

Abstract
Background
Mycoplasma pneumoniae (Mp) frequently colonizes the airways of patients with chronic asthma and likely contributes to asthma exacerbations. We previously reported that mice lacking surfactant protein A (SP-A) have increased airway hyperresponsiveness (AHR) during M pneumoniae infection versus wild-type mice mediated by TNF-α. Mast cells (MCs) have been implicated in AHR in asthma models and produce and respond to TNF-α.
Objective
Determine the contribution of MC/TNF interactions to AHR in airways lacking functional SP-A during Mp infection. Methods: Bronchoalveolar lavage fluid was collected from healthy and asthmatic subjects to examine TNF-α levels and M pneumoniae positivity. To determine how SP-A interactions with MCs regulate airway homeostasis, we generated mice lacking both SP-A and MCs (SP-A−/−KitW-sh/W-sh) and infected them with M pneumoniae.
Results
Our findings indicate that high TNF-α levels correlate with M pneumoniae positivity in human asthmatic patients and that human SP-A inhibits M pneumoniae–stimulated transcription and release of TNF-α by MCs, implicating a protective role for SP-A. MC numbers increase in M pneumoniae–infected lungs, and airway reactivity is dramatically attenuated when MCs are absent. Using SP-A−/−KitW-sh/W-sh mice engrafted with TNF-α−/− or TNF receptor (TNF-R)−/− MCs, we found that TNF-α activation of MCs through the TNF-R, but not MC-derived TNF-α, leads to augmented AHR during M pneumoniae infection when SP-A is absent. Additionally, M pneumoniae– infected SP-A−/−KitW-sh/W-sh mice engrafted with TNF-α−/− or TNF-R−/− MCs have decreased mucus production compared with that seen in mice engrafted with wild-type MCs, whereas burden was unaffected.
Conclusion
Our data highlight a previously unappreciated but vital role for MCs as secondary responders to TNF-α during the host response to pathogen infection.
Keywords: Mast cells, TNF, Mycoplasma species, airway hyperres-ponsiveness, mucus
Mycoplasma pneumoniae is well established as a human pathogen, adheres tenaciously to the airway epithelia,1,2 and infects approximately 2 million persons annually with M pneumoniae–induced respiratory tract infections. Although asthma can be caused by a myriad of triggering stimuli, such as air pollutants and allergens, symptoms are often exacerbated by colonization with M pneumoniae. Several studies have examined the relationship between M pneumoniae and asthma and suggest as many as 50% of patients with stable asthma might be colonized with M pneumoniae.3-5 Bronchoscopic biopsy samples from asthmatic patients with positive test results for M pneumoniae airway colonization show significant increases in mast cell (MC) numbers compared with those seen in patients with negative test results for M pneumoniae burden.5
MCs are highly granular immune cells that reside in all tissues exposed to the environment, are intricately involved in allergic responses, and more recently have been described as sentinels of the innate immune system during bacterial infections.6 Depending on the type and strength of stimulus, MCs are able to discharge very specific mediators, including a variety of cytokines and chemokines, only minutes after the initial encounter. In addition to being newly synthesized in the ensuing hours after stimulation, some specific mediators, such as TNF-α, are stored in a preformed manner and can be immediately released from granular stores on activation of MCs. MCs also express receptors for a variety of cytokines and chemokines and can therefore respond to intrinsic signals during infection.7 Engagement of these receptors leads to the release of inflammatory mediators commonly associated with asthma, such as histamine, tryptases, and leukotrienes, and can also alter the response of MCs to degranulation stimuli.8-11
Primary pulmonary defense is supplied by the pulmonary innate immune system, which is comprised of phagocytes and natural killer cells, as well as soluble mediators, such as surfactant proteins. A major protein constituent of surfactant is surfactant protein A (SP-A). SP-A has well-established roles in modulating innate immunity in the lung.12-14 SP-A binding moieties have been identified on M pneumoniae, and SP-A inhibits M pneumoniae growth in vitro.15,16 We have previously shown that M pneumoniae–infected SP-A−/− mice have significantly augmented airway hyperresponsiveness (AHR), mucus production, and cellular inflammation compared with wild-type (WT) mice.17 Inhibition of TNF-α by a transcriptional inhibitor protected SP-A−/− mice from M pneumoniae–induced phenotypes, but the cell type responsible for the TNF-α–mediated response during M pneumoniae infection was not elucidated.
Previously, we demonstrated that SP-A binds to MCs,18 which led to our current hypothesis that MCs might be critical to SP-A–mediated regulation of airway reactivity and inflammation. Here we report novel information on a key role played by airway MCs in modulation of AHR in SP-A−/− mice infected with M pneumoniae. Using mice deficient in SP-A and MCs (SP-A−/−KitW-sh/W-sh mice) engrafted with either TNF-α−/− or TNF receptor (TNF-R)−/− MCs, we found that augmented airway responses during M pneumoniae infection is a result of TNF-α interactions with MCs through the TNF-R and not MC-derived TNF-α. Therefore we describe a previously overlooked contribution of MCs as secondary responders to TNF-α during M pneumoniae infection.
METHODS
M pneumonia preparation
M pneumoniae from American Type Culture Collection (catalog no. 15531) was prepared as described previously.17
Mice
An inbred strain of SP-A−/− mice was generated, as previously described.19 Please see the Methods section in this article’s Online Repository at www.jacionline.org for additional information. All protocols were approved by the Institutional Animal Use and Care Committee at Duke University.
M pneumoniae inoculations
Mice 8 to 12 weeks of age were anesthetized by means of intraperitoneal injection of 10 μL/g body weight of a 12% ketamine (100 mg/mL) and 5% xylazine (20 mg/mL) mix and were infected with either 50 μL of sterile saline or 50 μL of 1 × 108 M pneumoniae units in sterile saline by means of intranasal instillation.
Airway physiology
Direct measurements of respiratory mechanics in response to methacholine were made by using the Flexivent system (SCIREQ, Montreal, Quebec, Canada) and reported as total pulmonary resistance (RT) in centimeters of H2O per milliliter per second, as described previously.17 See the Methods section in this article’s Online Repository for more information.
Isolation and analysis of bronchoalveolar lavage fluid from mice
Mice were euthanized and the lungs lavaged either 12 or 72 hours post Mp infection. For more information on isolation and analysis of bronchoalveolar lavage (BAL) fluid from mice, please see the Methods section in this article’s Online Repository.
Isolation of BAL fluid from human subjects
Fiberoptic bronchoscopy was performed on subjects after achievement of conscious sedation, as previously described.20 Asthma subjects met the National Asthma Education and Prevention Program criteria for asthma.21 Please see the Methods section in this article’s Online Repository for complete descriptions.
MC culture and reconstitution
Bone marrow was collected from C57BL/6, TNF-α−/−, or TNF-R−/− mice and cultured in the presence of 5 ng/mL recombinant IL-3 and 5 ng/mL recombinant stem cell factor (R&D Systems, Minneapolis, Minn), as previously described.22 Please see the Methods section in this article’s Online Repository for complete descriptions.
M pneumoniae quantitation
RT-PCR was performed with primers for the M pneumoniae–specific P-1 adhesion gene, as previously described,17 to assess the M pneumoniae burden. Please see the Methods section in this article’s Online Repository for complete descriptions.
Gravity fixation and immunohistochemistry methods
For more information on gravity fixation and immunohistochemistry methods, please see the Methods section in this article’s Online Repository.
In vitro MC assays
WT MCs were plated on a 24-well plate, and human SP-A, isolated as previously described,12 was added at a concentration of 10 to 40 μg/mL for 1 hour at 37°C. M pneumoniae was added at a multiplicity of infection of 100:1. Please see the Methods section in this article’s Online Repository for complete descriptions.
RESULTS
Association between asthmatic patients displaying high TNF-α levels in BAL fluid and M pneumoniae infection
Our previous observations showed that M pneumoniae–infected SP-A−/− mice have enhanced M pneumoniae colonization, increased levels of TNF-α, and significantly increased AHR compared with WT mice.17 Because some asthmatic patients have positive test results for M pneumoniae in the airway, we determined whether M pneumoniae positivity associated with increased TNF-α levels in BAL samples from human asthmatic patients. Lavage samples were assessed for TNF-α levels, and based on the results, before assessing M pneumoniae positivity, the samples from asthmatic subjects were grouped into a low TNF-α–producing group (defined as less than 15-fold over the average TNF-α levels in healthy subjects) and a high TNF-α–producing group (defined as greater than 45-fold over the average TNF-α levels in healthy subjects). There were significantly higher TNF-αlevels in both the “low asthmatic” and the “high asthmatic” groups compared with those seen in the healthy subjects (Fig 1, A), and there was also a significant difference between the 2 groups of asthmatic subjects. Cells from the lavage samples of each subject were then analyzed for M pneumoniae positivity; the investigator was blinded to the TNF-α levels. None of the healthy subjects had positive results for M pneumoniae, whereas approximately 10% of asthmatic subjects with low TNF-α levels versus about 50% of asthmatic subjects with high TNF-α levels had positive test results for M pneumoniae (Fig 1, B). In addition, we performed the reverse analysis comparing TNF-α levels in M pneumoniae–positive versus M pneumoniae–negative BAL samples and found that the presence of M pneumoniae was associated with significantly increased TNF-α levels (Fig 1, C). Although previous studies have shown that a subset of asthmatic patients are colonized with M pneumoniae and that certain asthmatic subjects have increased TNF-α levels in BAL fluid, ours is the first to establish a temporal link between these 2 phenotypes.3,4,23,24
FIG 1.
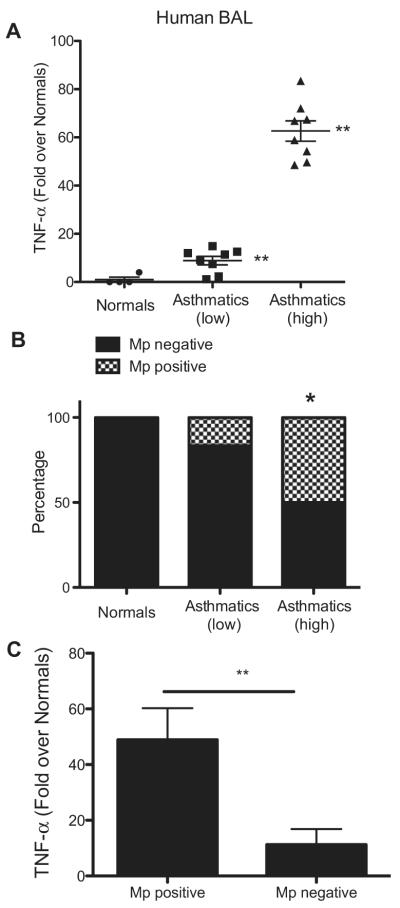
Mycoplasma pneumoniae (Mp) positivity and TNF-α levels in asthmatic samples. A, TNF-α in human BAL fluid (fold over healthy subjects). **P < .01, asthmatic patients (high) versus asthmatic patients (low) and both asthma groups versus healthy subjects. B, RT-PCR for M pneumoniae–specific P1 adhesin and dilution of samples into SP-4 broth to verify M pneumoniae positivity in subjects. *P < .05, asthmatic patients (high) versus healthy subjects and asthmatic patients (low). C, TNF values in M pneumoniae–positive and M pneumoniae–negative samples. **P < .01.
Recruitment of MCs into the lungs of M pneumoniae–infected mice
The above data show that TNF-α levels increase during M pneumoniae infection, and an association between M pneumoniae infection and the number of MCs in airway biopsy specimens from human asthmatic patients has been shown.4 Because MCs are potent reservoirs of TNF-α, we sought to determine the contribution of MCs in our model of M pneumoniae infection. MCs were localized to the subepithelial layer of the large airways and trachea in uninfected WT mice, as previously reported,6,25 and were comparable between uninfected lungs of WT and SP-A−/− mice (Fig 2, A and F). The number of MCs per lung section increased significantly in WTand SP-A−/− mice 72 hours after M pneumoniae infection (Fig 2, B and F). MCs are often adjacent to the basement membrane of the airway epithelium in M pneumoniae–infected mice (Fig 2, C), and positively stained granules, which are consistent with MC degranulation, were detected in M pneumoniae–infected lungs (Fig 2, D); no granules were detected in saline-treated lungs. MCs were detected in the lung parenchyma in close proximity to alveolar type II cells, which are known to produce and secrete SP-A (Fig 2, E). We found no significant effect of the lack of SP-A on the number or localization of MCs during M pneumoniae infection.
FIG 2.

MCs in Mycoplasma pneumoniae (Mp)–infected lung tissue. Mice were instilled with saline (A) or M pneumoniae (B-E) and lungs were analyzed by means of immunohistochemistry 3 days after infection for MCs. Fig 2, C, MCs adjacent to the large airway. Fig 2, D, Extracellular MC granules (arrow). Fig 2, E, MCs in the lung parenchyma. F, Total number of tissue MCs assessed in WT versus SP-A−/− lungs (n = 5 each). *P < .05.
MCs promote increased AHR and inflammation in M pneumoniae–infected SP-A−/− mice
Our previous findings suggested that during M pneumoniae infection, SP-A plays an important role in maintaining airway homeostasis by limiting an overzealous TNF-α response. Because MCs play key roles in asthma, we sought to determine the role of MCs in airway reactivity during M pneumoniae infection. Similar to published reports in other models, KitW-sh/W-sh mice had reduced AHR compared with that seen in WT mice, although it was significantly increased over that seen in saline-treated KitW-sh/W-sh control animals (Fig 3, A). To determine whether MCs play a role in increased AHR when SP-A is absent, we developed a mouse deficient in both MCs and SP-A (SP-A−/−KitW-sh/W-sh). As we previously reported, SP-A−/− mice treated with M pneumoniae have significantly greater AHR than WT mice.17 We found that by removing the MCs from the SP-A−/− mice (SP-A−/−KitW-sh/W-sh), AHR was reduced to baseline levels (Fig 3, B). These data indicate that MCs play a key role in the enhanced AHR observed in SP-A−/− mice.
FIG 3.

Contribution of MCs to AHR and inflammation. A and B, Three days after Mycoplasma pneumoniae (Mp), AHR to methacholine challenge was measured by using the Flexivent system. Mean percentage over baseline for highest methacholine dose is shown in parentheses. **P < .01 and ^P < .01, 3 independent experiments per graph. C and D, Cells in BAL fluid 12 hours (Fig 3, C) or 72 hours (Fig 3, D) after infection. *P < .05 versus WT M pneumoniae unless otherwise noted. E, Total protein in BAL fluid, n ≥ 12 mice per group.
Examination of BAL fluid cells from M pneumoniae–infected mice revealed an increase in the number of polymorphonuclear leukocytes-neutrophils (PMNs), the main responder to M pneumoniae infection,23 in SP-A−/− and SP-A−/−KitW-sh/W-sh mice compared with that seen in WT mice 12 hours after infection (Fig 3, C). By 72 hours, the majority of BAL fluid cells were macrophages. SP-A−/−KitW-sh/W-sh mice had significantly fewer macrophages compared with SP-A−/− mice but similar levels of neutrophils (Fig 3, D).
As a general indicator of lung injury, we measured total protein levels in the BAL fluid of the mice 72 hours after M pneumoniae infection. There were no differences between the groups, indicating that the changes in AHR are not associated with changes in total protein levels in the BAL fluid (Fig 3, E).
Recovery of AHR in SP-A−/−KitW-sh/W-sh mice reconstituted with WT MCs
To assess further the role of MCs in the increased AHR observed in M pneumoniae–infected SP-A−/− mice, we performed a series of reconstitution experiments. Retro-orbital injection of bone marrow–derived MCs results in engraftment of the lung with significant numbers of MCs.22 We repeated the M pneumoniae infection in MC-engrafted mice and assessed AHR. Engraftment of SP-A−/−KitW-sh/W-sh mice with MCs was verified by means of immunohistochemistry throughout the lung tissue (Fig 4, A and B). Engraftment of SP-A−/−KitW-sh/W-sh mice with WT MCs restored high levels of AHR to methacholine challenge, as expected in M pneumoniae–infected mice (Fig 4, C). These data indicate that the loss of MCs contributed to the decrease in AHR observed in SP-A−/−KitW-sh/W-sh mice compared with that seen in mice lacking only SP-A. TNF-α levels in BAL fluid were measured 12 hours after infection, and as previously reported, SP-A−/− mice had a significant increase in TNF-α levels compared with levels seen in WT mice.17 However, there was no difference in TNF-α levels observed in the SP-A−/− mice with MCs (SP-A−/− and SP-A−/−KitW-sh/W-sh mice engrafted with WT MCs) or without MCs (SP-A−/−KitW-sh/W-sh), and all were significantly increased over levels seen in WT infected mice (Fig 4, D).
FIG 4.

AHR in WT bone marrow–derived MC–reconstituted infected mice. A, Engraftment of MCs (arrows) verified by means of immunohistochemistry in lung tissue 1 month after injection. B, Quantification of engraftment. C, AHR to methacholine challenge was measured by using the Flexivent system. Mean percentage over baseline for highest methacholine dose is shown in parentheses. **P < .01, #P < .01, and ^P < 0.05, 3 independent experiments per graph. D, TNF-α in BAL fluid 12 hours after infection. *P < .05 versus WT Mycoplasma pneumoniae group, n ≥ 12 mice per group. Mp, Mycoplasma pneumoniae.
SP-A negatively regulates MC TNF-α responses to M pneumoniae
Purified bone marrow–derived MCs were stimulated with M pneumoniae to determine whether SP-A plays a role in limiting M pneumoniae–induced MC release of TNF-α. TNF-α released by the MCs stimulated with M pneumoniae peaked 12 to 14 hours after stimulation, and addition of exogenous human SP-A significantly inhibited this release (Fig 5, A). Analysis of MC RNA extracted after 1 hour of M pneumoniae stimulation indicated that exogenous SP-A added at a physiologic concentration (10 μg/mL) inhibited M pneumoniae–stimulated transcription of TNF-α (Fig 5, B). These findings suggest that SP-A can regulate the release and production of TNF-α in response to M pneumoniae infection.
FIG 5.
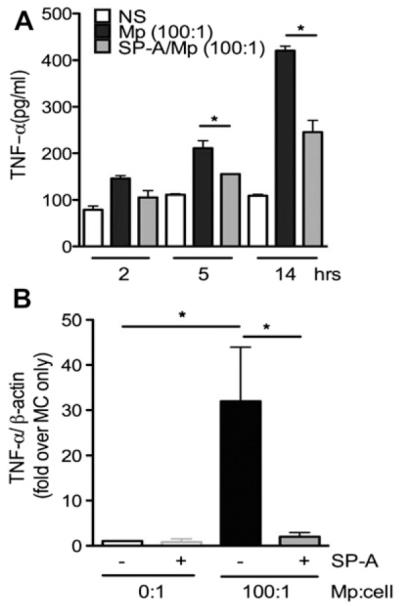
TNF-α production from Mycoplasma pneumoniae (Mp)–stimulated MCs. Nonstimulated (NS) or M pneumoniae–stimulated (multiplicity of infection, 100:1) bone marrow–derived MCs with and without SP-A (40 μg/mL) were analyzed over 14 hours (A) or 1 hour (B). Supernatants were analyzed for TNF-α release by means of ELISA; cell pellets were assessed by using RT-PCR for TNF-α transcription. *P < .05. Data are representative of 3 independent experiments.
Recovery of AHR in SP-A−/−KitW-sh/W-sh mice reconstituted with TNF-α−/− but not TNF-R−/− bone marrow–derived MCs
The above data revealed a key role for MCs in the increased AHR seen in SP-A−/− mice, and as previously published, inhibition of TNF-α can attenuate this response.17 High levels of TNF-α were measured in mice lacking MCs, suggesting that although MCs might be important in the augmented responses, MC-derived TNF-α might not be the key contributor to the increased levels of TNF-α. To better understand the contribution of TNF-α during M pneumoniae infection, we engrafted SP-A−/−KitW-sh/W-sh mice with TNF-α−/− and TNF-R−/− MCs. Mice reconstituted with TNF-α−/− MCs and infected with M pneumoniae demonstrated an enhancement in reactivity to methacholine that was comparable with the responsiveness observed in mice reconstituted with WT MCs (Fig 6, A). In contrast, SP-A–sufficient mice (KitW-sh/W-sh) engrafted with TNF-α−/− MCs continued to have attenuated AHR during M pneumoniae infection compared with that seen in mice engrafted with WT MCs (see Fig E1 in this article’s Online Repository at www.jacionline.org). However, M pneumoniae–infected SP-A−/−KitW-sh/W-sh mice receiving TNF-R−/− MCs had a significantly reduced response to methacholine compared with that seen in mice reconstituted with either WT or TNF-α−/− MCs. These data indicate that MC-derived TNF-α is a contributing factor in SP-A–sufficient mice but not the factor contributing to the high AHR in SP-A−/− mice infected with M pneumoniae. TNF-α acting on MCs through the TNF-R is critical in regulating AHR during M pneumoniae infection in SP-A−/− mice.
FIG 6.

Contribution of MC TNF-R to AHR and mucus. Mice were engrafted with TNF-α−/− or TNF-R−/− bone marrow–derived MCs. A, AHR to methacholine challenge was measured by using the Flexivent system. Mean percentage over baseline for highest methacholine dose is shown in parentheses. **, ^, #, $P < .01, 3 independent experiments per graph, n ≥ 12 mice per group. B and C, PAS-stained airways in infected mice (original magnification ×10) were scored. ***P < .001, **P < .01, and *P < .05; n = 7-12 sections per group. Mp, Mycoplasma pneumoniae.
Mucus production in M pneumoniae–infected SP-A−/− lungs is dependent on the MC TNF-α axis
We sought to determine the effect of MCs on production of mucus in SP-A−/− mice. As has been previously reported,17 WT infected mice have little mucus production in their large airways, whereas SP-A−/− mice demonstrate increases in airway periodic acid–Schiff staining (PAS; Fig 6, B). In SP-A−/−KitW-sh/W-sh mice PAS staining was barely detectable in the large airways, mimicking what was seen in the WT mice and suggesting a role for MCs in mucus production during M pneumoniae infection in SP-A−/− mice. Engraftment with WT MCs resulted in dramatic mucus production, although there was less detectable airway staining for PAS in mice that had been engrafted with either TNF-α−/− or TNF-R−/− MCs (Fig 6, C).
MCs contribute to the reduction of M pneumoniae burden in lung tissue
To assess the role of MCs in controlling M pneumoniae burden in the lung, we used RT-PCR to measure the levels of M pneumoniae P1 adhesin in the lung tissue 72 hours after M pneumoniae infection. As we reported previously, SP-A−/− mice have a significant increase in M pneumoniae burden compared with that seen in WT mice.17 In SP-A−/−KitW-sh/W-sh mice we found an even greater increase in M pneumoniae burden in the lungs. When these mice were engrafted with MCs, the M pneumoniae burden levels were reduced to those of the SP-A−/− mice (Fig 7), indicating that MCs are essential in controlling the M pneumoniae burden within the lung tissue.
FIG 7.
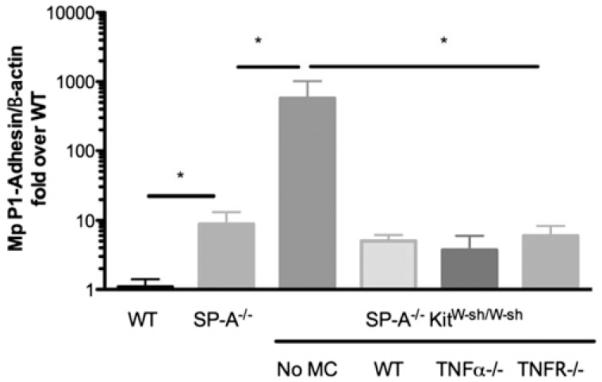
Contribution of MCs to Mycoplasma pneumoniae (Mp) clearance. M pneumoniae burden in lung tissue at day 3 was measured by using RT-PCR for the M pneumoniae–specific P1 adhesin gene relative to β-actin and is expressed as fold over WT M pneumoniae–infected mice. *P < .05, n ≥ 12 mice per group.
DISCUSSION
Not only do the findings presented here further the understanding of how the lung responds to M pneumoniae infection, they also suggest a novel role of the TNF-R on MCs in mediating responses to pathogens in the absence of SP-A or when SP-A is dysfunctional. In support of clinical data, we found increased MC numbers in the lung during M pneumoniae infection in mice.4 In addition, MCs are required for the enhanced AHR and mucus production observed in the SP-A−/− M pneumoniae–infected mice. SP-A−/−KitW-sh/W-sh mice continue to have high levels of TNF-α in their BAL fluid yet have lower AHR compared with SP-A−/− mice, indicating a pivotal role for MC products in mediating airway reactivity that is not dependent on MC TNF-α. When these mice (SP-A−/−KitW-sh/W-sh) are reconstituted with either WT or TNF-α−/− MCs, AHR levels are increased during methacholine challenge; however, an AHR phenotype is not re-established when TNF-R−/− MCs are engrafted. This strongly suggests that the M pneumoniae–induced increase in AHR observed in the SP-A−/− mouse is critically and specifically dependent on stimulation of MC TNF-α receptors.
Our data support a model in which SP-A has a profound effect on TNF-α and show that in the absence of SP-A, TNF-α acts on MCs through the TNF-R to induce increased mucus production and AHR (Fig 8). It has been previously reported by our laboratory and others that SP-A directly affects the production of TNF-α by macrophages stimulated with Toll-like receptor 4 agonists, and we show here that SP-A inhibits M pneumoniae–induced TNF-α production by MCs in vitro.14,26,27 It is also possible that SP-A affects TNF-α production by airway epithelial cells or PMNs. We speculate that the increased TNF-α that occurs in the absence of SP-A interacts in the submucosa with the TNF-R on MCs, causing release of a myriad of prestored and newly synthesized mediators. Many mediators from degranulating MCs, including cytokines, chemokines, lipid mediators, and histamine, might play a key role in the pathophysiology of the asthmatic lung (reviewed by Reuter et al28). Additionally, MC proteases stimulate tissue remodeling and enhance mucus secretion.29 We hypothesize that it is one of these mediators, or several working in concert, that causes AHR and mucus production in the presence of a robust TNF-α response during M pneumoniae infection.
FIG 8.
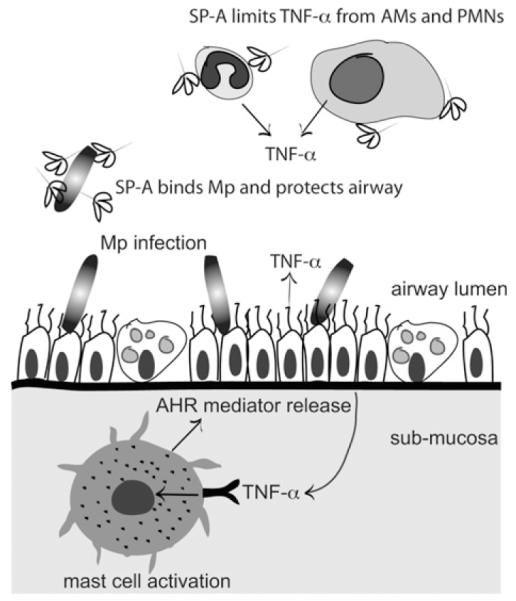
Protective role of SP-A against Mycoplasma pneumoniae (Mp)–induced AHR. SP-A binds M pneumoniae in the large airway and prevents overcolonization. If SP-A is dysfunctional, more M pneumoniae burden results in TNF-α overproduction from epithelial cells, alveolar macrophages(AMs), and/or neutrophils (PMNs). TNF-α encounters tissue MCs in the submucosa, which are stimulated through the TNF-R to release mediators that contribute to high levels of AHR.
Although the link between MC degranulation and mucus production has been shown (reviewed by Hart29), the direct contribution of MC-derived TNF-α in the regulation of mucus hyper-secretion remains incompletely defined. Here we show novel findings that although engraftment of WT MCs results in dramatic increases in PAS-positive airway cells, engraftment of either TNF-α−/− MCs or with TNF-R−/− MCs resulted in significantly fewer PAS-positive cells. These findings suggest a critical role for the TNF-mediated pathway in MCs for the increased presence of mucopolysaccharides in airway epithelium during M pneumoniae infection, which can contribute to the exacerbations experienced by a subset of asthmatic patients who become colonized with M pneumoniae.
The importance of MC-derived TNF-α in lung pathophysiology has been described by several laboratories. Using the well-established ovalbumin model of asthma, Nakae et al30 found that engraftment of KitW-sh/W-sh mice with TNF-α−/− MCs resulted in decreased lymphocyte recruitment and TH2 cytokine production compared with that seen in KitW-sh/W-sh mice engrafted with WT MCs.30,31 In a footpad model of Escherichia coli infection, MC-derived TNF-α was shown to be important for lymph node hypertrophy.32 On the basis of these studies, we anticipated that TNF-α produced by MCs in the lung would be pivotal in mediating the host response to M pneumoniae. Indeed, SP-A–sufficient KitW-sh/W-sh mice engrafted with TNF-α−/− MCs had attenuated AHR compared with those engrafted with WT MCs. However, SP-A−/−KitW-sh/W-sh mice engrafted with TNF-α−/− MCs maintained a high level of increased AHR. The differing role of MC-derived TNF-α in SP-A–sufficient versus SP-A–deficient lung environments is likely attributed to a compensatory mechanism that might be partially explained by increased PMN recruitment in the TNF-α−/− MC–engrafted SP-A−/−KitW-sh/W-sh mice compared with that seen in WT MC–engrafted SP-A−/−KitW-sh/W-sh mice. This additional increase in PMN recruitment could lead to heightened TNF-α production, therefore masking the importance of MC-derived TNF-α. Our data are interesting in that they show that TNF-α acting through the TNF-R on MCs is critical in modulating the host response to M pneumoniae when SP-A is absent.
Previous murine and clinical studies indicated a key role for MCs in Mycoplasma species infections.33,34 We tested the ability of MCs to kill M pneumoniae directly using in vitro techniques, and although we found that MCs were unable to kill M pneumoniae in samples directly (data not shown), our findings in vivo further strongly support the involvement of MCs in the resolution of M pneumoniae infection, albeit through indirect mechanisms. When MCs were reconstituted into the MC-deficient mice, the M pneumoniae burden was decreased nearly to the level detected in MC-sufficient mice. Interestingly, we found no difference in burden between mice engrafted with WT, TNF-α−/−, or TNF-R−/− MCs, indicating that neither signaling through the MC TNF-R nor MC-derived TNF-α is necessary for M pneumoniae resolution.
The current understanding of MCs and TNF-α interactions in asthmatic patients is not complete, in part because of the complexity of the multiple phenotypes that encompass human asthma. Researchers have delved into the aspect of how MC-derived TNF-α affects the lung and asthmatic phenotypes, although to the best of our knowledge, no studies have examined the effect of TNF-α acting on MCs to initiate degranulation and activation of newly synthesized mediators in vivo. In addition, a recent report by Wang et al35 concluded that asthmatic SP-A binds less to M pneumoniae membranes and is unable to protect airway epithelial cells from M pneumoniae stimulation in vitro. Although anti-TNF treatment has met with mixed results,24,36-39 our results suggest that targeting MCs might prove to be a safer and more effective way of reducing symptoms in asthmatic patients, who might have dysfunctional surfactant, with M pneumoniae infections. Historically, during microbial infections and asthma, the contributions of MCs to the inflammatory response have been primarily attributed to their ability to directly interact with allergens, pathogens, or both and respond by releasing a panoply of prestored and recently synthesized mediators. Our findings provide evidence that MCs can also respond to locally produced cytokines, such as TNF-α, and therefore serve as secondary responders, bolstering responses initiated by activated epithelial cells, other inflammatory cells, or both.
Supplementary Material
FIG E1. AHR in KitW-sh/W-sh–engrafted mice. Mice were engrafted with MCs for 1 month. AHR to methacholine challenge was assessed by using the Flexivent system. **P < .01. Mp, Mycoplasma pneumoniae.
Key messages.
High TNF-α levels associate with M pneumoniae positivity in asthmatic patients, and human SP-A inhibits M pneumoniae–stimulated transcription and release of TNF-α by MCs.
MC numbers increase in M pneumoniae–infected lungs, and airway reactivity is dramatically attenuated when MCs are absent from SP-A−/− mice.
Using SP-A−/−KitW-sh/W-sh mice engrafted with TNF-α−/− or TNF-R−/− MCs, we find that TNF-α activation of MCs through the TNF-R and not MC-derived TNF-α leads to augmented AHR during M pneumoniae infection in SP-A−/− mice.
Acknowledgments
We thank Dr Laura Hale and Paula Greer for the generous gift of GFP:TNF−/− mice, Kathy Evans for human SP-A isolation and preparation, and Michele Kislan for technical assistance. We also thank Drs Cong Jin, Chris Shelburne, and David Jacoby for helpful discussions about the manuscript.
Supported by National Institutes of Health grants F32-HL091642 (to J.G.L.) and PO1-AI81672 (to J.R.W., M.K., and W.M.F.).
B. J. Hsia, J. G. Ledford, and N. L. Lugogo receive research support from the National Institutes of Health. M. Kraft receives research support from GlaxoSmithKline, Merck, Eumedics, Novartis, Genentech, and Asthmatx (Boston Scientific).
Abbreviations used
- AHR
Airway hyperresponsiveness
- BAL
Bronchoalveolar lavage
- MC
Mast cell
- PAS
Periodic acid–Schiff
- PMN
Polymorphonuclear leukocyte-neutrophil
- RT
Pulmonary resistance
- SP-A
Surfactant protein A
- TNF-R
TNF receptor
- WT
Wild-type
Footnotes
Disclosure of potential conflict of interest: The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Baseman JB, Tully JG. Mycoplasmas: sophisticated, reemerging, and burdened by their notoriety. Emerg Infect Dis. 1997;3:21–32. doi: 10.3201/eid0301.970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krause DC, Balish MF. Cellular engineering in a minimal microbe: structure and assembly of the terminal organelle of Mycoplasma pneumoniae. Mol Microbiol. 2004;51:917–24. doi: 10.1046/j.1365-2958.2003.03899.x. [DOI] [PubMed] [Google Scholar]
- 3.Kraft M, Cassell GH, Henson JE, Watson H, Williamson J, Marmion BP, et al. Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am J Respir Crit Care Med. 1998;158:998–1001. doi: 10.1164/ajrccm.158.3.9711092. [DOI] [PubMed] [Google Scholar]
- 4.Kraft M, Hamid Q. Mycoplasma in severe asthma. J Allergy Clin Immunol. 2006;117:1197–8. doi: 10.1016/j.jaci.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Martin RJ, Kraft M, Chu HW, Berns EA, Cassell GH. A link between chronic asthma and chronic infection. J Allergy Clin Immunol. 2001;107:595–601. doi: 10.1067/mai.2001.113563. [DOI] [PubMed] [Google Scholar]
- 6.Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol. 2004;4:787–99. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 7.Juremalm M, Nilsson G. Chemokine receptor expression by mast cells. Chem Immunol Allergy. 2005;87:130–44. doi: 10.1159/000087640. [DOI] [PubMed] [Google Scholar]
- 8.Fifadara NH, Aye CC, Raghuwanshi SK, Richardson RM, Ono SJ. CCR1 expression and signal transduction by murine BMMC results in secretion of TNF-alpha, TGFbeta-1 and IL-6. Int Immunol. 2009;21:991–1001. doi: 10.1093/intimm/dxp066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pushparaj PN, Manikandan J, Tay HK, H’Ng SC, Kumar SD, Pfeilschifter J, et al. Sphingosine kinase 1 is pivotal for Fc epsilon RI-mediated mast cell signaling and functional responses in vitro and in vivo. J Immunol. 2009;183:221–7. doi: 10.4049/jimmunol.0803430. [DOI] [PubMed] [Google Scholar]
- 10.van Overveld FJ, Jorens PG, Rampart M, de Backer W, Vermeire PA. Tumour necrosis factor stimulates human skin mast cells to release histamine and tryptase. Clin Exp Allergy. 1991;21:711–4. doi: 10.1111/j.1365-2222.1991.tb03200.x. [DOI] [PubMed] [Google Scholar]
- 11.Hughes JM, Stringer RS, Black JL, Armour CL. The effects of tumour necrosis factor alpha on mediator release from human lung. Pulm Pharmacol. 1995;8:31–6. doi: 10.1006/pulp.1995.1004. [DOI] [PubMed] [Google Scholar]
- 12.Brinker KG, Garner H, Wright JR. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L232–41. doi: 10.1152/ajplung.00187.2002. [DOI] [PubMed] [Google Scholar]
- 13.Stamme C, Walsh E, Wright JR. Surfactant protein A differentially regulates IFN-gamma- and LPS-induced nitrite production by rat alveolar macrophages. Am J Respir Cell Mol Biol. 2000;23:772–9. doi: 10.1165/ajrcmb.23.6.4083. [DOI] [PubMed] [Google Scholar]
- 14.McIntosh JC, Mervin-Blake S, Conner E, Wright JR. Surfactant protein A protects growing cells and reduces TNF-alpha activity from LPS-stimulated macrophages. Am J Physiol Lung Cell Mol Physiol. 1996;271:L310–9. doi: 10.1152/ajplung.1996.271.2.L310. [DOI] [PubMed] [Google Scholar]
- 15.Piboonpocanun S, Chiba H, Mitsuzawa H, Martin W, Murphy RC, Harbeck RJ, et al. Surfactant protein A binds Mycoplasma pneumoniae with high affinity and attenuates its growth by recognition of disaturated phosphatidylglycerols. J Biol Chem. 2005;280:9–17. doi: 10.1074/jbc.M411570200. [DOI] [PubMed] [Google Scholar]
- 16.Kannan TR, Provenzano D, Wright JR, Baseman JB. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect Immun. 2005;73:2828–34. doi: 10.1128/IAI.73.5.2828-2834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ledford JG, Goto H, Potts EN, Degan S, Chu HW, Voelker DR, et al. SP-A preserves airway homeostasis during Mycoplasma pneumoniae infection in mice. J Immunol. 2009;182:7818–27. doi: 10.4049/jimmunol.0900452. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 18.Malherbe DC, Erpenbeck VJ, Abraham SN, Crouch EC, Hohlfeld JM, Wright JR. Surfactant protein D decreases pollen-induced IgE-dependent mast cell degranulation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L856–66. doi: 10.1152/ajplung.00009.2005. [DOI] [PubMed] [Google Scholar]
- 19.Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, et al. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci U S A. 1996;93:9594–9. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraft M, Cassell GH, Pak J, Martin RJ. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest. 2002;121:1782–8. doi: 10.1378/chest.121.6.1782. [DOI] [PubMed] [Google Scholar]
- 21.Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma—summary report 2007. J Allergy Clin Immunol. 2007;120(suppl):S94–138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 22.Jin C, Shelburne CP, Li G, Potts EN, Riebe KJ, Sempowski GD, et al. Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation. J Clin Invest. 2011;121:941–55. doi: 10.1172/JCI43584. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 23.Martin RJ, Chu HW, Honour JM, Harbeck RJ. Airway inflammation and bronchial hyperresponsiveness after Mycoplasma pneumoniae infection in a murine model. Am J Respir Cell Mol Biol. 2001;24:577–82. doi: 10.1165/ajrcmb.24.5.4315. [DOI] [PubMed] [Google Scholar]
- 24.Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–8. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkman GL. The mast cell in normal human bronchus and lung. J Ultrastruct Res. 1968;23:115–23. doi: 10.1016/s0022-5320(68)80035-8. [DOI] [PubMed] [Google Scholar]
- 26.Alcorn JF, Wright JR. Surfactant protein A inhibits alveolar macrophage cytokine production by CD14-independent pathway. Am J Physiol Lung Cell Mol Physiol. 2004;286:L129–36. doi: 10.1152/ajplung.00427.2002. [DOI] [PubMed] [Google Scholar]
- 27.Sano H, Sohma H, Muta T, Nomura S, Voelker DR, Kuroki Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J Immunol. 1999;163:387–95. [PubMed] [Google Scholar]
- 28.Reuter S, Stassen M, Taube C. Mast cells in allergic asthma and beyond. Yonsei Med J. 2010;51:797–807. doi: 10.3349/ymj.2010.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart PH. Regulation of the inflammatory response in asthma by mast cell products. Immunol Cell Biol. 2001;79:149–53. doi: 10.1046/j.1440-1711.2001.00983.x. [DOI] [PubMed] [Google Scholar]
- 30.Nakae S, Ho LH, Yu M, Monteforte R, Iikura M, Suto H, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol. 2007;120:48–55. doi: 10.1016/j.jaci.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 31.Kim YS, Ko HM, Kang NI, Song CH, Zhang X, Chung WC, et al. Mast cells play a key role in the development of late airway hyperresponsiveness through TNF-alpha in a murine model of asthma. Eur J Immunol. 2007;37:1107–15. doi: 10.1002/eji.200636612. [DOI] [PubMed] [Google Scholar]
- 32.McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, et al. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol. 2003;4:1199–205. doi: 10.1038/ni1005. [DOI] [PubMed] [Google Scholar]
- 33.Xu X, Zhang D, Lyubynska N, Wolters PJ, Killeen NP, Baluk P, et al. Mast cells protect mice from Mycoplasma pneumonia. Am J Respir Crit Care Med. 2006;173:219–25. doi: 10.1164/rccm.200507-1034OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michels NM, Chu HW, LaFasto SC, Case SR, Minor MN, Martin RJ. Mast cells protect against airway Mycoplasma pneumoniae under allergic conditions. Clin Exp Allergy. 2010;40:1406–13. doi: 10.1111/j.1365-2222.2010.03488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Voelker DR, Lugogo NL, Wang G, Floros J, Ingram JL, et al. Surfactant protein A is defective in abrogating inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2011;301:L598–606. doi: 10.1152/ajplung.00381.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 37.Erin EM, Leaker BR, Nicholson GC, Tan AJ, Green LM, Neighbour H, et al. The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthma. Am J Respir Crit Care Med. 2006;174:753–62. doi: 10.1164/rccm.200601-072OC. [DOI] [PubMed] [Google Scholar]
- 38.Rouhani FN, Meitin CA, Kaler M, Miskinis-Hilligoss D, Stylianou M, Levine SJ. Effect of tumor necrosis factor antagonism on allergen-mediated asthmatic airway inflammation. Respir Med. 2005;99:1175–82. doi: 10.1016/j.rmed.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 39.Morjaria S, Deleuze-Masquefa C, Lafont V, Gayraud S, Bompart J, Bonnet PA, et al. Impairment of TNF-alpha production and action by imidazo[1,2-alpha] quinoxalines, a derivative family which displays potential anti-inflammatory properties. Int J Immunopathol Pharmacol. 2006;19:525–38. doi: 10.1177/039463200601900308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIG E1. AHR in KitW-sh/W-sh–engrafted mice. Mice were engrafted with MCs for 1 month. AHR to methacholine challenge was assessed by using the Flexivent system. **P < .01. Mp, Mycoplasma pneumoniae.