over a dozen common mutations, many patients with autosomal recessive disease have compound heterozygote mutations, with a nearly unlimited number of possible combinations given the 180 identified pathogenic mutations.
Infant and childhood onset polymerase gamma diseases
The clinical expression of myocerebrohepatopathy is distinct from Alpers-Huttenlocher syndrome [12, 14]. Myocerebrohepatopathy is much less common than Alpers-Huttenlocher syndrome. It has its onset usually by six months of life, much younger than what is seen in Alpers-Huttenlocher syndrome. These infants usually present with liver failure and lactic acidosis, and then may develop an encephalopathy, not necessarily involving seizures. The liver involvement in myocerebrohepatopathy is often the most devastating symptom without the hepatic pathology found in Alpers-Huttenlocher syndrome [Table 1; 7]. Alpers-Huttenlocher syndrome and myocerebrohepatopathy are therefore sometimes confused with each other due to rapid progression leading to early death [1, 5, 9, 12]. The clues that distinguish these polymerase gamma entities include the presence of medically intractable seizures, associated devastating encephalopathy, and if they live long enough a distinctive pathologically defined liver involvement in Alpers-Huttenlocher syndrome [5, 11]. However, liver involvement may be early and variable in the course of the Alpers-Huttenlocher syndrome [5], making the distinction difficult in some cases.
Alpers-Huttenlocher Syndrome
The hallmark clinical features of Alpers-Huttenlocher syndrome are intractable seizures, developmental regression and liver dysfunction. This triad of clinical hepatocerebral symptoms and seizures, when combined with 2 of 11 other findings, constitute the clinical diagnosis of Alpers-Huttenlocher syndrome (Table 2).
Table 2.
Diagnostic criteria for Alpers-Huttenlocher syndrome.
|
Adopted from [9]
The most common age of onset is between 2 – 4 years, with a range of 3 months to 8 years [5, 20, 25]. The age of Alpers-Huttenlocher syndrome onset is bimodal with a second peak onset between 17 – 24 years, with a range of 10 – 27 years [26 - 28]. Infants and children with Alpers-Huttenlocher syndrome are healthy until disease onset, although some have identified non-specific developmental delays. Age of onset is influenced, in part, by specific mutations within the polymerase gamma gene, other genes [29], and environmental factors such as intercurrent viral infections and certain medications like valproic acid. Changes within the polymerase gamma nucleotide sequence can induce changes in the enzyme activity of polymerase gamma that are influenced by the environment, called ecogenetic structural nucleotide variant changes, which can alter age of onset, severity of disease, and organ involvement of Alpers-Huttenlocher syndrome [12, 14]. The environment influence of concurrent viral illness can also induce Alpers-Huttenlocher syndrome onset as well as stepwise progression of this disorder. The full set of mechanisms for this variability remains uncertain.
Harding [5, 28] described the later age of onset and this second peak of Alpers-Huttenlocher syndrome onset has been confirmed by other reports describing polymerase gamma mutations in this population [21, 26, 27]. The age range of this latter group of Alpers-Huttenlocher syndrome patients is 17 – 24 years of age. As further described in the genetics section, the majority of patients in the older aged onset group have homozygous recessive mutations in polymerase gamma, homozygous p.A467T or p.W748S transitions in the linker region. An attractive hypothesis is that the later aged onset is due to homozygous mutations in the linker region. However, recently a compound heterozygous p.T851A and p.R1047W has been reported [26]. So, mutation location is, in part, responsible for alterations in polymerase gamma activity and phenotype, however there are clearly other modifying factors responsible.
ORGAN INVOLVEMENT
Organ involved in Alpers-Huttenlocher syndrome are primarily those that require large amounts of energy and prone to oxidative damage, which include: brain, peripheral nervous system, liver, and gastrointestinal tract.
Nervous system involvement
Seizures are the most dramatic central nervous symptom of Alpers-Huttenlocher syndrome. In about 50% of patients, seizures are the heralding symptom. Once seizures appear, the tempo of the disease becomes rapidly progressive. Death usually occurs within 4 years of onset [26 - 28]. In other cases, disease progression is slower [14, 21]. The etiology for the variation in disease progression is unknown. The first association of the use of valproic acid causing rapid onset of liver disease in a child with Alpers-Huttenlocher syndrome was in 1992 [30], and soon lead to the understanding that the clinical variables associated with valproate-induced liver failure, were in fact the clinical features of Alpers-Huttenlocher syndrome. Therefore, valproic acid (and divalproex sodium) have likely altered the natural history of Alpers-Huttenlocher syndrome and may have given the impression of a universal rapidly progressive disease. Since the ability to rapidly identify patients with Alpers-Huttenlocher syndrome and the acceptance that valproate is contraindicated in this disorder, an analysis of seizure frequency, duration, and medication use (without valproic acid exposure) needs to be performed to understand the natural history of disease progression without this variable.
The electroencephalogram (EEG) findings and seizure semiology can vary from patient to patient, and change as the disease progresses in any patient. Some patients have febrile convulsions, which may indicate co-morbid seizure networks in some patients [31]. Often, the initial afebrile seizure semiology and EEG are not well-described, however in those patients with well documented EEG studies and witnessed seizures, occipital lobe findings are most common. Focal asymmetric occipital-predominant seizures present as visual and tactile hallucinations, nausea and vomiting, dysautonomia, headache and eye nystagmus auras and/or seizures, often together with focal and/or generalized motor seizures [31, 32]. The EEG, during the initial period of seizure manifestations, often presents with occipital predominant epileptiform discharges with concomitant focal slowing of the frequency (Figure 1). Epileptiform wave morphologies are described as spike/polyspikes and may be unilateral or bilateral over the posterior head region [31-34]. As the disease progresses, most patients have repeated episodes of status epilepticus and epilepsia partialis continua [9, 11, 20, 21, 31, 35 - 37]. Over time, seizures become more resistant to medical therapy and individual seizures have longer duration. Myoclonus and myoclonic seizures become more prominent and become almost continuous, and both are refractory to medical treatment. Death is not unusual in the setting of persistent seizures and myoclonus. In any patient with an epileptic encephalopathy without adequate explanation, Alpers-Huttenlocher syndrome should be considered and valproate avoided unless genetic testing of polymerase gamma is normal, as for unclear reasons valproic acid induces liver dysfunction that results in liver failure [11, 30, 31, 38, 39]. Seizures and the lack of effective medical control is a major morbidity, with death due to uncontrolled seizures commonly seen.
Figure 1.

Electroecephalogram (EEG) epoch of a 7-year old female with Alpers-Huttenlocher syndrome. This figure demonstrates regional slowing in the parieto-occipital regions bilaterally with epileptiform discharges within the right occipital region. The scale legend depicted represents time (1 second interval) and amplitude (microvolts).
The predominance of neuronal loss in the calcarine and striate cortex presents clinically as blindness. Visual loss may be transitory early in the course of Alpers-Huttenlocher syndrome but usually becomes permanent at some point during the disease progression. Microscopic changes show spongiosis, neuronal loss and astrocytosis, which extends and progresses into the depth of the cortex [5]. Early in disease, central volume loss exceeds cortical volume loss and can lead to the appearance of hydrocephalus ex vacuo [9]. In the terminal phases of the disease, the entire cortex becomes a thin gliotic scar with resultant cerebral atrophy. These pathological changes likely correlate with the evolving encephalopathy as the disease progresses. Medically intractable seizures produce hippocampus sclerosis. The cerebellum is variably involved, often with Purkinje cell dropout and Bergman gliosis with preserved granular cell layer [11]. The degree of ataxia likely relates to the loss of Purkinje cells.
Most patients are healthy before the onset of symptoms, but some can demonstrate non-specific neurological symptoms, such as clumsiness, migraine headaches, progressive ataxia or mild medically controlled seizures before explosive seizure onset [21, 26, 31, 35]. Migraine headaches can be associated with visual hallucination auras due to occipital lobe involvement [21, 35].
Other nervous system manifestations are reported in Alpers-Huttenlocher syndrome. The predominance of neuronal cell loss in the calcarine and striate cortex induces cortical blindness. Visual loss may be initially transitory but as the disease progresses may become complete. Cranial nerve dysfunction of abnormal eye movements, and delayed acquisition of smooth pursuits (delayed maturation of cranial nerves and eye movement networks within the cortex and cerebellum) are due to central and peripheral neuronal dysfunction. The peripheral nervous system is also involved in most patients with disease progression.
Evaluating sensory neuropathy in young children is difficult and often escapes identification until muscle stretch reflexes are lost. The objective loss of pain and temperature, vibratory sensation, and light touch sensation are usually late findings. In older patients with recessive polymerase gamma mutations, sensory peripheral neuropathy was demonstrated to be due to loss of the peripheral axonal branches of the dorsal root ganglia neurons, secondary to the neuronal death [40]. The same mechanism is likely occurring in younger children, but rigorous testing in the younger population of Alpers-Huttenlocher syndrome has not been reported. However, sensory neuropathy has been reported as an early finding demonstrating its presence in younger patients [31]. Ataxia can be an early symptom and is universally seen during the course of the disease due to cerebellar, sensory nerve ascending pathways and/or cortical tract involvement [31, 34, 36, 41].
Liver involvement
There are environmental exposures that can accelerate liver dysfunction. Valproic acid exposure inducing severe liver dysfunction has been one of the defining features of AHS [11, 30 - 32, 39]. Liver dysfunction occurs without valproate exposure, and is part of the defining natural history of this illness, as Huttenlocher et al. [3] early description of the disease through those patients described by Harding [5, 28], liver involvement without valproic acid exposure has been one of the defining symptoms. Furthermore, the histochemical changes induced by valproic acid are identical to that found without exposure (Table 1) [5, 7]. The pathological features in Alpers-Huttenlocher syndrome are distinctive and differ from other chemically induced or toxic liver disorders [42, 43]. Like other organ involvement, the expression of liver dysfunction varies between patients; however exposure to valproic acid seems to the catalyst for liver dysfunction, regardless of the patient’s inherited predisposition or early or late liver involvement. Unfortunately, the exact mechanism of liver alterations remains elusive. If a patient lives long enough, most all patients will eventually develop liver involvement and subsequent failure.
In the situation where the patient is placed on valproate, liver dysfunction usually begins within 2 – 3 months, but may be delayed by up to 6 months. Liver dysfunction is usually heralded by hypoglycemia, decreased albumin synthesis, reduced synthesis of coagulation factors, and mild transaminase elevation. Early recognition of liver dysfunction may stop the progression to failure. One study demonstrated that the use of intravenous levocarnitine reversed the course of early valproate-induced hepatic failure [44]. In rare cases, liver dysfunction induced by valproate can be reversed by drug withdrawal [32, 45].
Hypoglycemia can be an early symptom of Alpers-Huttenlocher syndrome as it is with several other electron transport chain disorders, especially during liver immaturity in the first 1 – 2 years of life [46]. Onset in the first year of life, with normal liver biopsy has been described [31]. Slightly older patients have been described with fasting hypoglycemia [47]. Unexplainably, the hypoglycemic events may resolve over time. The precise etiology remains unclear but likely reflects a secondary impairment of mitochondrial fatty acid oxidation due to electron transport chain dysfunction. Coagulopathies may occur with or without frank cirrhosis in Alpers-Huttenlocher syndrome. These are the result of hepatic biosynthetic failure of vitamin K dependent clotting factors II, VII, IX, and X. However, it is not uncommon in the intensive care unit to observe prothrombotic episodes of deep venous (e.g. subclavian or femoral) thrombosis at the site of venous line placement in Alpers-Huttenlocher syndrome patients. This can be from a relative deficiency in the liver synthesis of anti-coagulant proteins C, S, or anti-thrombin III.
Gastrointestinal involvement
Many patients develop swallowing dysfunction, delayed gastric emptying, and intestinal dysmotility, which all worsen as the disease progresses. These children often require placement of a gastric tube for nutrition in the mid-stage of the disorder. The use of continuous gastric tube feedings is often required because normal bolus feedings are not tolerated due to poor gastric and intestinal motility. Disease progression often results is complete gastrointestinal dysmotility and many patients require jujenal tube feedings and eventually total parental nutrition. A complete loss of the longitudinal muscularis propria external muscle layer in the gastrointestinal tract was found in 2 patients with Alpers-Huttenlocher syndrome at autopsy, first presenting at age 2 and 13 years (Saneto, unpublished data). Mitochondrial DNA depletion within the muscularis propria was demonstrated in one patient with polymerase gamma mutations compatible with Alpers-Huttenlocher syndrome in an infant who died at 20 days of life [48]. A similar pathological finding has been reported in another mitochondrial DNA depletion syndrome, mitochondrial neurogastrointenstinal encephalomyopathy disease [49]. The reason for the specific nature of the loss of the external muscle layer of the gastrointestinal tact is unknown, but the mitochondrial DNA depletion within smooth muscle of the muscularis propria of both diseases may be responsible.
Pancreatitis may also occur in Alpers-Huttenlocher syndrome. It is tempting to invoke pancreatitis to valproic acid exposure. Especially, since valproic acid induction of pancreatitis is a well-known side effect in patients with epilepsy [50]. However, Harding [5] reported pancreatitis in patients not exposed to valproic acid. The mechanism of pancreatitis in Alpers-Huttenlocher syndrome remains unknown.
Cardiac involvement
Cardiomyopathy and congestive failure occur in perhaps 10% of the children with Alpers-Huttenlocher syndrome. While not common, this complication can further compromise liver function when significant right-sided failure is present.
DIAGNOSIS OF ALPERS-HUTTENLOCHER SYNDROME
Genetic testing
Sequencing polymerase gamma should be performed if Alpers-Huttenlocher syndrome is suspected. Although screening for the few most common mutations may capture most cases, most experts now recommend full sequencing. There are more than 60 mutations in polymerase gamma reported to be associated with Alpers-Huttenlocher syndrome, so the number and combination of mutations makes full sequencing of the gene the most sensitive and specific method in confirming the diagnosis. The use of gene sequencing has more importance in Alpers-Huttenlocher syndrome than in other biochemical disorders because there are no specific or sensitive biochemical markers for polymerase gamma disease. Some evidence exist that 3-methylglutaconic aciduria and electron transport chain involvement may have more severely affected disease [51]. Muscle tissue may show a mosaic histochemical staining pattern of cells negative for cytochrome c oxidase and enhanced trichrome staining (ragged-red fibers). However, neither of these findings is diagnostic for Alpers-Huttenlocher syndrome.
Electron transport chain enzymology
Depression of electron transport chain enzymatic activities in muscle and liver are not specific for Alpers-Huttenlocher syndrome and are an insensitive method of diagnosis, especially early in the course of the illness. Reports have demonstrated a variable pattern of electron transport chain defects, ranging from completely normal to single and multiple electron transport chain deficiencies [24, 38]. The variability of electron transport chain complex activity findings is likely related to the stage of disease and the tissue tested. As the disease progresses, abnormal electron transport chain activities become more pronounced as the catalytic subunits of the various complexes encoded by mitochondrial DNA limit enzyme activity. Therefore, analysis of electron transport chain should not be used to screen or diagnose Alpers-Huttenlocher syndrome.
Mitochondrial DNA content
The mitochondrial DNA copy number in Alpers-Huttenlocher syndrome patients is 3% – 40% of normal [11, 18, 22, 22]. The use of mitochondrial DNA content may seem useful for diagnosis, but it is not sensitive or specific for Alpers-Huttenlocher syndrome because mitochondrial DNA content may be normal early in the disease process. In addition, mitochondrial DNA depletion may be seen in other diseases having muscle and/or liver involvement making diagnosis based on mitochondrial DNA content misleading (Table 3). Muscle and liver mitochondrial DNA depletion may lag behind disease symptoms. Some mutations do not alter mitochondrial DNA content but the fidelity of the transcript and render the mitochondrial DNA useless [52]. In this latter case, the mitochondrial DNA content will be normal. If mitochondrial DNA content is measured, only liver and muscle tissue should be used, as polymerase gamma mutations do not always induce mitochondrial DNA depletion in blood cells [53]. As the disease progresses, almost all Alpers-Huttenlocher patients will begin to demonstrate mitochondrial DNA depletion. Rare exceptions have been reported in patients with Alpers-Huttenlocher syndrome who had mitochondrial DNA deletions without mitochondrial DNA depletion [54]. Therefore, the finding of mitochondrial DNA depletion may be helpful in the diagnosis, but its absence cannot be used to exclude Alpers-Huttenlocher syndrome.
Table 3.
Disorders of Mitochondrial Maintenance (mitochondrial DNA depletion disorders)
| Gene | Age on Onset | CNS Features | Systemic Features | Depletion Organ(s) |
|---|---|---|---|---|
| POLG1 | Infancy- Adult |
Ataxia, Seizures, Dementia, Blindness PEO, Neuropathy |
Liver failure* GI dysmotility Vomiting, Myopathy |
Liver, muscle |
| DGUOK | Neonatal | Dystonia, Nystagmus Hypotonia |
Liver failure | Liver |
| MPV17 | Neonatal- Adult |
Ataxia, Neuropathy Dystonia, Hypotonia |
Liver failure, MR, Scoliosis Corneal scarring |
Liver |
| Twinkle | Infancy | Athetosis, Seizures, Myopathy, MR, PEO Psychosis, Neuropathy Ataxia, Hypotonia |
Liver failure | Liver |
| TK2 | Infancy- Childhood |
Seizures, PEO | Myopathy | Muscle |
| RRM2B | Neonatal | Microcephaly, MR Hearing loss, Hypotonia |
Myopathy, Tubulopathy, Nephrocalcinosis |
Muscle, kidney |
| SUCLG1 | Neonatal | Hypotonia, Severe early encephalopathy |
Lactic acidosis Hepatomegaly |
Liver, muscle |
| SUCLA2 | Neonatal- Infancy |
Dystonia, Hearing loss, Encephalopathy PEO, Neuropathy, MR, Hypotonia |
Muscle |
POLG1: polymerase gamma 1; DGUOK: deoxyguanosine kinase; MPV17: encodes a small mitochondrial membrane protein of unclear function; TWINKLE: mitochondrial DNA helicase; TK2: thymidine kinase; RRM2B: p53-dependent ribonucleotide reductase; SUCLG1: GTP-dependent succinyl-CoA synthase; SUCLA2: ATP-dependent succinyl-CoA synthase.
PEO: progressive external ophthalmoplegia; MR: mental retardation; GI: gastrointestinal
Liver failure is induced by valproic acid exposure but will occur without exposure. All mitochondrial DNA depletion syndromes are inherited as autosomal recessive traits. There are also autosomal dominant forms of polymerase gamma mutations that have adult onset.
Electroencephalogram and seizures
Well-described and specific EEG findings and seizure semiology early in the disease course may suggest Alpers-Huttenlocher syndrome as a diagnosis (Figure 1). Explosive seizures with asymmetric occipital lobe predominance of epileptiform discharges that evolve into status epilepticus and/or epilepsia partialis continua, combined with psychomotor delay occur frequently in Alpers-Huttenlocher syndrome, and not in other illnesses, so these findings should signal the possibility of Alpers-Huttenlocher syndrome [31 - 33]. Only a few epileptic syndromes including early-onset childhood epilepsy with occipital spikes (Panayiotopoulos syndrome), late-onset childhood epilepsy with occipital spikes (Gastaut type), Lafora disease, Celiac disease, Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes (MELAS), Myoclonus, Epilepsy with Ragged-red Fibers (MERRF), Epilepsy with bilateral occipital calcifications, idiopathic photosensitive occipital epilepsy, and malformations of cortical development have a posterior head predominance of spike/polyspike and wave epileptiform discharges [55]. This combination of epileptiform location and seizure semiology should also signal the clinician to confirm that polymerase gamma is normal before starting valproic acid.
Neuroimaging
Neuroimaging findings are non-specific but can be helpful in diagnosis. Computerized tomography (CT) and magnetic resonance (MR) imaging may be normal early in the course of the disease, but as the disease progresses MR imaging changes reflect acute and chronic pathological changes. Diffusion weighted imaging acquisition is most sensitive for demonstrating early Alpers-Huttenlocher syndrome lesions that cannot be seen by routine MRI [39]. The occipital location of EEG abnormalities and neuronal loss/gliosis can sometimes be seen on MR as hyperintensity on T2/FLAIR sequences in the occipital regions suggesting mitochondrial dysfunction [55]. Often when the seizures are uncontrolled, T2/FLAIR hyperintensities are prominent in the thalami and basal ganglia [32]. There may also be transient resolution of MR changes with normalization of the signal changes [56]. However, as the disease progresses, MR imaging demonstrate atrophy reflecting the pathological changes in the basal ganglia and brainstem induced by the disease [5, 54]. Some patients have cerebellar involvement, which corresponds to the prominent Purkinje cell loss seen in autopsy cases [5].
The diagnosis of Alpers-Huttenlocher syndrome takes a great deal of clinical acumen. The clinical expression of signs and symptoms vary in timing, intensity, and severity. Clinical suspicion is needed early in the course of diagnosis, as the sequence of symptoms may be distinctive for each patient. However, eventually the constellation of clinical symptoms will be expressed and polymerase gamma sequencing will confirm the diagnosis.
GENETICS OF ALPERS-HUTTENLOCHER SYNDROME
Polymerase gamma protein
The polymerase gamma protein is the only known DNA polymerase in the mammalian mitochondria and is responsible for mitochondrial DNA replication and repair [23, 57]. Polymerase gamma is encoded by the polymerase gamma gene on chromosome 15q25, synthesized in cytoplasm, and transported to its inner mitochondrial matrix. Here in the inner membrane it associates with other nuclear encoded proteins that make up the mtDNA replisome and nucleoids. Polymerase gamma has three distinct DNA activities; a 5′->3′ DNA polymerase, a 3′->5′ exonuclease, and a 5′-deoxyribose phosphate lyase activity. The exonuclease activity is located within the N-terminal region and is connected by a linker region to the C-terminal domain, which contains the 5′-3′ polymerase activity (Figure 2A). The polymerase region has three subdomains named palm, fingers, and thumb. The C-terminal domain contains the 5′-3′-polymerase activity as well as the 5′-deoxyribose phosphate lyase activity. The exact location of the 5′-deoxyribose phosphate lyase activity within the C-terminal region is unknown [58]. There are three highly conserved sequence motifs within the exonuclease region, I, II, and III, and three within the polymerase region, A, B, and C, which are essential for full polymerase gammaactivity [23, 57]. Within the linker region, there are two regions required for full activity, the accessory protein interacting domain (AID) located toward the N-terminal part of the linker region and the intrinsic processivity subdomain (IP) located distally in the linker region (toward the C-terminal).
Figure 2.
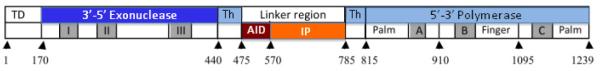
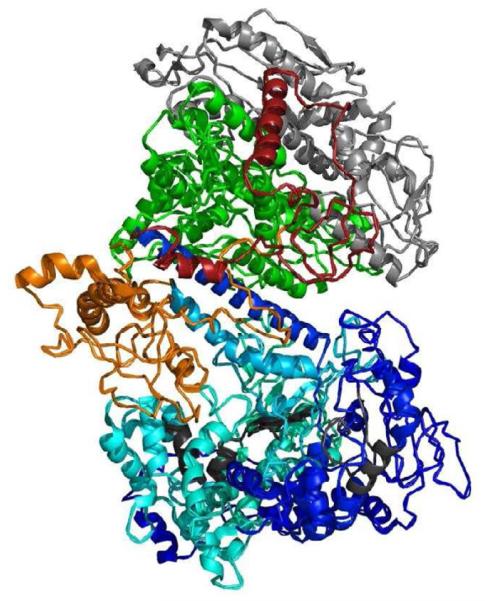
Functional and structural domains of the polymerase protein. A. Linear organization of the polymerase gamma protein. The N-terminal domain (NTD) region is the mitochondrial targeting sequence and is located at the N-terminal of the protein. The thumb subdomains (Th) of the protein and are found between the exonuclease and linker region as well as within the polymerase region [63]. The exonuclease domain contains essential motifs I, II, and III for its activity. The polymerase domain contains subdomains comprising the thumb (Th), palm, and finger, which contain motifs A, B, and C, respectively. There are two regions that make up the palm subdomain within the polymerase domain. The motifs located within the polymerase domain are critical for polymerase activity [23, 57]. The small arrows indicate position of the numbered amino acid position that begin a subdomain. B. Three dimensional structure of the human DNA polymerase holoenzyme [59]. The color scheme of the polymerase gamma catalytic subunit is the same as in A (light blue for the polymerase domain, dark blue for the exonuclease region, red for the accessory interacting domain (AID) domain and orange for the intrinsic processivity (IP) domain. The conserved motifs in the exonuclease (I, II, and III) and in the polymerase (A, B, and C) are colored in charcoal. The two accessory subunits are colored in green, for the proximal subunit, and grey for the distal subunit.
Holoenzyme
The holoenzyme of polymerase gamma is a heterotrimer comprised of one molecule of polymerase gamma associated with two molecules of polymerase gamma 2 [8, 59]. Polymerase gamma 2, the 55-kDa accessory protein enhances the processivity (the average number of nucleotides added by the enzyme per association-disassociation with the template DNA) of the holenzyme [60]. One site of polymerase gamma 2 association is with the linker region at the AID subdomain of the catalytic subunit (Figure 2B) [61]. This interaction increases the DNA-binding affinity of the holoenzyme. The second polymerase gamma 2 protein makes limited contact with polymerase gammaand is labeled the distal accessory site (Figure 2B) [60]. Interaction of polymerase gamma 2 to this distal site enhances the polymerization rate of the holoenzyme [61]. The replisome complex also comprises the mitochondrial DNA helicase PEO1 (Twinkle), a single stranded DNA binding protein, mtSSB, and a number of accessory proteins and transcription factors [23].
Polymerase gamma genetics
Alpers-Huttenlocher syndrome is an autosomal recessive disorder. Both copies of polymerase gamma are expressed in mammalian cells and mono-allelic expression of wild-type polymerase gamma is sufficient to avoid disease [62]. Homozygote dominant polymerase gamma mutations or even a dominant polymerase gamma mutation with a recessive mutation have not been described, suggesting that these possible mutation combinations are likely embryonic lethal. Although Alpers-Huttenlocher syndrome producing mutations are distributed along the entire length of the polymerase gamma gene, mutations tend to cluster within distinct tertiary regions within the gene [63]. Thus far, no dominant mutations have been reported to cause Alpers-Huttenlocher syndrome. Autosomal dominant mutations in polymerase gamma are only known to cause progressive external ophthalmoloplegia and occur primarily in the polymerase region. Recessive mutations within the polymerase region decrease polymerase activity, alter DNA-binding affinity, and lower catalytic efficiency [63]. Mutations in the linker region of the accessory interactive subdomain (AID) mediate interactions with polymerase gamma 2 and destabilize the polymerase gamma-DNA complex. When mutations are found in the intrinsic processivity region, they induce decreases in processivity, DNA binding and polymerase activity [64]. Mutations within the exonuclease region, specifically in the finger subdomain, confer partitioning of the DNA substrate between the polymerase and exonuclease sites, and predict to diminish the fidelity of the polymerase [63]. In fact, however, mutations in the exonuclease region have not shown to diminish exonuclease activity but confer their effect on polymerase activity [65, 66].
Recessive mutations in polymerase gamma can be present as homozygous or compound heterozygous mutations. In general, in trans compound heterozygous mutations usually induce a more severe phenotype, while homozygous recessive mutations are associated with milder and later-onset disease [23, 39]. Both types of recessive mutations are associated with early childhood and juvenile onset Alpers-Huttenlocher syndrome. However, for reasons that are not currently understood, either in trans compound heterozygous or homozygous mutations can be found in the severe early onset phenotype and milder juvenile onset Alpers-Huttenlocher syndrome patients. The reasons for such disparate genotype/phenotype variation within a single syndrome, Alpers-Huttenlocher syndrome, and the larger group of polymerase gamma syndromes are not completely understood.
A partial explanation for the phenotypic variability within Alpers-Huttenlocher syndrome may be the location of the mutation within the gene. The in trans compound heterozygote mutations are usually associated with more severe early onset or childhood disease. The combination of mutations within two distinct regions of polymerase gamma causes a compounding of the reduction in enzyme activity and thereby more severely compromising mitochondrial DNA replication [63]. The most common example are patients with the in trans compound heterozygote mutation within the linker region and polymerase region, p.A467T/p.W748S, which is expressed as decreased survival and increased incidence of liver failure compared to patients with homozygous mutations p.A467T/p.A467T and p.W748S/p.W748S [13, 21, 67]. Unfortunately, this attractive hypothesis cannot fully explain why these same homozygous mutations can also give rise to early onset Alpers-Huttenlocher syndrome or the same in trans compound heterozygote mutations to other distinct polymerase gamma syndromes with a wide range of disease onset and severity [17, 21]. So, although it is likely that combinations of mutations within distinct regions of polymerase gamma do, in part, determine Alpers-Huttenlocher syndrome phenotype, there are other yet unknown mechanisms involved.
Ecogenetic structural nucleotide variants
One possible emerging mechanism for phenotypic variability is the finding of silent nucleotide polymorphisms, ecogenetic structural nucleotide variants that depending on environmental or epigenetic situations can alter disease expression. When present, a particular polymerase gamma genotype would remain silent unless specific conditions are present that can either lead to disease or modify phenotype. The polymorphic nature of polymerase gamma suggests that the presence of ecogenetic structural nucleotide variants may exist [14]. Two of these ecogenetic structural nucleotide variants have emerged; within the Northern European population, p.E1143G and p.Q1236H have been described [20, 67, 68]. The p.E1143G has a frequency of approximately 4.5% and the p.Q1236H approximately 8.6% of the Northern European population, but are almost non-existent in the Asian, sub-Saharan African or African-American populations (http://ww.ncb/htm.nih.gov/SNP_ref.cgi?rs=23-7441). The presence of the ecogenetic structural nucleotide variant p.E1134G has been demonstrated to increase catalytic rate for incoming nucleotides as well as increased intrinsic stability in in-vitro studies [64]. When present in cis with certain pathological mutations, especially with the p.W748S mutation, p.E1143G can modify disease phenotype by enhancing enzyme activity [20, 67]. This same ecogenetic structural nucleotide variant can also increase the liver’s sensitivity to valproic acid exposure [68]. Thus, the ecogenetic structural nucleotide variants ENSV, p.E1143G can be a disease modifying by either enhancing enzyme activity or compromising liver function, depending on the genetic situation and environmental exposure. As with p.E1143G, the presence of p.Q1236H can induce liver sensitivity to valproic acid exposure. It is estimated that the presence of either the p.E1143G or p.Q1236H increases valproic acid sensitivity > 20 fold [68]. A question arises whether either ecogenetic structural nucleotide variant is wholly or partially responsible for induced liver failure induced by valproic acid exposure in Alpers-Huttenlocher syndrome. We have preliminary evidence that both p.E1143G and p.Q1236H are likely independent from other, yet unknown, polymerase gamma mutations that induce liver failure; 6 previous reported patients with Alpers-Huttenlocher syndrome and liver failure did not have either ecogenetic structural nucleotide variant [31; Wong personal communication]. There are rare reports of long-term use of valproic acid exposure with polymerase gamma mutation(s) without liver failure [21]. Whether other valproic acid sensitive ecogenetic structural nucleotide variants exist and the full understanding of valproic acid induced liver failure remain unclear.
Other ecogenetic structural nucleotide variants exist, such as the p.R964C mutation that induces high levels of lactic acidosis induced by mitochondrial toxicity when polymerase gamma is exposed to the nucleoside reverse-transcription inhibitor, stavudine [69]. This ecogenetic structural nucleotide variant has been shown to have a similar binding affinity for dTTP as wild type enzyme and only a small 33% decrease in dTTP incorporation efficiency. However, the p.R964C mutation induced a three fold decreased in discrimination of d4TTP incorporation over natural substrate (dTTP) in addition to impaired polymerase function, suggesting this as a mechanism of d4T-induced mitochondrial toxicity [70]. Whether other ecogenetic structural nucleotide variants exist that may alter polymerase gamma function and clinical symptoms remains unclear, but this mechanism of altering enzyme activity may be important in defining environmental alterations in clinic phenotype.
Other genes causing Alpers-Huttenlocher syndrome
Alterations in other genes that are part of the mitochondrial DNA replisome can induce Alpers-Huttenlocher syndrome. Two siblings have been described with in trans compound heterozygote mutations in the mitochondrial DNA helicase PEO1 [Twinkle; 29]. Autopsy in one of the siblings who died during a status epilepticus event revealed that the liver changes did not meet the strict criteria of histochemical and structural findings of Alpers-Huttenlocher syndrome (Table 1). However, it is likely that the young age of this patient’s death prevented full expression of the liver changes, as the other features of Alpers-Huttenlocher syndrome were present (Table 1).
EPIDEMIOLOGY OF POLYMERASE GAMMA MUTATIONS IN ALPERS-HUTTENLOCHER SYNDROME
The true prevalence of Alpers-Huttenlocher syndrome is unknown. Due to high mortality and early death, epidemiology parameters of incidence and prevalence are not informative. However, Alpers-Huttenlocher syndrome must be uncommon based on the following. It is estimated that the minimum birth frequency of children who will develop mitochondrial disease is approximately 1 in 5,000 [71]. Of this population, up to 25% will develop polymerase gamma disease [19]. Crude estimates from published reports suggest that up to 30% of patients with recessive polymerase gamma mutations have Alpers-Huttenlocher syndrome, however true figures are not known. A best estimate of birth prevalence of children that will ultimately develop Alpers-Huttenlocher syndrome is about 1 in 100,000 [72]. Clearly, more reliable population studies are needed to more reliably estimate incidence and prevalence of Alpers-Huttenlocher syndrome.
Population estimates of common mutations
The most common mutation reported inducing Alpers-Huttenlocher syndrome and associated polymerase gamma-spectrum disorders is the p.A467T mutation. The second most common mutation in Alpers-Huttenlocher syndrome and polymerase gamma-spectrum disorders is the p.W748S mutation. Elegant population studies have demonstrated that both mutations arose from a common ancestor within various populations of Northern European decent [73]. The carrier frequency of p.A467T is estimated to be as high as 0.6% in the Belgium and 1% in the Norway populations and the p.W748S frequency estimated to be 1:125 in Finland [35, 36, 73]. Other Alpers-Huttenlocher syndrome founder gene mutations have not been discovered in other ethnic populations. Whether a founder effect is exclusive to the Northern European ethnic population is not known, as most of the early mutational research has centered on the North European population. Polymerase gamma mutations inducing Alpers-Huttenlocher syndrome have been found in many other ethnic groups [31, 74, 75]. As more patients are described with polymerase gamma mutations it is possible that more “hot spot” regions of polymerase will be uncovered.
Gender
Males and females seem to be equally affected in Alpers-Huttenlocher syndrome [12, 14, 21, 38]. There is a non-significant slight increase in males in the infant age group and mild female predominance in the older age range having Alpers-Huttenlocher syndrome.
THERAPY
Treatment of Manifestations
Treatment is limited to symptom management and supportive care and family education should be addressed as soon as the family is able to absorb the diagnosis. The global perspective of care should be palliative even if death is not imminent. Quality of life issues, specifically the intensity of treatment that can be offered when major changes occur in disease status is a critical conversation. This illness progresses to a fatal encephalopathy or liver failure and varying levels of treatment is available and can be addressed openly with the family. Supportive care will include the placement of a gastrostomy feeding tube for medication, hydration, and/or nutrition. Different levels of supportive ventilation may include less invasive treatments such as continuous positive airway pressure (CPAP) or Bilevel positive airway pressure (BiPAP) assisted nasal ventilation or involve the placement of a tracheostomy and use of mechanical ventilation. Involvement of palliative care services can assist the care team in these discussions, as well as practical aspects of implementation. Most patients utilize rehabilitative services and rehabilitation units are often where family members learn to care for their loved ones with new gastrostomy tubes and ventilatory support. Occupational, physical and/or speech therapy is performed to maintain neurological function for as long as possible and to insure comfort when deterioration begins.
Consultation with a gastroenterologist early in the disease course is necessary to address feeding and nutritional issues, and to assess and manage liver pathology. Surgical placement of a gastric feeding tube when appropriate can maintain nutritional status and/or prevent aspiration of oral feedings, but in some situations, given the ultimate course of the illness, some families may wish not to take this course.
Ventilation disorders are common in Alpers-Huttenlocher syndrome and can exist long before hypoventilation is clinically obvious. Assessment of both daytime and nocturnal ventilatory function can be performed for evidence of central and/or obstructive apnea using polysomnography with measurement of pCO2 and monitoring by pulse oximetry. Tracheostomy placement and artificial ventilation may be performed, but again, some families may wish to allow the natural course of the illness proceed without this invasive and usually life-long approach.
Attempts should be made to control the seizures as best as possible. However, refractory epilepsy, especially epilepsia partialis continua, may be impossible to control with any treatment and the side-effects of treatment may outweigh any clinical benefit. There is no evidence that newer medications such as lamotrigine, topiramate, oxcarbazepine, or levetiracetam offer a better therapeutic benefit over the older medications (phenobarbital, phenytoin, carbamazepine, primidone), however, the newer medications tend to be less sedating, may require less processing by the liver, and may have fewer drug-drug interactions. Valproic acid (Depakene) and sodium divalproate (divalproex; Depakote) should be avoided. Because other anticonvulsants have also been implicated in accelerating liver deterioration, it is reasonable to monitor liver functions every two to four weeks after introducing any new medications.
Movement disorders are common and some such as chorea and athetosis may cause pain. Muscle relaxants and pain medications, including narcotics, would be advised in this circumstance. Some movement disorders can be treated with dopaminergic medication such as levodopa-carbidopa or tetrabenazine, and therefore a trial of either of these medications can be considered.
Standard treatment for liver failure can include small frequent meals or continuous feeding to compensate for impaired gluconeogenesis. In addition, reducing dietary protein to a minimum, use of non-absorbable sugars to create an osmotic diarrhea, and the use of conjugating agents to treat hyperammonemia may be helpful. Because levocarnitine may have some benefit in the setting of liver failure and because of its low toxicity, some recommend its use from the time of diagnosis.
The role of liver transplantation in treatment of liver failure Alpers-Huttenlocher syndrome is contraindicated. The multisystem organ involvement with the relentless progression in the context of neurocognitive impairment would exclude the isolated correction of liver failure with transplantation.
Recent reports suggest that cerebral spinal fluid folate deficiency can occur in Alpers-Huttenlocher syndrome [76]. Testing for cerebral folate deficiency requires a lumbar puncture for detection of 5-methyl-tetrahydrofolate concentration in cerebral spinal fluid. Cerebral folate deficiency would be treated with calcium Leucovorin, which crosses the blood brain barrier and increases cerebral folate levels. From a practical standpoint, cerebral folate deficiency can occur at any point in the disease process and monitoring cerebral spinal fluid folate a few times a year is not practical, so using empiric treatment is reasonable.
There are no standard of care guidelines available to suggest the frequency for which these tests should be obtained. Testing should be guided by clinical features and the proposed schedule should be modified if the clinical course is stable. Monitoring blood counts, electrolytes, liver enzymes (AST, ALT, GGT), liver function tests (preprandial serum glucose concentration, serum concentration of ammonia, albumin, bilirubin (free and conjugated) and cholesterol, and prothrombin time or INR as measure of coagulation factors) is reasonable every few months. There is no clear evidence that either lactic acid or plasma amino acids assist in management but some clinicians find value in these tests. Because of liver disease it is reasonable to monitor the plasma concentration of free and total carnitine, even in those on levocarnitine supplementation. Other periodic tests may include liver ultrasound, EEG, Brainstem Auditory Evoke Potential, swallowing evaluations and polysomography with pulmonary function tests.
CONCLUSION
Alpers-Huttenlocher syndrome is a mitochondrial disorder induced by autosomal recessive mutations in polymerase gamma that decrease mitochondrial DNA replication. Ultimately, polymerase gamma dysfunction induces a progressive depletion of mitochondrial DNA and evolving organ dysfunction. Premature death is universal, and often occurs within 4 years of the onset of symptoms. Alpers-Huttenlocher syndrome is exclusively an autosomal recessive disorder caused by homozygous or in trans compound heterozygous mutations in polymerase gamma. There is no precise phenotype to genotype correlation, although there are suggestions that location of mutations within specific regions of polymerase gamma may play a role in the phenotypic expression of the disorder. Clinically, most patients have a normal early development, with timing of onset and progression of disease that varies in presentation, severity, tempo of progression and sequence of organ involvement. However, the constellation of progressive clinical symptoms within brain, liver, and muscle remains the hallmark of the disease. Many questions remain: elucidation of mutations or ecogenetic structural nucleotide variants that alter disease phenotype, breath of external factors that can modify disease course and the natural history of various mutation combinations (phenotype/genotype).
The relentless progression and lack of treatment makes Alpers-Huttenlocher syndrome a challenging disease to treat and emotionally wrenching for families. There are no treatments that serve to modify the course of the illness and supportive treatment is recommended. There is a novel possible treatment medication that is making its way through the clinical trial pathway that may offer hope [77]. Some exciting findings in a mouse model of polymerase gamma disease has been recently published, suggesting that if diagnosed early enough, exercise might significantly delay disease manifestations [78]. Whether exercise can help modify the disease remains to be proved in humans, but the concept is exciting.
Since the molecular, clinical, and biochemical elucidation of Alpers-Huttenlocher syndrome over a decade ago, there is an enlarging body of information concerning the genetic and environmental elements of Alpers-Huttenlocher syndrome. There is much work to be done to understand how genotype influences phenotype, discovery of rational treatments, and better medical care for patients and families. Hopefully, through research and astute clinical judgment these facets of Alpers-Huttenlocher syndrome will be accomplished.
ACKNOWLEDGEMENTS
The authors wish to thank the patients and their families that have allowed us to participate in their medical care.
This work was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences ES 065078 (to WCC), NIH grant U54NS078059-01 and the Mitochondrial Research Guild at Seattle Children’s Hospital (to RPS), and the UCSD Christini Fund, the Wright Foundation, the Lennox Foundation, the Jane Botsford-Johnson Foundation and the Hailey’s Wish Foundation (to RKN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25:469–505. [Google Scholar]
- [2].Bullard WM. Diffuse cortical sclerosis of the brain in children. J Nerv Ment Dis. 1890;15:699–709. [Google Scholar]
- [3].Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976;33:186–192. doi: 10.1001/archneur.1976.00500030042009. [DOI] [PubMed] [Google Scholar]
- [4].Sandback U, Lerman P. Progressive cerebral poliodystrophy-Alpers’ disease: disorganized giant neuronal mitochondria on electron microscopy. J Neurol Neurosurg Psychiatry. 1972;35:749–755. doi: 10.1136/jnnp.35.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): A personal review. J Child Neurol. 1990;5:273–287. doi: 10.1177/088307389000500402. [DOI] [PubMed] [Google Scholar]
- [6].Eggar J, Harding BN, Boyd SG, Wilson J, Erdohazi M. Progressive neuronal degeneration of childhood (PNDC) with liver disease. Clin Pediatr. 1987;26:167–173. doi: 10.1177/000992288702600401. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen KV, Sharief FS, Chan SSL, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006;45:108–116. doi: 10.1016/j.jhep.2005.12.026. [DOI] [PubMed] [Google Scholar]
- [8].Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase gamma. Genomics. 1996;35:449–458. doi: 10.1006/geno.1996.0490. [DOI] [PubMed] [Google Scholar]
- [9].Naviaux RK, Nyhan WJ, Barshop BA, Poulton J, Markusic D, Karpinski NC, Hass RH. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers syndrome. Ann Neurol. 1999;25:54–58. doi: 10.1002/1531-8249(199901)45:1<54::aid-art10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- [10].Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterization by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- [11].Naviaux RK, Nguyen KV. POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- [12].Wong L-JC, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, Milone M, Cohen BH, Wical B, Ganesh J, Basinger AA, Burton BK, Swoboda K, Gilbert DL, Vanderver A, Saneto RP, Maranda B, Arnold G, Abdenur JE, Waters PJ, Copeland WC. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mut. 2008;29:E150–E172. doi: 10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang S, Wang J, Lee N-C, Milone M, Halberg MC, Schmitt ES, Craigen WJ, Zhang W, Wong L-JC. Mitochondrial DNA polymerase γ mutations: an ever expanding molecular and clinical spectrum. J Med Genet. 2011;48:669–681. doi: 10.1136/jmedgenet-2011-100222. [DOI] [PubMed] [Google Scholar]
- [14].Saneto RP, Naviaux RK. Polymerase gamma disease through the ages. Dev Disabil Res Rev. 2010;16:163–174. doi: 10.1002/ddrr.105. [DOI] [PubMed] [Google Scholar]
- [15].Cohen BH, Chinnery PF, Copeland WC. GeneReviews at Gene Tests: Medical Genetics Information Resource, Copyright. University of Washington; Seattle: 2010. Available from: http://www. genetests.org (database online) [Google Scholar]
- [16].Cohen BH, Naviaux RK. The clinical diagnosis of POLG disease and other mitochondrial DNA depletions disorders. Methods. 2010;51:364–373. doi: 10.1016/j.ymeth.2010.05.008. [DOI] [PubMed] [Google Scholar]
- [17].Blok MJ, van den Bosch BJ, Jongen E, Hendrickx A, de Die-Smulders CE, Hoogendijk JE, Brusse E, de Visser M, Poll-The BT, Bierau J, de Coo IF, Smeets HJ. The unfolding clinical spectrum of POLG mutations. J Med Genet. 2009;46:776–785. doi: 10.1136/jmg.2009.067686. [DOI] [PubMed] [Google Scholar]
- [18].Spinazzola A, Zeviani M. Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J Intern Med. 2009;265:174–192. doi: 10.1111/j.1365-2796.2008.02059.x. [DOI] [PubMed] [Google Scholar]
- [19].Chinnery PF, Zeviani M. 135th ENMC workshop: polymerase gamma and disorders of mitochondrial DNA synthesis, 21-23 September 2007. Naarden, The Netherlands. Neuromuscul Disord. 2008;18:257–267. doi: 10.1016/j.nmd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- [20].Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, Prokisch H, Lochmuller H, McFarland R, Ramesh V, Klopstock T, Freisinger P, Salvi F, Mayr JA, Santer R, Tesarova M, Zeman J, Udd B, Taylor RW, Turnbull D, Hanna M, Fialho D, Suomalainen A, Zeviani M, Chinnery PF. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- [21].Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthum S, Ferrari G, Aarseth JH, Bindoff LA. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006;129:1685–1692. doi: 10.1093/brain/awl097. [DOI] [PubMed] [Google Scholar]
- [22].Ferrari G, Lamanea E, Donati A, Filosto M, Briem E, Carrara F, Parini R, Simonati A, Santer R, Zeviani M. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-γ A. Brain. 2005;128:723–731. doi: 10.1093/brain/awh410. [DOI] [PubMed] [Google Scholar]
- [23].Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- [24].Polymerase Gamma gene mutation database. accessed at: http://tools.niehs.nih.gov/polg/
- [25].De Vries MC, Rodenburg RJ, Morava E, van Kaauwen EPM, ter Laak H, Mullaart RA, Snoeck IN, van Hasselt PM, Harding P, van den Heuvel LPW, Smeitink JAM. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur J Pediatr. 2007;166:229–234. doi: 10.1007/s00431-006-0234-9. [DOI] [PubMed] [Google Scholar]
- [26].Wiltshire E, Davidzon G, DiMauro S, Akman HO, Sadlier L, Haas L, Zuccollo J, McEwen A, Thorburn DR. Juvenile Alpers Disease. Arch Neurol. 2008;65:121–124. doi: 10.1001/archneurol.2007.14. [DOI] [PubMed] [Google Scholar]
- [27].Uusimaa J, Hinttala R, Rantala H, Paivarinta M, Herva R, Roytta M, Soini H, Moilanen JS, Remes AM, Hassinen I, Majamaa K. Homozygous W748S mutation in the POLG1 gene in patients with juvenile-onset Alpers syndrome. Epilepsia. 2008;49:1038–1045. doi: 10.1111/j.1528-1167.2008.01544.x. [DOI] [PubMed] [Google Scholar]
- [28].Harding BN. Progressive neurological degeneration of childhood with liver disease (Alpers’ Disease) presenting in young adults. J Neurol Neurosurg and Psych. 1995;58:320–325. doi: 10.1136/jnnp.58.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- [30].Bicknese AR, May W, Hickey WR, Dodson WE. Early childhood hepatocerebral degeneration misdiagnosed as valproate hepatotoxicity. Ann Neurol. 1992;32:767–775. doi: 10.1002/ana.410320610. [DOI] [PubMed] [Google Scholar]
- [31].Saneto RP, Lee I-C, Koenig MK, Bao X, Weng S-W, Naviaux RK, Wong L-JC. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure. 2010;19:140–146. doi: 10.1016/j.seizure.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wolf NI, Rahman S, Schmitt B, Taanman J-W, Duncan AJ, Harting I, Wohlrab G, Ebinger F, Rating D, Bast T. Status epilepticus in children with Alpers’ disease caused by POLG1 mutations: EEG and MRI findings. Epilepsia. 2009;50:1596–1607. doi: 10.1111/j.1528-1167.2008.01877.x. [DOI] [PubMed] [Google Scholar]
- [33].Engelsen BA, Tzoulis C, Karlsen B, Lillebo A, Laegreid LM, Aasly J, Zeviani M. Bindoff LA. POLG1 mutations cause syndromic epilepsy with occipital lobe predilection. Brain. 2008;131:818–828. doi: 10.1093/brain/awn007. [DOI] [PubMed] [Google Scholar]
- [34].Tulinius MH, Hagne I. EEG findings in children and adolescents with mitochondrial encephalomyopathies: a study of 25 cases. Brain and Develop. 1991;13:167–173. doi: 10.1016/s0387-7604(12)80024-6. [DOI] [PubMed] [Google Scholar]
- [35].Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, Van Goethem G, Lofgren A, Hackman P, Paetau A, Kaakkola S, Majamaa K, Varilo T, Udd B, Kaarainen H, Bindoff LA, Suomalainen A. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–441. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Winterhun S, Ferrara G, He L, Taylor RW, Zeviani M, Turnbull DM, Engelsen BA, Moen G, Bindoff LA. Autosomal recessive mitochondrial ataxia syndrome due to mitochondrial polymerase gamma mutation. Neurology. 2005;64:1204–1208. doi: 10.1212/01.WNL.0000156516.77696.5A. [DOI] [PubMed] [Google Scholar]
- [37].Gauthier-Villars M, Landrieu P, Cormier-Daire V, Jacquemin E, Chretien D, Rotig A, Rustin P, Munnich A, de Lonlay P. Respiratory chain deficiency in Alpers syndrome. Neuropediatr. 2001;32:150–152. doi: 10.1055/s-2001-16614. [DOI] [PubMed] [Google Scholar]
- [38].Davidzon G, Mancuso M, Ferraris S, Quinzii C, Hirano M, Peters HL, Kirby D, Thorburn DR, DiMauro S. POLG mutations and Alpers syndrome. Ann Neurol. 2005;57:921–923. doi: 10.1002/ana.20498. [DOI] [PubMed] [Google Scholar]
- [39].Nguyen KV, Ostergaard E, Ravin SH, Balslev T, Danielsen ER, Vardag A, McKiernan PJ, Gray G, Naviaux RK. POLG mutations in Alpers syndrome. Neurology. 2005;65:1493–1495. doi: 10.1212/01.wnl.0000182814.55361.70. [DOI] [PubMed] [Google Scholar]
- [40].Lax NZ, Whittaker RG, Hepplewhite PD, Reeve AK, Blakely EL, Jaros E, Ince PG, Taylor RW, Fawcett PRW, Turnbull DM. Sensory neuronopathy in patients harboring recessive polymerase γ mutations. Brain. 2012;135:62–71. doi: 10.1093/brain/awr326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Van Goethem G, Luoma P, Rantamaki M, Memar A, Kaakkola S, Hackman P, Krahe R, Lofgren A, Martin JJ, De Jonghe P, Suomalainen A, Udd B, Van Broeckhoven C. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. doi: 10.1212/01.wnl.0000140494.58732.83. [DOI] [PubMed] [Google Scholar]
- [42].Simonati A, Filosto M, Savio C, Tomelleri G, Tonin P, Dalla Bernardina B, Rizzuto N. Features of cell death in brain and liver, the target tissues of progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher disease) Acta Neuropathol. 2003;106:57–65. doi: 10.1007/s00401-003-0698-x. [DOI] [PubMed] [Google Scholar]
- [43].Boyd SG, Harden A, Egger J, Pampiglione G. Progressive neuronal degeneration of childhood with liver disease (“Alpers Disease”): characteristic neurophysiological features. Neuropediatr. 1986;17:75–80. doi: 10.1055/s-2008-1052505. [DOI] [PubMed] [Google Scholar]
- [44].Bohan TP, Helton E, McDonald I, Konig S, Gazitt S, Sugimoto T, Scheffner D, Cusmano L, Li S, Koch G. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology. 2001;56:1405–1409. doi: 10.1212/wnl.56.10.1405. [DOI] [PubMed] [Google Scholar]
- [45].McFarland R, Hudson G, Taylor RW, Green SH, Hodges S, McKieman PJ, Chinnery PF, Ramesh V. Reversible valproate hepatotoxicity due to mutations in mitochondrial DNA polymerase gamma (POLG1) Arch Dis Child. 2008;93:151–153. doi: 10.1136/adc.2007.122911. [DOI] [PubMed] [Google Scholar]
- [46].Fellman V, Kotarsky H. Mitochondrial hepatopathies in the newborn period. Semin Fetal Neonatal Med. 2011;16:222–228. doi: 10.1016/j.siny.2011.05.002. [DOI] [PubMed] [Google Scholar]
- [47].Mochel F, Slama A, Touati G, Desguerre I, Giurgea I, Rabier D, Brivet M, Rustin P, Saudubray J-M, DeLonlay P. Respiratory chain defects may present only with hypoglycemia. J Clin Endocrinol Metab. 2005;90:3780–3785. doi: 10.1210/jc.2005-0009. [DOI] [PubMed] [Google Scholar]
- [48].Giordano C, Powell H, Leopizzi M, de Curtis M, Travaglini C, Sebastiani M, Gallo P, Taylor RW, d’Amari G. Fatal congenital myopathy and gastrointestinal pseudo-obstruction due to POLG1 mutations. Neurology. 2009;72:11103–1105. doi: 10.1212/01.wnl.0000345002.47396.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Giordano C, Sebastiani M, De Giorgio R, Travaglini C, Tancredi A, Valentino ML, Bellan M, Cossarizza A, Hirano M, d’Amati G, Carelli V. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am J Pathol. 2008;173:1120–1128. doi: 10.2353/ajpath.2008.080252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bai HX, Ma MH, Orabi AI, Latif SU, Bhandari V, Husain SZ. Novel characterization of drug-associated pancreatitis in children. J Pediatr Gastroenterol Nutr. 2011;53:423–428. doi: 10.1097/MPG.0b013e318228574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wortmann SB, Rodenburg RJT, Jonckheere A, de Vries MC, Huizing M, Heldt K, van den Heuvel LP, Wendel U, Kluijtmans LA, Engelke UF, Wevers RA, Smeitink JAM, Morava E. Biochemical and genetic analysis of 3-mtehylglutaconic aciduria type IV: a diagnostic strategy. Brain. 2009;132:136–146. doi: 10.1093/brain/awn296. [DOI] [PubMed] [Google Scholar]
- [52].Taanman J-W, Rahman S, Pagnamenta AT, Morris AA, Bitner-Glindzicz M, Wolf NI, Leonard JV, Clayton PT, Schapira AH. Analysis of mutant DNA polymerase γ in patients with mitochondrial DNA depletion. Hum Mut. 2009;30:248–254. doi: 10.1002/humu.20852. [DOI] [PubMed] [Google Scholar]
- [53].Dimmock D, Tang L-Y, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- [54].Kollberg G, Moslemi A-R, Darin N, Nennesom I, Bjarnadottir I, Uvebrant P, Holme E, Melberg A, Tulinius M, Oldfers A. POLG1 mutations associated with progressive encephalopathy in childhood. J Neuropathol Exp Neurol. 2006;65:463–469. doi: 10.1097/01.jnen.0000229987.17548.6e. [DOI] [PubMed] [Google Scholar]
- [55].Taylor I, Scheffer IE, BErkovic SF. Occipital epilepsies: identification of specific and newly recognized syndromes. Brain. 2003;126:753–769. doi: 10.1093/brain/awg080. [DOI] [PubMed] [Google Scholar]
- [56].Saneto RP, Friedman S, Shaw DWW. Neuroimaging in mitochondrial disease. Mitochondrion. 2008;8:396–413. doi: 10.1016/j.mito.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Ann Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- [58].Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Identification of 5′deoxyribose phosphate lyase activity to human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U.S.A. 1998;95:12244–12248. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lecrenier N, Van Der Gruggen P, Poury F. Mitochondrial DNA polymerase from yeast to man: a new family of polymerases. Gene. 1997;185:147–152. doi: 10.1016/s0378-1119(96)00663-4. [DOI] [PubMed] [Google Scholar]
- [60].Lee Y-S, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–324. doi: 10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lee Y-S, Molineux IJ, Yin YW. A single mutation in human mitochondrial DNA polymerase pol gammaA affects both polymerization and proofreading activities, but only as a holoenzyme. J Biol Chem. 2010;285:28105–28116. doi: 10.1074/jbc.M110.122283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chan SSL, Longley MJ, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. JBC. 2005;36:31341–31346. doi: 10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- [63].Euro L, Farnum GA, Palin E, Suomalainen A, Kaguni LS. Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol γ. Nucleic Acids Res. 2011;39:9072–9084. doi: 10.1093/nar/gkr618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chan SSL, Longley M, Copeland WC. Modulation of the W748S mutation in DNA polymerase gamma by the E1143G polymorphism in mitochondrial disorders. Hum Mol Genet. 2006;15:3473–3483. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Stumpf JD, Bailey CM, Spell D, Stillwagon M, Anderson KS, Copeland WC. mip1 containing mutations associated with mitochondrial disease causes mutagenesis and depletion of mtDNA in Saccharomyces cerevisiae. Hum Mol Genet. 2010;19:2123–33. doi: 10.1093/hmg/ddq089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Szczepanowska K, Foury F. A cluster of pathogenic mutations in the 3′-5′ exonuclease domain of DNA polymerase gamma defines a novel module coupling DNA synthesis and degradation. Hum Mol Genet. 2010;19:3516–3529. doi: 10.1093/hmg/ddq267. [DOI] [PubMed] [Google Scholar]
- [67].Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787:312–319. doi: 10.1016/j.bbabio.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Stewart JD, Horvath R, Baruffini E, Ferrero I, Bulst S, Watkins PB, Fontana RJ, Day CP, Chinnery PF. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010;52:1791–1796. doi: 10.1002/hep.23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yamanaka H, Gatanaga H, Kosalaraksa P, Matsuoka-Aizawa S, Takahashi T, Kimura S, Oka S. Novel mutation of human DNA polymerase γ associated with mitochondrial toxicity induced by anti-HIV treatment. J Infect Dis. 2007;195:1419–1425. doi: 10.1086/513872. [DOI] [PubMed] [Google Scholar]
- [70].Bailey CM, Kasiviswanathan R, Copeland WC, Anderson KS. R964C mutation of DNA polymerase γ imparts increased stavudine toxicity by decreasing nucleoside analog discrimination and impairing polymerase activity. Antimicrob Agents and Chemother. 2009;53:2610–2612. doi: 10.1128/AAC.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metabol. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- [72].Naviaux RK. Alpers syndrome. Orphan Net http://www.orpha.net/consor/cgibin/OC_Exp.php?lng=EN&Expert=726.
- [73].Hakonen AH, Davidzon G, Salemi R, Bindoff LA, Van Goethem G, DiMauro S, Thorburn DR, Suomalainen A. Abundance of the POLG disease mutations in Europe, Australia, New Zealand, and the United States explained by single ancient European origin. Eur J Hum Genet. 2007;15:779–783. doi: 10.1038/sj.ejhg.5201831. [DOI] [PubMed] [Google Scholar]
- [74].Mohamed K, FathAllah W, Ahmed E. Gender variability in presentation with Alpers’ syndrome: a report of eight patients for the UAE. J Inherit Metab Dis. 2011;34:439–441. doi: 10.1007/s10545-011-9278-8. [DOI] [PubMed] [Google Scholar]
- [75].Gonzalez-Vioque E, Bazquez A, Ferandez-Moreira D, Bornstein B, Bautista J, Arpa J, Navarro C, Campos Y, Fernandez-Moreno MA, Garesse R, Arenas J, Martin MA. Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch Neurol. 2006;63:107–111. doi: 10.1001/archneur.63.1.107. [DOI] [PubMed] [Google Scholar]
- [76].Hasselmann O, Blau N, Ramaekers VT, Quadros EV, Sequeira JM, Weissert M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet and Metab. 2010;99:58–61. doi: 10.1016/j.ymgme.2009.08.005. [DOI] [PubMed] [Google Scholar]
- [77].Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Barnes AA, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91–102. doi: 10.1016/j.ymgme.2011.10.009. [DOI] [PubMed] [Google Scholar]
- [78].Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnoplosky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. PNAS. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]