

Abstract
Objectives
The aim of this study was to evaluate the links between connexin43 (Cx43) expression, myocardial conduction velocity, and ventricular tachycardia in a model of healed myocardial infarction.
Background
Post-infarction ventricular arrhythmias frequently cause sudden death. Impaired myocardial conduction has previously been linked to ventricular arrhythmias. Altered connexin expression is a potential source of conduction slowing identified in healed scar border tissues. The functional effect of increasing border-zone Cx43 has not been previously evaluated.
Methods
Twenty-five Yorkshire pigs underwent anterior infarction by transient left anterior descending coronary artery occlusion, followed by weekly testing for arrhythmia inducibility. Twenty animals with reproducibly inducible sustained monomorphic ventricular tachycardia were randomized 2:1:1 to receive AdCx43, Adβgal, or no gene transfer. One week later, animals underwent follow-up electrophysiologic study and tissue assessment for several functional and molecular measures.
Results
Animals receiving AdCx43 had less electrogram fractionation and faster conduction velocity in the anterior-septal border zone. Only 40% of AdCx43 animals remained inducible for ventricular tachycardia, while 100% of controls were inducible after gene transfer. AdCx43 animals had 2-fold higher Cx43 protein levels in the anterior-septal infarct border, with similar percents of phosphorylated and intercalated disk-localized Cx43 compared with controls.
Conclusions
These data mechanistically link Cx43 expression to slow conduction and arrhythmia susceptibility in the healed scar border zone. Targeted manipulation of Cx43 levels improved conduction velocity and reduced ventricular tachycardia susceptibility. Cx43 gene transfer represents a novel treatment strategy for post-infarction arrhythmias.
Keywords: connexin, gene therapy, myocardial infarction, ventricular tachycardia
Sudden cardiac arrest remains the leading cause of death in developed countries. Previous research has shown that the majority of cases of sudden cardiac arrest occur in the setting of acute or chronic ischemic heart disease (1). Ventricular tachycardia (VT) is a common cause of sudden cardiac arrest in patients with healed myocardial infarction (MI) scars, and electrophysiologic study (EPS) generally localizes the VT circuit to the MI scar border (2). The underlying arrhythmia mechanism for scar-related VT is re-entry through this border zone. Wavelength theory suggests that re-entry should be extinguished by increasing either conduction velocity (CV) or tissue refractory properties. We previously eliminated arrhythmia inducibility by delaying repolarization (3). Here, we focus on the conduction side of the equation.
Factors previously shown to impede conduction through healed scar include reduced intercellular coupling and discontinuities in tissue architecture (2,4). The major factors controlling intercellular coupling are gap junction density and function. Connexin43 (Cx43) is the predominate ventricular gap junction protein (5). Previous studies have shown reduced border-zone Cx43 expression and gap junction localization, persisting chronically after MI (4,6). In the subacute phase, days after MI, these Cx43 changes paired with reductions in cellular excitability have been linked to CV slowing and VT (7,8), but longitudinal study has shown that the determinants of cellular excitability (resting membrane potential, action potential amplitude, and upstroke velocity) returned to normal within 2 weeks of infarction (9). Although these investigators and others have implicated gap junction remodeling in the pathogenesis of VT coming from the healed infarct border, it has thus far been challenging to directly evaluate the association between reduced Cx43 and VT in chronic scar. Here, we address this issue using our previously validated ventricular gene transfer methods and porcine model of healed MI and inducible VT.
Methods
For full details of methods, please see the Online Appendix. Briefly, 25 Yorkshire pigs (weight 25 to 30 kg) were studied as follows. At the start of the study, animals underwent infarction and defibrillator implantation. Weekly thereafter, animals underwent noninvasive EPS through their defibrillators. Four weeks after MI, animals with repeatedly inducible sustained monomorphic VT (SMVT) at both 3-week and 4-week post-MI EPS were randomized 2:1:1 to gene transfer with AdCx43, Adβgal, or no virus. Immediately before and 1 week after gene transfer, animals underwent invasive EPS. After the follow-up EPS, animals were sacrificed, and their hearts were harvested for optical mapping, histology, and Cx43 analyses. The Adβgal virus controls and the no-virus controls were compared as a single group with the AdCx43 animals by prospective design. Continuous variables were analyzed using Student t tests or 2-way analysis of variance as appropriate. VT inducibility was assessed using chi-square tests. All statistical tests were conducted at the 0.05 significance level. Data are presented as mean ± SD (or as medians and ranges because of the lack of a normal distribution and the very small numbers for the post hoc substudy within the AdCx43 group).
Results
Twenty-five pigs started the study. Five were excluded before gene transfer: 2 died during infarction, 1 died suddenly during the first day after infarction, 1 developed a refractory defibrillator infection, and 1 had no inducible SMVT. We could induce ventricular fibrillation (VF) in 10% to 25% of animals each week with programmed stimulation (including the pre-MI and post–gene transfer EPS). Unlike the reproducibility that we saw in SMVT induction, VF was only a sporadic finding and was not consistently induced every week in any animal. Overall, the pre–gene transfer observations in this study (MI survival, VF and SMVT inducibility) were similar to our previously published experience with the porcine healed MI–VT model (3,10).
Twenty animals met our prospective enrollment criteria of reproducibly inducible SMVT at both the 3-week and 4-week post-MI EPS. These animals received coronary artery and venous infusion of AdCx43 (10 animals), Adβgal (5 animals), or no virus (5 animals) immediately after the 4-week post-MI EPS. Repeat electrophysiologic testing 1 week after gene transfer revealed that only 4 of the 10 AdCx43 animals continued to have any inducible VT, while all 10 controls remained inducible (p = 0.01) (Fig. 1).
Figure 1. Arrhythmia Inducibility.
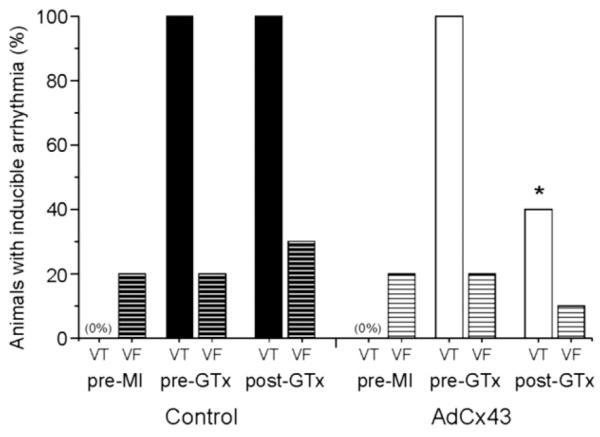
The bar graph shows the percent of animals with sustained monomorphic ventricular tachycardia (VT) or ventricular fibrillation (VF) induced at the pre–myocardial infarction (MI), pre–gene transfer (GTx), and 1-week post-GTx time points. The percent of animals with sustained monomorphic VT was significantly reduced in the AdCx43 group relative to controls. There were no statistically significant differences in VF inducibility. *p = 0.01.
Functional impact of increasing Cx43 in healed scar border
We used 2 methods to determine the functional effects of Cx43 gene transfer: paced fractionated electrogram duration, an in vivo measure of electrogram fractionation correlated with local conduction properties (11), and ex vivo optical mapping that has been shown to reliably measure tissue CV (12). Because of limitations in equipment availability for these methods, we could perform paced fractionated electrogram duration only in the first 10 animals and optical mapping in the second 10 animals of the study (5 Cx43 animals and 5 controls per measure). Both CV measures showed conduction improvement with Cx43 overexpression (Fig. 2). Paced fractionated electrogram duration shortened from the pre–gene transfer to the post– gene transfer EPS in AdCx43 animals but not controls (−13 ± 5 ms vs. +5 ± 4 ms, p = 0.04). Likewise, optical mapping showed significantly faster CV in anteroseptal tissues from AdCx43 animals compared to controls (50 ± 10 cm/s vs. 31 ± 12 cm/s, p = 0.02).
Figure 2. Functional Effects of Connexin43 GTx.

(A) The left panel shows the change in paced fractionated electrogram duration (PFED) comparing pre–gene transfer (GTx) to post-GTx electrophysiologic study. The right panel shows electrogram examples. (B) Transmural conduction velocity (CV) was faster in the AdCx43 group than in controls (left panel). Example transmural isochronal maps are shown in the right panel. The wedges were paced from the endocardial border. The blue coloration marks the region of earliest activation, and the red-colored area is the last activated along the transmural face of the tissue wedge (n = 5 per group). †p > 0.05.
Because CV also would be affected by differences in fibrosis, we tested for this potential confounder to our CV results by quantifying fibrosis in Masson’s trichrome–stained microsections from the optically mapped tissue wedges where we had measured CV. As expected in this scarred region, fibrosis was extensive and considerably variable between animals, but we found no significant differences in percent fibrosis between groups (23 ± 8% in AdCx43 animals vs. 14 ± 4% in controls, p = 0.35). These data further connect our measured CV improvement to increased Cx43 expression rather than between-group differences in other factors.
Cx43 expression and location
We assessed anterior-septal border-zone Cx43 expression using Western blot analysis (Figs. 3A and 3B). The Cx43 antibody recognized total Cx43, so we distinguished the differentially phosphorylated variants by their molecular weights. AdCx43 animals had 2-fold higher levels of total Cx43 compared with controls (3.4 ± 0.4 vs. 1.7 ± 0.4, p = 0.01), with similar distribution across the individual phosphorylated bands (AdCx43 animals: nonphosphorylated 21 ± 3%, P1 40 ± 1%, and P2 39 ± 2%; controls: nonphosphorylated 20 ± 3%, P1 37 ± 2%, and P2 43 ± 3%). We evaluated connexin localization using confocal immunohistochemistry (Figs. 3C and 3D). Cx43 was highly lateralized in the border-zone tissues of all animals, but we saw a similar percent of the total expressed Cx43 localized to the intercalated disk in both groups (26 ± 6% of total Cx43 in AdCx43 animals vs. 25 ± 4% in controls). Overall, these data suggest similar processing of transgenic and endogenous Cx43 with similar relative percents of phosphorylated and intercalated disk-localized Cx43 in all animals but an absolute increase in phosphorylated or intercalated disk-localized Cx43 in the AdCx43 animals due to the bulk effect of increased total Cx43 expression.
Figure 3. Cx43 Expression in the Anterior-Septal Border Zone.

(A) Representative immunoblots of connexin43 (Cx43) and beta-actin from control (C) and AdCx43 (Cx) animals. Phosphorylated Cx43 bands (P1 and P2) and nonphosphorylated Cx43 (NP) are depicted. (B) The bar graph shows quantification of band intensity for Cx43, normalized to beta-actin expression. Bar height indicates total Cx43, and the relative distribution of Cx43 in the NP, P1, and P2 bands is demarcated by the colored bands. (C) Immunofluorescent labeling of Cx43 and pan-cadherin from anterior-septal border-zone samples. (D) Percent of Cx43 co-localized with pan-cadherin indicating intercalated disk localization (p = NS). *p = 0.01.
Electrocardiography, echocardiography, and clinical observations
On a daily basis, we assessed activity level, appetite, respiration, and hydration. We also weighed the animals before infarction, gene transfer, and sacrifice. Throughout the study, AdCx43 and control animals did not differ for any of these measures. We compared electrocardiographic and echocardiographic measurements and found no significant differences either between groups or within groups comparing before and after gene transfer (Table 1). Limitations to efficacy. In an attempt to better understand factors affecting efficacy, we performed post hoc analyses within the AdCx43 group, comparing the 6 AdCx43 animals rendered uninducible with the 4 AdCx43 animals with continued VT inducibility. The data suggested that uninducible animals had increased total connexin, intercalated disk localized connexin, and CV relative to those that remained inducible, although there was considerable overlap in the data range between the 2 groups (Table 2). In our limited investigation of fibrosis (evaluated only in the tissue wedges in which we measured CV), uninducible animals also had higher fibrosis content.
Table 1.
Electrocardiographic and Echocardiographic Data
| Controls |
AdCx43 Animals |
|||||
|---|---|---|---|---|---|---|
| Variable | Pre-MI | Pre-GTx | Post-GTx | Pre-MI | Pre-GTx | Post-GTx |
| Electrocardiography | ||||||
| P-wave duration (ms) | 64 ±1 | 75 ±2 | 74 ±3 | 70 ±3 | 80 ±3 | 78 ± 5 |
| PR interval (ms) | 107 ± 4 | 115 ± 4 | 115 ± 4 | 118 ± 2 | 121 ± 4 | 124 ± 6 |
| QRS duration (ms) | 62 ±4 | 84 ±4 | 78 ±3 | 67 ±2 | 74 ±3 | 77 ± 2 |
| QTc interval (ms) | 410 ± 14 | 422 ± 14 | 411 ± 11 | 454 ± 12 | 429 ± 10 | 431 ± 7 |
| RR interval (ms) | 560 ± 30 | 422 ± 14 | 529 ± 30 | 628 ± 25 | 522 ± 29 | 544 ± 36 |
| Echocardiography | ||||||
| LVEF | 69 ±2 | 39 ±4 | 66 ±2 | 38 ± 2 | ||
| LV internal diameter-diastole | 3.8 ± 0.1 | 4.9 ± 0.2 | 3.4 ± 0.1 | 4.5 ± 0.1 | ||
| Septal width in diastole | 0.9 ± 0.1 | 0.6 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.1 | ||
| LV posterior width in diastole | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.9 ± 0.1 | ||
Values are mean ± SD. The pre-MI study was performed immediately before the infarct procedure, the pre-GTx study was performed immediately the GTx procedure (post-MI week 4), and the post-GTx study was performed 1 week after GTx (post-MI week 5). Echocardiography was not performed at the pre-GTx time point. There were no significant differences comparing pre-GTx with post-GTx within groups or comparing between the control and AdCx43 groups.
GTx = gene transfer; LV = left ventricular; LVEF = left ventricular ejection fraction; MI = myocardial infarction; QTc = Bazett’s corrected QT interval.
Table 2.
Comparison Within thr AdCx43 Group Between Noninducible and Continued Inducible Animals After Gene Transfer
| Noninducible |
Continued Inducible |
|||
|---|---|---|---|---|
| Median | Range | Median | Range | |
| Immunohistochemistry | ||||
| Cx43 at intercalated disk (%) | 0.31 | 0.19 to 0.42 | 0.17 | 0.12 to 0.38 |
| Western blot | ||||
| Total Cx43 (relative to beta-actin) | 3.44 | 2.58 to 4.3 | 2.86 | 2.8 to 4.6 |
| Cx43 in nonphosphorylated band (% of total) | 24 | 23.6 to 25.1 | 18 | 11.7 to 25.2 |
| Cx43 in P1 band (% of total) | 38 | 37.9 to 38.7 | 41 | 38.9 to 42.8 |
| Cx43 in Ps band (% of total) | 37 | 36.1 to 38.4 | 41 | 35.8 to 45.4 |
| Electrophysiologic study | ||||
| VT configurations before gene transfer | 1 | 1 to 3 | 2 | 2 to 3 |
| PFED at sacrifice | 56 | 35 to 82 | 64 | 40 to 118 |
| Ex vivo optical mapping | ||||
| Conduction velocity | 0.56 | 0.52 to 0.59 | 0.43 | 0.36 to 0.61 |
| Echocardiography | ||||
| EF at sacrifice (%) | 38 | 33 to 42 | 36 | 35 to 45 |
| Change in EF from baseline (%) | −25 | −22 to −27 | −29 | −26 to −31 |
| LVID at sacrifice (cm) | 4.43 | 4.1 to 4.8 | 4.55 | 4.5 to 4.6 |
| Septal wall thickness at sacrifice (cm) | 0.67 | 0.65 to 0.69 | 1.00 | 0.74 to 1.09 |
| Histology | ||||
| Fibrosis (% of total tissue area) | 37 | 20 to 53 | 12 | 3 to 27 |
Because of the small numbers and post hoc nature, these data were not statistically analyzed and are presented as descriptive only.
Cx43 = connexin43; EF = ejection fraction; LVID = left ventricular internal dimension and end-diastole; PFED = paced fractionated electrogram duration; VT = ventricular tachycardia.
We evaluated VT characteristics in the AdCx43 animals. Twelve-lead configurations of the induced VTs before gene transfer were not distinctly different between successes and failures within the AdCx43 group. Before gene transfer, animals rendered uninducible had an average of 1.3 ± 0.8 distinct VTs per animal, and continually inducible animals had 2.3 ± 0.5 VTs per animal. In the post– gene transfer study, 3 of the 4 treatment failures had only 1 inducible VT per animal, but the remaining animal now had 5 inducible configurations. One of the pre–gene transfer VT configurations remained inducible in 3 of the 4 failures. The other failure no longer had either of the 2 pre–gene transfer VT configurations but rather an entirely different configuration. The animal with 5 VT configurations had 4 new configurations in addition to 1 of the 2 configurations seen before gene transfer.
Discussion
Post-infarct patients have a risk for ventricular arrhythmias that persists at a measurable level indefinitely. Post hoc analysis of the Multicenter Automatic Defibrillator Implantation Trial II showed that the highest mortality benefit and incidence of appropriate defibrillator shocks occurred years after incident MI (13). Limitations of currently available treatment options for ventricular arrhythmias have motivated us and others to search for novel strategies to solve this problem. A fundamental step in this development process has been to define the pathophysiological alterations underlying the target arrhythmia that might be amenable to correction.
Mechanism of ventricular arrhythmias after MI
Surprisingly little information is available about cellular and molecular mechanisms of ventricular arrhythmias after infarct scar healing, which we define according to the time course described by Holmes et al. (14) in large mammals as completion of the necrotic (0 to 7 days after MI) and fibrotic (7 to 21 or 28 days) phases and entry into the remodeling phase that lasts indefinitely. Most of the data available on post-infarct VT mechanisms come from dogs studied 4 to 5 days after infarction. At that early time point, the considerable changes in border-zone myocyte electrophysiology include decreased sodium and L-type calcium current densities with altered kinetics, decreased transient and delayed repolarizing potassium currents, and both reduced and lateralized Cx43 expression (7,8,15–20). The data from dogs 4 to 5 days after MI are exceedingly important in helping us understand, and eventually improve therapy for, arrhythmias occurring in the days to weeks after infarction. A problem with applying these findings to the healed scar is that many of the ion channel alterations are resolved over the succeeding weeks after infarction (9,21), so these observations are insufficient to explain VT arising weeks, months, or years after MI.
What then is known about arrhythmias in the healed scar? De Bakker et al. (2) mapped VT circuits in vivo during cardiac surgical procedures for 72 patients with healed MIs. They localized the vast majority of the VTs to focal areas in the border zone >1.4 cm2 in diameter (countering a common misconception that re-entrant VT circuits are necessarily large, encircling the entire infarct scar). They compared the tachycardia activation sequence with tissue histology and found that VT consistently traveled through areas where extensive fibrosis infiltrated between myocytes (2,22,23). In separate work around the same time, Smith et al. (4) found that the border-zone tissues had marked alterations in the cellular distribution of connexins. Rather than a normal localization at intercalated disks along the ends of myocytes, they described a lateralization of connexins across the entire cell membrane in border-zone myocytes. Smith et al. did not map VT circuits, and they described the connexin finding as a general property of border-zone myocytes. These data led to what currently are the 2 prevailing hypotheses for post-infarct VT mechanism: that the VT circuit is a “zigzag course” through surviving myocytes in a maze of fibrosis, independent of cellular electrophysiology, or that VT occurs as a function of reduced connectivity and potentially altered myocyte excitability or refractory properties.
We previously have reported validation data on the porcine post-MI VT model demonstrating that the scar has healed at post-MI week 4 and that the model shares many common elements with VT seen chronically after infarction in humans (10). The principal findings of our present study are that Cx43 expression is decreased and intracellular localization is lateralized. These alterations correlated with heterogenous conduction by in vivo measures and slowed CV during ex vivo testing. Gene transfer–mediated overexpression of Cx43 increased the absolute amount of phosphorylated and intercalated disk-localized Cx43, improved CV, and reduced VT inducibility. These data solidly connect Cx43 expression and function to ventricular CV and arrhythmia mechanism. Our inability to completely eliminate VT with this intervention suggests that other factors (e.g., pattern or level of fibrosis, cellular excitability, and/or repolarization) are involved as well.
Potential causes of treatment failure
An important consideration for future attempts to refine the CV strategy for VT elimination will be defining the causes of treatment failure. As a first step in this process, we compared AdCx43 animals with successful elimination of VT with those that remained inducible. Because we did not prospectively design the comparison between successes and failures in the AdCx43 group and we had very small numbers to work with, we did not statistically analyze these data. Looking at data trends between the 2 groups, we saw suggestions of decreased Cx43 expression and intercalated disk localization, increased Cx43 phosphorylation, decreased CV, decreased fibrosis, and an increased number of baseline VTs per animal in the treatment failures. Looking at VT configurations, we saw that most of the treatment failures had continued inducibility of at least 1 pre–gene transfer VT after therapy.
On the basis of these observations, possible explanations for treatment failure include inadequate gene transfer, altered Cx43 processing after gene transfer, incorrect targeting of the gene transfer, increased mass or complexity of the border-zone region in the failures, or the need to reach a critical CV threshold to extinguish re-entry. Another potential explanation given the complex anatomy created by interdigitation of fibrosis, surviving normally functioning myocytes, and potentially of surviving impaired myocytes is that the outcome of the connexin gene transfer strategy depends critically on having sufficient viable cardiomyocytes capable of successful gene transfer, expression, and improved function within the targeted slow conduction zone. Differences in total Cx43 expression and CV support inadequate gene transfer as a cause. Phosphorylation and localization differences suggest the possibility of altered protein processing. The persistence of pre– gene transfer VT configurations suggests that the tissue target might have been missed. The larger number of baseline VTs per animal and reduced fibrosis (i.e., greater myocyte mass within the border zone) in the treatment failures suggests that improving CV might be insufficient to disrupt VT in areas with more complex anatomic or functional defects. Because all of the AdCx43 animals had improved CV relative to the controls, the lower median CV in the treatment failures may indicate the need to surpass a CV threshold to terminate re-entry, although 1 animal had persistent VT inducibility despite a CV of 63 cm/s, suggesting that a CV threshold would not be a complete explanation for failure. Prospective study with a much larger dataset is required to definitively answer this question.
Study limitations
As in any research study, our data have certain limitations that need to be considered when interpreting our results. Where possible, we validated our findings with multiple measurements, but we could not comprehensively evaluate every bit of border zone to fully define percent fibrosis or to eliminate the possibility of any continued necrotic or inflammatory elements. We relied on prior research for some of these conclusions. We also were limited in both post-infarction and post– gene transfer study durations because of practical issues of expense, pig size, and the time course of gene expression with adenovirus vectors. Longer term expression (e.g., with adeno-associated virus or lentivirus vectors) would be needed for translational studies or clinical use of this treatment strategy (24).
Another limitation of our study is whether the absence of VT on EPS predicts long-term success. Certainly, EPS is questionable as a screening tool to identify post-infarct patients at risk for sudden death (25), but the available research suggests an entirely different situation in patients with known VT substrate. In radiofrequency ablation patients who started procedures with readily inducible VT and who were then rendered completely uninducible by ablation, 80% to 95% remained free of VT on 2-year to 5-year follow-up (26–28). These data suggest that the elimination of VT inducibility is a valid end point in a population with known, reproducible VT.
Safety issues
In limited safety analysis, we saw no spontaneous arrhythmias and no abnormalities of repolarization in animals treated with AdCx43. We also saw no change in left ventricular ejection fraction, no clinical signs of heart failure, or other adverse events. One concern that comes from a strategy of increased cellular connectivity is that we might cause recovery of conduction in previously nonconducting areas and create new zones of slow conduction. We did have 2 animals (of 10) with new VT configurations after gene transfer, so we cannot exclude this possibility. Better in vivo mapping methods to more precisely define the areas of slow or absent conduction before gene transfer with comparison with results after gene transfer would be needed to fully evaluate this possibility. Those methods are beyond our current capabilities.
Another theoretical concern is that forced increases in cellular connectivity may allow the spread of toxins or death signals during active ischemia. The published research on connexins and myocardial ischemia is complex. Properly functioning connexins are essential for the protective effects of ischemic preconditioning (29). Connexin function and preconditioning have also been associated with a reduction in ischemic arrhythmias (30). However, closure of connexins during ischemia reduces infarct size in situations in which preconditioning has not occurred (31). In 1 study, infarct size decreased with transgenic expression of connexin32, a connexin that does not close in response to ischemia (32). Another study used gene transfer methods at the time of ischemia, and thus gene expression a few days after the ischemic insult, and found increased infarct size (33). We did not assess recurrent ischemia after gene transfer in this study, but our overexpression of wild-type Cx43 and evidence of normal processing and function suggest that the gene transfer-induced Cx43 proteins would close during ischemia in the same way that endogenous proteins do.
Conclusions
Our data show that Cx43 gene transfer to the healed scar border resulted in overexpression of functional connexins with phosphorylation and localization characteristics that mirrored protein produced from endogenous gene expression. Increasing local connexin expression improved CV and reduced VT inducibility, thus establishing a mechanistic link between connexin expression and post-infarct VT previously not possible in studies that could observe but not intervene on Cx43 expression levels. An added benefit to the CV strategy in comparison with manipulating repolarization is the absence of any theoretical possibility of triggered arrhythmias in cells after gene transfer. Open questions about the long-term durability of this effect, the safety in the setting of recurrent ischemia, and the possibility of creating new slow conduction zones require further study. With resolution of these issues, connexin manipulation to improve CV could become a novel therapy for arrhythmias originating in the border of healed infarct scar.
Supplementary Material
Acknowledgments
Defibrillators and leads for this study were donated by Boston Scientific Corporation.
This study was funded by the National Institutes of Health (grant R01HL67148 to Dr. Donahue) and the American Heart Association (post-doctoral fellowship to Dr. Greener). Dr. Donahue has received research supplies from St. Jude Medical, Boston Scientific, and Medtronic; and research grant support from St. Jude Medical.
Abbreviations and Acronyms
- CV
conduction velocity
- Cx43
connexin43
- EPS
electrophysiologic study
- MI
myocardial infarction
- SMVT
sustained monomorphic ventricular tachycardia
- VF
ventricular fibrillation
- VT
ventricular tachycardia
Footnotes
All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
APPENDIX For an expanded Methods section and supplemental figures and references, please see the online version of this article.
REFERENCES
- 1.Roberts WC. Coronary arteries in fatal acute myocardial infarction. Circulation. 1972;45:215–30. doi: 10.1161/01.cir.45.1.215. [DOI] [PubMed] [Google Scholar]
- 2.de Bakker J, van Capelle F, Janse M, et al. Reentry as a cause of ventricular tachycardia in patients with chronic ischemic heart disease: electrophysiologic and anatomic correlation. Circulation. 1988;77:589–606. doi: 10.1161/01.cir.77.3.589. [DOI] [PubMed] [Google Scholar]
- 3.Sasano T, McDonald AD, Kikuchi K, Donahue JK. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat Med. 2006;12:1256–8. doi: 10.1038/nm1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith J, Green C, Peters N, Rothery S, Severs N. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–21. [PMC free article] [PubMed] [Google Scholar]
- 5.Davis LM, Kanter HL, Beyer EC, Saffitz JE. Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. J Am Coll Cardiol. 1994;24:1124–32. doi: 10.1016/0735-1097(94)90879-6. [DOI] [PubMed] [Google Scholar]
- 6.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–75. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 7.Baba S, Dun W, Cabo C, Boyden P. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation. 2005;112:2386–96. doi: 10.1161/CIRCULATIONAHA.105.534784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabo C, Yao J, Boyden PA, et al. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72:241–9. doi: 10.1016/j.cardiores.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Ursell PC, Gardner PI, Albala A, Fenoglio JJ, Jr., Wit AL. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ Res. 1985;56:436–51. doi: 10.1161/01.res.56.3.436. [DOI] [PubMed] [Google Scholar]
- 10.Sasano T, Kelemen K, Greener ID, Donahue JK. Ventricular tachycardia from the healed myocardial infarction scar: validation of an animal model and utility of gene therapy. Heart Rhythm. 2009;6:S91–7. doi: 10.1016/j.hrthm.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res. 2000;47:11–22. doi: 10.1016/s0008-6363(00)00096-1. [DOI] [PubMed] [Google Scholar]
- 12.Fouts K, Fernandes B, Mal N, Liu J, Laurita KR. Electrophysiological consequence of skeletal myoblast transplantation in normal and infarcted canine myocardium. Heart Rhythm. 2006;3:452–61. doi: 10.1016/j.hrthm.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Wilber DJ, Zareba W, Hall WJ, et al. Time dependence of mortality risk and defibrillator benefit after myocardial infarction. Circulation. 2004;109:1082–4. doi: 10.1161/01.CIR.0000121328.12536.07. [DOI] [PubMed] [Google Scholar]
- 14.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–53. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 15.Dun W, Boyden P. Diverse phenotypes of outward currents in cells that have survived in the 5-day-infarcted heart. Am J Physiol. 2005;289:H667–73. doi: 10.1152/ajpheart.00180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao J, Hussain W, Patel P, Peters N, Boyden P, Wit A. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circ Res. 2003;92:437–43. doi: 10.1161/01.RES.0000059301.81035.06. [DOI] [PubMed] [Google Scholar]
- 17.Jiang M, Cabo C, Yao J, Boyden P, Tseng G. Delayed rectifier K currents have reduced amplitudes and altered kinetics in myocytes from infarcted canine ventricle. Cardiovasc Res. 2000;48:34–43. doi: 10.1016/s0008-6363(00)00159-0. [DOI] [PubMed] [Google Scholar]
- 18.Pu J, Boyden P. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997;81:110–9. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- 19.Lue W, Boyden P. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Circulation. 1992;85:1175–88. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
- 20.Peters N, Coromilas J, Severs N, Wit A. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–96. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 21.Dun W, Baba S, Yagi T, Boyden P. Dynamic remodeling of K+ and Ca2+ currents in cells that survived in the epicardial border zone of canine healed infarcted heart. Am J Physiol. 2004;287:H1046–54. doi: 10.1152/ajpheart.00082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bakker J, van Capelle F, Janse M, et al. Slow conduction in the infarcted human heart. “Zigzag” course of activation. Circulation. 1993;88:915–26. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 23.de Bakker J, Coronel R, Tasseron S, et al. Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: role of the arrangement of surviving cardiac fibers. J Am Coll Cardiol. 1990;15:1594–607. doi: 10.1016/0735-1097(90)92832-m. [DOI] [PubMed] [Google Scholar]
- 24.Bauer A, McDonald AD, Nasir K, et al. Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation. 2004;110:3115–20. doi: 10.1161/01.CIR.0000147185.31974.BE. [DOI] [PubMed] [Google Scholar]
- 25.Buxton AE, Lee KL, DiCarlo L, et al. for the Multicenter Unsustained Tachycardia Trial Investigators. Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. N Engl J Med. 2000;342:1937–45. doi: 10.1056/NEJM200006293422602. [DOI] [PubMed] [Google Scholar]
- 26.O’Donnell D, Bourke J, Furniss S. Standardized stimulation protocol to predict the long-term success of radiofrequency ablation of postinfarction ventricular tachycardia. Pacing Clin Electrophysiol. 2003;26:348–51. doi: 10.1046/j.1460-9592.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 27.Rothman S, Hsia H, Cossu S, Chmielewski I, Buxton A, Miller J. Radiofrequency catheter ablation of postinfarction ventricular tachycardia: long-term success and the significance of inducible nonclinical arrhythmias. Circulation. 1997;96:3499–508. doi: 10.1161/01.cir.96.10.3499. [DOI] [PubMed] [Google Scholar]
- 28.Della Bella P, De Ponti R, Uriarte JA, et al. Catheter ablation and antiarrhythmic drugs for haemodynamically tolerated post-infarction ventricular tachycardia; long-term outcome in relation to acute electrophysiological findings. Eur Heart J. 2002;23:414–24. doi: 10.1053/euhj.2001.2804. [DOI] [PubMed] [Google Scholar]
- 29.Li G, Whittaker P, Yao M, Kloner RA, Przyklenk K. The gap junction uncoupler heptanol abrogates infarct size reduction with preconditioning in mouse hearts. Cardiovasc Pathol. 2002;11:158–65. doi: 10.1016/s1054-8807(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 30.Papp R, Gonczi M, Kovacs M, Seprenyi G, Vegh A. Gap junctional uncoupling plays a trigger role in the antiarrhythmic effect of ischaemic preconditioning. Cardiovasc Res. 2007;74:396–405. doi: 10.1016/j.cardiores.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Miura T, Ohnuma Y, Kuno A, et al. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;286:H214–21. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Sinovas A, Sanchez JA, Gonzalez-Loyola A, et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J Physiol. 2010;588:1139–51. doi: 10.1113/jphysiol.2009.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prestia KA, Sosunov EA, Anyukhovsky EP, et al. Increased cell-cell coupling increases infarct size and does not decrease incidence of ventricular tachycardia in mice. Front Physiol. 2011;2:1. doi: 10.3389/fphys.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.