

Abstract
The efficacy of therapeutic modalities in chronic myeloid leukemia (CML) depends on both genetic and epigenetic mechanisms. This review focuses on epigenetic mechanisms involved in the pathogenesis of CML and in resistance of tumor cells to tyrosine kinase inhibitors leading to the leukemic clone escape and propagation. Regulatory events at the levels of gene regulation by transcription factors and microRNAs are discussed in the context of CML pathogenesis and therapeutic modalities.
Keywords: CML, Epigenetics, microRNA, Tyrosine kinase inhibitors
Introduction
It has been over half a century since a specific chromosomal aberration – the Philadelphia chromosome (Ph chromosome), was found in chronic myeloid leukemia (CML) [1], but still many questions regarding molecular mechanisms of pathogenesis in CML remain unanswered. The CML genetics provided detailed analyses of the Ph chromosome that is a result of a reciprocal translocation t(9;22)(q34;q11) accompanied by breaks on long arms of chromosome 9(9q34) proximally from the ABL gene and on long arms of chromosome 22(q11) with BCR (Breakpoint Cluster Region) [2]. The hybrid gene BCR-ABL product translates a chimeric protein with strong and constitutive tyrosine kinase activity that phosphorylates target proteins to facilitate expansion of hematopoietic stem and progenitor cells. The end of the last century was marked by significant progress in CML treatment following the introduction of the tyrosine kinase inhibitor (TKI) imatinib (IM) that binds to the kinase domain (KD) of BCR-ABL and inhibits its tyrosine kinase activity [3]. Despite the high efficiency of imatinib therapy, still approximately 30 % of patients develop resistance to imatinib resulting in first line therapy failure. Imatinib resistance due to the mutations in KD of BCR-ABL can be bypassed by 2nd line TKIs such as dasatinib or nilotinib [4]. Resistance to the 2nd line therapy also develops and not surprisingly is also associated with specific KD mutations [5]. Genetic mechanisms at the level of KD sequence integrity must be important; but other mechanisms of primary or acquired resistance to TKI are also now being studied including additional clonal aberrations, BCR-ABL overexpression, and TKI bioavailability. However, apart from BCR-ABL, there are other genetic or epigenetic alterations that are still unknown as they may contribute to CML stem cells survival during a long term TKI therapy that successfully inhibits the BCR-ABL activity but is not curative. One can imagine that TKI therapy may in the future benefit from being combined with other agents in order to achieve deep and long term molecular responses.
Abnormal epigenetic regulation of the expression of CML-associated genes may play a critical role in its pathogenesis and in the mechanisms modulating therapeutic responsiveness. Epigenetics is thought to involve well recognized regulatory mechanisms of gene expression such as DNA methylation or covalent post-translational modifications of histone core proteins that lead to changes in the chromatin accessibility for mRNA transcription regulation [6, 7]. As well as nuclear events, other mechanisms including non-coding RNA-mediated (by microRNAs, miRs) specific mRNA silencing at the levels of translation and RNA stability are also considered to be very powerful epigenetic mediators to modulate CML expression profiles and phenotypic outcomes. MicroRNAs are able to control hundreds of mRNAs and thus they control broad physiological and pathological features including tumor aggressiveness. Unlike mRNAs, microRNAs are stable and therefore can be routinely quantitated and potentially may also serve as disease biomarkers.
This review summarizes the role of epigenetics in CML, and focuses on DNA methylation and histone modification as well as post-transcriptional effects of microRNAs in the pathogenesis of CML from diagnosis and throughout treatment.
DNA Methylation in CML
The methylation of CpG islands is an active enzymatic and transcription-inhibiting control mechanism that balances the levels of gene expression that is frequently dysregulated in hematological malignancies. A large number of genes (mostly tumor suppressors) are inactivated by hypermethylation of CpG islands mainly in the promoter regions while some genes (such as oncogenes) are hypomethylated. This phenomenon has been documented to play a critical role in both solid tumors and leukemias [8].
ABL1 (v-abl Abelson murine leukemia viral oncogene homolog 1) methylation at its Pa promoter represents a likely marker of CML pathogenesis [9, 10]. The frequency of methylation in chronic phase (CP) CML however ranges from 26 % [11] to 77 % [12], 78 % [10] and 81 % [13•]. Sun et al. [14] confirmed the high incidence of Pa methylation in CP bone marrow (BM) samples in contrast to normal BM. They observed copies of ABL1 promoter Pa to be methylated 20–60 % in BM from 7 CP CML patients at diagnosis. No Pa methylation was detected in normal BMs or colonies derived from them. On the other hand, most colonies from CP CML patients were methylated at the Pa. The authors suggested that ABL1 Pa methylation was an early marker of CML in BM. Asimakopoulos et al. [9] demonstrated that in accelerated phase (AP) CML, methylation is likely to be an allele-specific process, since each progenitor cell carries both methylated and unmethylated alleles. This paragraph documents the promising but also quite highly disputed significance of the ABL1 hypermethylation in CML.
One of the most frequently studied genes in leukemias is the cell cycle regulating gene p15 (CDKN2B). Cyclin-dependent kinase (CDK) inhibitor p15 (p15Ink4b) together with CDK4 or CDK6 are known to negatively regulate cyclin D transcription leading to inhibition of cell cycle progression. Abnormal hypermethylation of p15 gene regions has been associated with the disease progression in myelodysplastic syndrome (MDS) [15, 16] and with the poor outcome in acute myelogenous leukemia (AML) [17]. The clinical importance of p15 methylation in AML patients is not conclusive [18, 19]. Similarly, the significance of p15 methylation in CML patients is not fully understood as the p15 promoter in CML patients is hypomethylated [20, 21], while others observed p15 hypermethylation in 18 % and 24 % of patient samples, respectively [12, 22]. To conclude, p15 methylation in CML patients requires additional work to fully understand its clinical relevance.
Ras association domain-containing protein 1 (RASSF1A) is another candidate gene for DNA methylation. Avramouli et al. [23] however did not observe in 41 CML patients in different stages of the disease any methylation of RASSF1A that is normally involved in cell cycle control. In contrast, RASSF1A was methylated in the CML-derived K562 cell line. Methylation of RASSF1A is not likely to be critical step in the pathogenesis and/or the progression of primary CML samples, although it seems to have clinical importance in Hodgkin disease [24] and in epithelial cancers [25].
Methylation of additional genes was later associated with progression of CML and potential resistance to TKI treatment. For instance, two genes involved in development, TFAP2A (transcription factor AP-2 alpha) and EBF2 (early B-cell factor 2), were found to be more methylated in blast crisis (BC) compared to CP in the study of 55 CP CML and 8 BC CML samples [25]. The autophagy related 16-like 2 gene ATG16L2 was methylated in 69 % of CML patients. Patients with methylated ATG16L2 had a lower probability of achieving a major molecular response (MMR) at 12 or 18 months compared with the unmethylated cases at baseline [25].
One of the candidate tumor suppressor genes, often methylated in many malignancies, is the death-associated protein kinase 1 DAPk1. The DAPk subfamily is part of the Ser/Thr kinase family, whose members show significant homology in their catalytic domains and have cell death-associated functions such as the capability of inducing apoptosis. Qian et al. [26] examined the methylation pattern of DAPk1 in 49 CML patients at different stages of the disease (29 patients in CP, 3 in AP and 17 in BC). Aberrant methylation of DAPk1 was observed in 51 % of CML cases. A correlation between gender and methylation status of DAPk1 and also with the CML stages was found. These results indicate that DAPk1 methylation may play a significant role in the CML progression.
Recently, a significant methylation in CML progressed to AP or BC was found near five genes that were selected based on genome wide methylation studies in K562 cells, OSCP1 and TFAP2E, or were detected to be hypermethylated in patients with myeloproliferative neoplasms, PGRA and PGRB; CDKN2B (p15) was found to be deleted in K562 [13]. Moreover, a significant increase in the number of methylated genes in advanced stages of the disease was also noted. On the other hand, no significant differences in overall survival between patients in CP with 0–4 hypermethylated genes compared to CP with 4–10 hypermethylated genes were observed.
The methylation status of additional genes observed in several studies with CML patients in different stages of disease is summarized in Table 1.
Table 1.
DNA methylation status in CML patients
| Gene | Methylation status by CML stages % | ||
|---|---|---|---|
| CP | AP | BC | |
| Calcitonin [27] | 6 | 63 | 92 |
| HIC1 [28] | 50 | – | 100 |
| ER [29] | 50 | – | 100 |
| PDLIM4 [13•] | 13 | 29 | 33 |
| HOXA4 [30] | 60 | – | 91 |
| HOXA5 [30] | 33 | – | 87 |
| DDIT3 [31] | 68 | 100 | 53 |
CP Chronic phase; AP Accelerated phase; BC Blast crisis
DNA Methylation Inhibitors (DNMTi) in CML
Nowadays, several drugs, especially demethylating agents, targeting DNA methyl transferase 1, are being utilized in AML and MDS [32] and are strongly considered also in CML. The most abundantly tested DNMTi are 5-azacitidine and 5-aza-2´-deoxycitidine (decitabine). These agents are incorporated into both DNA and RNA during replication or transcription, respectively, and inhibit effects of DNA methyltransferase (DNMT) [33]. Kantarijan et al. [34] reported the results of decitabine treatment in 130 CML patients not previously treated with imatinib. The patients were treated with decitabine at 100 mg/m2 over 6 hours every 12 hours x 5 days (1000 mg/m2) in the first 13 patients, 75 mg/m2 in the subsequent 33 patients, and 50 mg/m2 in the remaining 84 patients. Hematological response rates were 28 % in BC, 55 % in AP, and 63 % in CP. Severe and prolonged myelosuppression was one of the side effects during this therapy. A median of three courses were required to achieve optimal response suggesting that the decitabine effect on leukemia cells is slow but continuous. A phase I study of low-dose prolonged exposure to decitabine treatment in 50 patients (44 with AML/MDS, 5 CML, and 1 acute lymphoblastic leukemia (ALL)) with myeloid malignancies has shown that most of the hematologic and/or cytogenetic responses were achieved at a dose of 15 mg/m2 for 10 days [35, 36]. Decitabine was also used in a phase II trial in 25 CML patients previously treated with imatinib that developed resistance [21] at a dose of 15 mg/m2 intravenously daily, 5 days a week for 2 weeks in combination with imatinib 600 mg orally daily in 18 and 10 patients with CML in AP and BC, respectively. The combination therapy was well tolerated with mild toxicities limited to myelosuppression. Similarly to the phase I trial, where decitabine was applied alone, the overall hematologic response rate was 30 % in BC and 50 % in AP. Moreover, the hematologic response rate was higher in patients without any mutation in the BCR-ABL kinase domain (KD) than in those with mutations. Based on these findings, the authors suggested that decitabine cannot efficiently destroy tumor cell resistant to imatinib.
Hydralazine and valproate are now being tested for their potential to inhibit DNA methylation and histone deacetylation, although they are commonly used as an antihypertensive and epileptic drug, respectively. Additionally, they have shown promising results in combination with cytotoxic chemotherapy or alone in the treatment of several solid and hematologic neoplasias, including MDS [37, 38]. Cervera et al. [39] evaluated the effect of these agents on the response rates and their role in reverting the resistance to IM in CML patients with acquired resistance who had no access to 2nd generation TKI [39]. Eight patients received hydralazine 182 mg (fast acetylators) or 83 mg (slow acetylators) daily, magnesium valproate 30 mg/kg t.i.d. and the same dose of IM at the time of progression. Mild neurologic toxicity was the only side effect during the treatment and no toxicity of grade 3/4 was observed. All but one of the patients achieved hematologic and cytogenetic responses or at least stabilization of disease and the overall survival was 63 % at 18 months.
Therapy Modulating Histone Modifications in CML
Histone modifications are an integral part of gene regulation involving transcription as well as other cellular processes within the nucleus. Histone modifications involve acetylation, methylation, phosphorylation [40] and other posttranslational modifications that mark sites of chromatin accessibility, DNA integrity and repair, transcription and replication [41]. Each of the core histones is strongly conserved throughout evolution and can be modified at arginine-(R) or lysine-(K) residues and also at other less studied residues [42]. Histone methylation and acetylation are the most investigated modifications that are reversible. In general, malignant tissues demonstrate not only methylation of DNA, but also changes in histone modification resulting in chromatin condensation rate with effects on proliferation, differentiation and finally on tumor suppression. Pro- and anti-apoptotic genes and cell cycle genes are also widely studied targets of histone modifications in cancer cells [43, 44]. Specific enzymatic machinery is involved in gene regulatory effects involving specific histone acetylation including histone deacetylases (HDAC) and histone acetyltransferases (HATs). In CML, histone modifications have been studied since HDAC inhibitors were developed.
HDAC inhibitors, that may have the potential to restore normal acetylation of histone proteins and transcription factors, can be classified into several groups: aliphatic acids (phenylbutyrate), cyclic peptides (romidepsin), benzamides (entinostat), electrophilic ketones, and hydroxamates (vorinostat) [33, 45]. At present, there also exist additional compounds such as panobinostat. In addition, valproic acid (VA), is also considered to have inhibition effects on HDAC. The results of the therapy with VA in combination with hydralazine are mentioned above.
Several HDAC inhibitors can target genes involved in the growth arrest and apoptosis, including one of the most studied inhibitors, suberoylanilide hydroxamic acid (SAHA, vorinostat), which has been approved by FDA for the treatment of cutaneous T-cell lymphoma [46]. Nimmanapalli et al. [47] observed that the treatment with SAHA alone induced the expression of p21 and/or p27, but decreased the mRNA and protein level of BCR-ABL in K562 and LAMA-84 cells. Moreover, co-treatment with SAHA and IM caused stronger down-regulation of the levels compared to either drug alone. The BCR-ABL protein level was also notably decreased in CD34+ cells derived from BC CML patients treated with IM and the apoptosis was induced. It is possible that the SAHA treatment enhances the cytotoxicity of IM and that it may show activity against imatinib-refractory BC CML [47]. These results are in accordance with the study of Fiskus et al. [48] evaluating the effects of vorinostat and dasatinib (2nd generation TKI) co-treatment, which caused attenuation of the BCR-ABL protein level and induction of apoptosis in imatinib-sensitive and imatinib-resistant cells as well as in primary CML cells from patients in advanced stages of the disease. This mechanism of how hydroxamates function on CML cells remains unclear. It is suggested that hydroxamates (LAQ824, LBH589, and vorinostat) may induce acetylation and inhibition of non-histone protein Heat Shock Protein 90 (a chaperone for BCR-ABL), and thus promotes polyubiquitinylation and proteasomal degradation of BCR-ABL [48, 49].
MicroRNAs in CML
As pointed out above, the posttranscriptional level is as important as chromatin level in guidance of the epigenetic regulations. MicroRNAs are evolutionarily conserved tiny RNA molecules with potentially great regulatory functions in many biological processes in humans, animals and plants. Their regulatory functions are processed through the complementarities of their “seed sequence“ and 3’UTR of the targeted gene. If the complementarities are incomplete, the translation into a protein is blocked. In the case of perfect complementarities, the RNA-induced silencing complex (RICS) induces mRNA degradation.
The number of known microRNAs continuously increases. Up to now, more than 1000 pre-microRNAs have been annotated in the human genome. Based on bioinformatic predictions, it is expected that more than a half of gene products in the human genome may be regulated by microRNAs, with each microRNA capable of regulating hundreds of genes [50].
When studying microRNAs associated with CML, several important issues arose:
(i) what is the microRNA expression profile specificity in CML pathogenesis ?, (ii) can some microRNAs be regulated by BCR-ABL or do microRNAs target BCR-ABL ?, and finally (iv) what are the microRNA targets in CML and are they are involved in CML-associated processes ?
We and others demonstrated that the expression profiles of mature microRNAs are typical for CML including both mixed as well as highly purified peripheral blood (PB)-derived cell populations [51, 52, 53•]. In spite of the different biological materials and microarray platforms used in the studies, the published microRNA expression profiles clearly distinguished: CML patients from healthy individuals [51] and more importantly also found differences between clinically defined CML phases [53•], and between patients either responding or not responding to TKI therapy [54•]. MicroRNA-based expression profiles may reflect a transformation from CP to AP or BC of CML [53•]. Despite the different blood cells and methods used for the microRNA expression profiling, significant overlap exists, and it may be crucial in understanding the aberrant CML hematopoiesis as well as the treatment resistance.
One of the hallmarks of CML is a decreased expression of miR-150 (in comparison to healthy individuals) in bone marrow-derived mononuclear cells (MNC) and CD34+ cells [51] and in MNC [52] and total leukocytes of PB [53•] at CML diagnosis. A significantly reduced amount of miR-150 was found in MNC [52] and total leukocytes of PB also in AP and BC [53•]. Moreover, miR-150 was significantly increased and recovered to the physiological level following the relatively short therapy with imatinib [52]. This is consistent with the data showing a normal level of miR-150 in CML patients-responders to imatinib [53•]. Additionally, we found a uniform profile of the significant decrease of miR-150 expression in MNC and polymorphonuclear cells (PMNC) fractionated from peripheral blood of six CML patients at diagnosis in comparison to the levels in MNC and PMNC of healthy individuals. After short treatment with imatinib (5–7 months from imatinib start), the amount of miR-150 significantly increased to normal levels in MNC and PMNC (unpublished data).
In several papers, the amount of miR-146a was found to be significantly decreased at CML diagnosis [51, 52, 53•]. It was shown that miR-146a expression was normalized after imatinib treatment [52, 53•]. A significantly decreased expression of miR-10a was reported in MNC and CD34+ cells in CML patients at diagnosis in comparison to healthy controls [51]. Aberrantly decreased levels of miR-10a were confirmed in MNC of PB at diagnosis [52]. However, after short treatment with imatinib, the microRNA levels were normalized.
We may find several discrepancies in the microRNA expression profiles in CML such as the miR-17-92 cluster also known as oncomir-1. The MiR-17-92 is a polycistronic cluster of six microRNAs with their increased expression levels in CD34+ cells of CML patients at diagnosis. In contrast, their levels were physiologically expressed in CD34+ blastic cells [55]. Interestingly, the expression levels of pri-miR-17-92 were found increased in CD34+ cells at the CML diagnosis and blast crisis [55]. We showed that the levels of miR-17-92 were significantly increased in leukocytes of PB in blast crisis while levels were normal or slightly increased at diagnosis [53]. In addition, Flamant et al. [52] found a decrease in expression of oncomir-1 in MNC PK 14 days after starting Imatinib. Recently, in a study with a small cohort of patients (n = 17), it was suggested that the miR-451 expression level found in total leukocytes of PB at diagnosis may be associated with the response to IM therapy. In addition, it was shown that the miR-451 level may inversely correlate with the BCR-ABL transcript level in some CML patients [56]. However, expression of miR-451 appears in this study to be relatively more heterogeneous. It was suggested that besides BCR-ABL, other mediators may be involved in the regulation of miR-451 expression [56]. Such heterogeneous expression of miR-451 expression among total leukocytes from CML patients may be explained by our unpublished data on the MNC and PMNC fractions from peripheral blood of six CML patients at diagnosis and after a short imatinib treatment (5–7 months). We observed that the expression of miR-451 was significantly reduced at diagnosis in PMNC, while in MNC, the transcript levels remained normal. After short treatment with imatinib, accompanied by a BCR-ABL transcript level reduction, the miR-451 transcript levels were normalized in PMNC and significantly increased in MNC in comparison to healthy PMNC and MNC, respectively. Thus, the expression of miR-451 found in total leukocytes is an average of expression in MNC and PMNC. In the case of miR-451, the expression profile is rather dependent on the cell type and its abundance within MNCs.
Bioinformatic Prediction of miRNA Targets in CML
We and others showed that in silico predicted targeted genes of selected microRNAs may be associated with CML pathogenesis. The increased levels of 13 miRNAs (miR-19a, miR-19b, miR-17, miR-20, miR-92a, miR-106a, miR-221, miR-222, miR-126, miR-146a, miR-181a, miR-181b, let7c, miR-155) and decreased levels of 4 miRNAs (miR-103, miR-150, miR-451, miR-144) in PB cells from blast crisis were used to uncover signaling pathways and their role in CML (Fig. 1) [53•]. Some of the predicted proteins are involved in mitogen-activated pathway (MAP) kinase signaling that functionally interact with BCR-ABL. The miRNA may also regulate PI3K (phosphatidylinositol-3-kinase) pathway. The gene encoding PI3K is the predicted target of miR-181a, miR-221 and miR-19a (their levels were decreased in CML patients treated with imatinib) [53•]. Decreased levels of the above mentioned miRNAs may contribute to the increased levels of PI3K and thus in the PI3K/AKT/mTOR signaling pathway and it may potentially regulate resistance development in CML. AKT1 is a member of the anti-apoptotic pathway of PI3K that is associated with the BCR-ABL directed transformation and with the response to the TKI treatment. The PI3K/AKT/mTOR pathway is activated in imatinib naive cells, but under imatinib treatment, it may contribute to the resistant phenotype [57]. Recent work of Rokah et al. [58] supplemented the predicted genes in the annotated CML pathways being targets of miR-155, miR-31 and miR-564 that were found to be aberrantly expressed in K562 cell lines in comparison to blood cells of healthy individuals (also see Fig. 1).
Fig. 1.
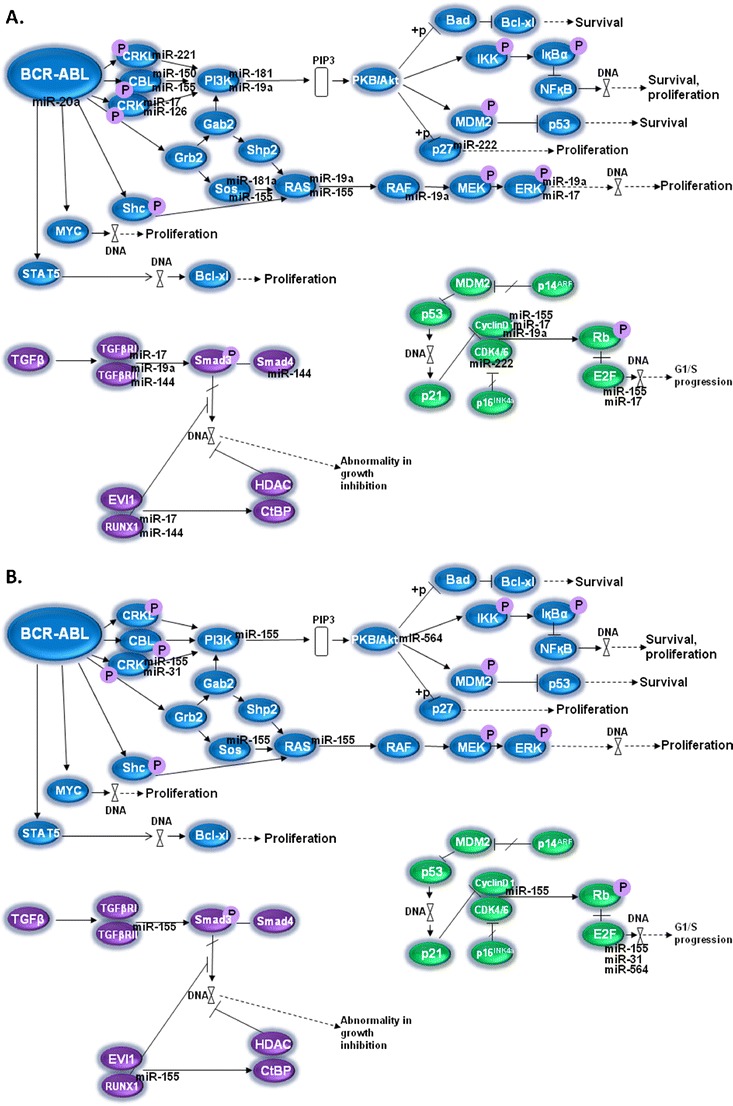
Scheme of CML hematopoietic stem cell signaling pathways in which predicted targets of microRNAs associated with CML are involved. A. microRNAs identified by Machova Polakova et al. [53•]: miR-103, miR-150 and miR-144 were found to be decreased in peripheral blood cells of blast crisis, while other illustrated micoRNAs were increased; B. microRNAs identified by Rokah et al. [58]: all four illustrated microRNAs were found to be decreased in the K562 cell line. MAPK pathway is shown in blue, Transforming growth factor beta signaling pathway in purple and cell cycle with p53 pathway in green. Note: the scheme is modified according to the hsa05220 of the KEGG database (Kanehisa M, Goto S, Furumichi M, et al.: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38:D355-D360.)
MMP16, which drives a membrane transmigration of cancer cells, and NFASC were predicted as targets of deregulated miRNAs in CML: miR-150 and miR-146a. For the other two deregulated molecules, miR-142-3p and miR-199b-5p, the following targets were predicted: ACVR2A, ANK3, ARHGEF12, C18orf25, CCNJ, MAP3K11, MYH9, MYO5A, RBM47, RHEB, RNF38, and ZNF618 [52]. Genes encoding proteins that may be important in CML are also ARHGEF12 and MYH9. The ARHGEF12 is known as the LRG encoding Rho guanin factor gene essential for nucleotide exchange and was firstly identified as the fusion partner of MLL in acute myeloid leukemia. The MYH9 encodes the heavy chain of non-muscle myosin and its upregulation was described in myeloid cells during differentiation [58]. To conclude this part, targets of microRNAs may be involved in clinically relevant tasks in CML, but a detailed understanding of these targets will require additional studies.
Function of miRNAs Regulated by BCR-ABL
Two important groups of miRNAs may play a significant role in the CML pathogenesis: (i) miRNAs that are downregulated by BCR-ABL and (ii) miRNAs that are upregulated by BCR-ABL.
Several miRNAs are regulated by BCR-ABL in the Ph+ cell lines:
Pri-miR-17-92 transcription is directly regulated by c-MYC and also by BCR-ABL [55]. The inhibition of c-MYC using anti-c-MYC specific shRNA together with imatinib treatment induced the highest reduction in the amount of pri-miR-17-92 in K562 cells. As a consequence, nearly complete arrest of proliferation was observed following the inhibition of c-MYC expression. The inhibitory effect on growth of K562 through c-MYC silencing was partially reverted by the overexpression of ectopic construct encoding miR-17-19b. Altogether, the data support the existence of the BCR-ABL / c-MYC / miR-17-92 pathway in CML pathogenesis.
MiR-130a is mediated by BCR-ABL in K562 cells [59]. After the BCR-ABL silencing in K562, the levels of miR-130a and miR-130b significantly decreased, whereas the expression of their putative target, the growth negative regulator CCN3, significantly increased. To prove that CCN3 is the target of miR130a/b, the HL-60 cell line was transfected individually with both miRNAs showing that the expression of the tumor suppressor gene CCN3 was repressed with a different intensity. Finally, overexpression of miR-130a (but not miR-130b) significantly inhibited protein expression of its target CCN3.
The expression levels of miR-150 significantly increased following the inhibition of the BCR-ABL activity by imatinib treatment in the Ph+ cell line MOLM7 [53•]. The MYB is a proved target gene of miR-150 and represents a transcription factor for the proliferation and survival of normal and leukemic blasts. Experiments on a murine model of BC CML showed that c-MYB is required for BCR-ABL driven leukemogenesis [60]. Thus, the reduction of miR-150 might contribute to the up-regulation of c-MYB. Moreover, an observed significant inverse correlation of miR-150 and MYB and with BCR-ABL expressions and a significant positive correlation of MYB with the BCR-ABL transcript levels support the above mentioned conclusions [53•]. The network between BCR-ABL, miR-150 and MYB needs to be further clarified and tested for some expected clinical benefit.
The differentiation arrest in the myeloid blastic phase of CML depends on the BCR-ABL-MAPK induced activity of hnRNP E2. The hnRNP E2 is a poly(rC)-binding protein that controls the translation of specific mRNAs. The induced activity of hnRNP E2 in CML blast crisis silences miR-328, thus the pro-apoptotic effect is arrested through the PIM1 overexpression. PIM1 is an oncogene encoding a serine/threonine kinase, inducing cell proliferation and survival [61]. In association with the described BCR-ABL-MAPK-hnRNP E2- miR-328 pathway, the authors rediscovered a novel function for microRNA – a decoy activity. The hnRNP E2 suppresses the translation and synthesis of the transcription factor CCAAT/enhancer binding protein (C/EBP), alpha (CEBPA, that is essential for myeloid differentiation) through the C-rich element in 5’UTR mRNA of CEBPA. The MiR-328 interacts outside its “seed sequence” with hnRNP E2 with a higher affinity than between hnRNP E2 and mRNA CEBPA. The interaction between miR-328 and hnRNP E2 enables the translation of CEBPA, thus a myeloid differentiation is induced. Moreover, there is a feedback loop as C/EBPalpha induces the transcription of pri-miR-328 that might be prevented by hnRNP E2 through the BCR-ABL induced MAPK hnRNP E2 signaling pathway [61].
MicroRNAs may Directly Target BCR-ABL
The identification of miRNAs that directly target the expression of BCR-ABL may play a role in the microRNA-based therapy in CML. A recent study showed that normal ABL, and also BCR-ABL, are targets of miR-203 [62]. The predicted duplexes, formed between the miR-203 “seed sequence” and 3’UTR of ABL, showed complete complementarity. The 3’UTR region of ABL is present in the BCR-ABL fusion gene. It was observed that the upstream region of the gene encoding miR-203 was hypermethylated in Ph+ leukemias, resulting in the absence of miR-203. After the reintroduction of miR-203 into the CML cell lines, K562 and KCL-22, expression of miR-203 was restored. Additionally, it was accompanied by an ABL and BCR-ABL decrease and dramatic reduction in the cell proliferation. It was shown that epigenetic inactivation through promoter region methylation of miR-203 might allow expression of ABL and BCR-ABL to escape the regulatory control. Epigenetic regulation of miR-203 expression represents an example of aberrant hypermethylation of the promoter region of genes encoding microRNAs leading to an inappropriate increase of oncogene levels.
Conclusion
Significant progress in CML treatment has been achieved since the introduction of first and 2nd line tyrosine kinase inhibitors. Despite the high efficiency of the TKI therapy, some patients developed resistance due to mutations in the BCR-ABL kinase domain, resulting in therapy failure. Other mechanisms of resistance are not fully understood. Abnormal epigenetic regulation of the expression of important genes may play a critical role in the pathogenesis of CML and its resistance to therapy. Since epigenetic changes have the potential to be modulated, new drugs targeting different epigenetic mechanisms are strongly considered for the therapy of CML especially those that developed resistance to imatinib including demethylated agents and HDAC inhibitors. MicroRNAs are also potentially very important in the pathogenesis of CML. In the future, microRNA-based therapy is planned to be one of the approaches in CML management. This requires that we improve the chemical design of microRNA antisense oligonucleotides and microRNA mimics and we must to develop efficient delivery methods for microRNAs. Lastly, recent progress in biotechnology and bioinformatics provides novel promising tools (such as next generation sequencing) for de novo epigenetic factors characterization and deeper understanding of the epigenetic mechanisms to be successfully utilized for personalized CML therapy in the future.
Acknowledgments
Disclosure
K.M.P. and J.K. are supported by IGA MZČR NT11555.
T.S. is supported by grants GACR P305/12/1033 and FR-TI2/509 and also in part by institutional funding from UNCE 204021, PRVOUK-P24/LF1/3, SVV-2012-264507 and Operational Program Research and Development for Innovations (CZ.1.05/1.1.00/02.0109).
Contributor Information
Katerina Machova Polakova, Phone: +420-22-1977181, FAX: +420-22-1977371, Email: katerina.machova@uhkt.cz.
Jitka Koblihova, Phone: +420-22-1977181, FAX: +420-22-1977371, Email: jitka.koblihova@uhkt.cz.
Tomas Stopka, Phone: +420-22-4965970, FAX: +420-22-4965916, Email: tstopkal@lf1.cuni.cz.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;142:1497. doi: 10.1126/science.144.3623.1229. [DOI] [PubMed] [Google Scholar]
- 2.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–93. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 3.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–66. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 4.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 5.Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118:1208–15. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Matzke M, Matzke AJ, Kooter JM. RNA: guiding gene silencing. Science. 2001;293:1080–83. doi: 10.1126/science.1063051. [DOI] [PubMed] [Google Scholar]
- 8.Bonifer C, Cockerill PN. Chromatin mechanisms regulating gene expression in health and disease. Adv Exp Med Biol. 2011;711:12–25. doi: 10.1007/978-1-4419-8216-2_2. [DOI] [PubMed] [Google Scholar]
- 9.Asimakopoulos FA, Shteper PJ, Krichevsky S, et al. ABL1 methylation is a distinct molecular event associated with clonal evolution of chronic myeloid leukemia. Blood. 1999;94:2452–60. [PubMed] [Google Scholar]
- 10.Issa JP, Kantarjian H, Mohan A, et al. Methylation of the ABL1 promoter in chronic myelogenous leukemia: lack of prognostic significance. Blood. 1999;93:2075–80. [PubMed] [Google Scholar]
- 11.Ben-Yehuda D, Krichevsky S, Rachmilewitz EA, et al. Molecular follow-up of disease progression and interferon therapy in chronic myelocytic leukemia. Blood. 1997;90:4918–23. [PubMed] [Google Scholar]
- 12.Nguyen TT, Mohrbacher AF, Tsai YC, et al. Quantitative measure of c-abl and p15 methylation in chronic myelogenous leukemia: biological implications. Blood. 2000;95:2990–92. [PubMed] [Google Scholar]
- 13.Jelinek J, Gharibyan V, Estecio MR, et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS One. 2011;6:e22110. doi: 10.1371/journal.pone.0022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun B, Jiang G, Zaydan MA, et al. ABL1 promoter methylation can exist independently of BCR-ABL transcription in chronic myeloid leukemia hematopoietic progenitors. Cancer Res. 2001;61:6931–37. [PubMed] [Google Scholar]
- 15.Uchida T, Kinoshita T, Hotta T, Murate T. High-risk myelodysplastic syndromes and hypermethylation of the p15Ink4B gene. Leuk Lymphoma. 1998;32:9–18. doi: 10.3109/10428199809059242. [DOI] [PubMed] [Google Scholar]
- 16.Tien HF, Tang JH, Tsay W, et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol. 2001;112:148–54. doi: 10.1046/j.1365-2141.2001.02496.x. [DOI] [PubMed] [Google Scholar]
- 17.Wong IH, Ng MH, Huang DP, Lee JC. Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes: potential prognostic implications. Blood. 2000;95:1942–49. [PubMed] [Google Scholar]
- 18.Deneberg S, Grövdal M, Karimi M, et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24:932–41. doi: 10.1038/leu.2010.41. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez S, Suela J, Valencia A, et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS One. 2010;5:e12197. doi: 10.1371/journal.pone.0012197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman JG, Civin CI, Issa JP, et al. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 1997;57:837–41. [PubMed] [Google Scholar]
- 21.Oki Y, Kantarjian HM, Gharibyan V, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109:899–906. doi: 10.1002/cncr.22470. [DOI] [PubMed] [Google Scholar]
- 22.Uehara E, Takeuchi S, Yang Y, et al. Aberrant methylation in promoter-associated CpG islands of multiple genes in chronic myelogenous leukemia blast crisis. Oncol Lett. 2012;3:190–92. doi: 10.3892/ol.2011.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avramouli A, Tsochas S, Mandala E, et al. Methylation status of RASSF1A in patients with chronic myeloid leukemia. Leuk Res. 2009;33:1130–32. doi: 10.1016/j.leukres.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Murray PG, Qiu GH, Fu L, et al. Frequent epigenetic inactivation of the RASSF1A tumor suppressor gene in Hodgkin's lymphoma. Oncogene. 2004;23:1326–31. doi: 10.1038/sj.onc.1207313. [DOI] [PubMed] [Google Scholar]
- 25.Dunwell T, Hesson L, Rauch TA, et al. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer. 2010;9:44. doi: 10.1186/1476-4598-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian J, Wang YL, Lin J, et al. Aberrant methylation of the death-associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur J Haematol. 2009;82:119–23. doi: 10.1111/j.1600-0609.2008.01178.x. [DOI] [PubMed] [Google Scholar]
- 27.Nelkin BD, Przepiorka D, Burke PJ, et al. Abnormal methylation of the calcitonin gene marks progression of chronic myelogenous leukemia. Blood. 1991;77:2431–34. [PubMed] [Google Scholar]
- 28.Issa JP, Zehnbauer BA, Kaufmann SH, et al. HIC1 hypermethylation is a late event in hematopoietic neoplasms. Cancer Res. 1997;57:1678–81. [PubMed] [Google Scholar]
- 29.Issa JP, Zehnbauer BA, Civin CI, et al. The estrogen receptor CpG island is methylated in most hematopoietic neoplasms. Cancer Res. 1996;56:973–77. [PubMed] [Google Scholar]
- 30.Strathdee G, Holyoake TL, Sim A, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res. 2007;13:5048–55. doi: 10.1158/1078-0432.CCR-07-0919. [DOI] [PubMed] [Google Scholar]
- 31.Wang YL, Qian J, Lin J, et al. Methylation status of DDIT3 gene in chronic myeloid leukemia. J Exp Clin Cancer Res. 2010;29:54. doi: 10.1186/1756-9966-29-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholz B, Marschalek R. Epigenetics and blood disorders. Br J Haematol. 2012;158:307–322. doi: 10.1111/j.1365-2141.2012.09193.x. [DOI] [PubMed] [Google Scholar]
- 34.Kantarjian HM, O'Brien S, Cortes J, et al. Results of decitabine (5-aza-2'deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer. 2003;98:522–28. doi: 10.1002/cncr.11543. [DOI] [PubMed] [Google Scholar]
- 35.Issa JP, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–40. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 36.Issa JP, Gharibyan V, Cortes J, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–56. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- 37.Arce C, Pérez-Plasencia C, González-Fierro A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:e98. doi: 10.1371/journal.pone.0000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Candelaria M, Herrera A, Labardini J, et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. . Ann Hematol. 2011;90:379–87. doi: 10.1007/s00277-010-1090-2. [DOI] [PubMed] [Google Scholar]
- 39.Cervera E, Candelaria M, López-Navarro O, et al. Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2012;12:207–12. doi: 10.1016/j.clml.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–53. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 41.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 42.Gronbaek K, Muller-Tidow C, Perini G, et al. A critical appraisal of tools available for monitoring epigenetic changes in clinical samples from patients with myeloid malignancies. Haematologica. 2012 Apr 4. [DOI] [PMC free article] [PubMed]
- 43.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 44.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 45.Mithraprabhu S, Grigoriadis G, Khong T, Spencer A. Deactylase inhibition in myeloproliferative neoplasms. Investig New Drugs. 2010;28(Suppl 1):S50–7. doi: 10.1007/s10637-010-9590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 47.Nimmanapalli R, Fuino L, Stobaugh C, et al. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101:3236–39. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 48.Fiskus W, Pranpat M, Balasis M, et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clin Cancer Res. 2006;12:5869–78. doi: 10.1158/1078-0432.CCR-06-0980. [DOI] [PubMed] [Google Scholar]
- 49.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 50.Hansen TB, Kjems J, Bramsen JB. Enhnacing miRNA annotation confidence in miRBase by continuous cross dataset analysis. RNA Biol. 2011;8:378–83. doi: 10.4161/rna.8.3.14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agirre X, Jiménez-Velasco A, José-Enériz ES, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6:1830–40. doi: 10.1158/1541-7786.MCR-08-0167. [DOI] [PubMed] [Google Scholar]
- 52.Flamant S, Ritchie W, Guilhot J, et al. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica. 2010;95:1325–33. doi: 10.3324/haematol.2009.020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Machová Poláková K, Lopotová T, Klamová H, et al. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:41. doi: 10.1186/1476-4598-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.José-Enériz ES, Román-Gómez J, Jiménez-Velaso A, et al. MicroRNA expression profiling in imatinib-resistant chronic myeloid leukemia patients without clinically significant ABL1-mutations. Mol Cancer. 2009;8:69. doi: 10.1186/1476-4598-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venturini L, Battmer K, Castoldi M, et al. Expression of the miR-19-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007;109:4399–4405. doi: 10.1182/blood-2006-09-045104. [DOI] [PubMed] [Google Scholar]
- 56.Scholl V, Hassan R, Zalcberg IR. miRNA-451: A putative predictor marker of Imatinib therapy response in chronic myeloid leukemia. Leuk Res. 2012;36:119–21. doi: 10.1016/j.leukres.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 57.Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates resistance development. Leukemia. 2005;19:1774–82. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 58.Rokah OH, Granot G, Ovcharenko A, et al. Downregulation of miR-31, miR-155, and miR-564 in chronic myeloid leukemia cells. PLoS One. 2012;7:e35501. doi: 10.1371/journal.pone.0035501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suresh S, McCallum L, Lu W, et al. MicroRNAs 130a/b are regulated by BCR-ABL and downregulate expression of CCN3 in CML. J Cell Commun Signal. 2011;5:183–191. doi: 10.1007/s12079-011-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lidonnici MR, Corradini F, Waldron T. Requirement of c-Myb for p210BCR/ABL-dependent transformation of hematopoietic progenitors and leukemogenesis. Blood. 2008;111:4771–79. doi: 10.1182/blood-2007-08-105072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eiring AM, Harb JG, Neviani P, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140:652–65. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bueno MJ, Castro IP. Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]