

Abstract
Chiari malformation type I (CMI) is a disorder characterized by hindbrain overcrowding into an underdeveloped posterior cranial fossa (PCF), often causing progressive neurological symptoms. The etiology of CMI remains unclear and is most likely multifactorial. A putative genetic contribution to CMI is suggested by familial aggregation and twin studies. Experimental models and human morphometric studies have suggested an underlying paraxial mesoderm insufficiency. We performed a case-control association study of 303 tag single nucleotide polymorphisms (SNP) across 58 candidate genes involved in early paraxial mesoderm development in a sample of 415 CMI patients and 524 sex-matched controls. A subgroup of patients diagnosed with classical, small-PCF CMI by means of MRI-based PCF morphometry (n = 186), underwent additional analysis. The genes selected are involved in signalling gradients occurring during segmental patterning of the occipital somites (FGF8, Wnt, and retinoic acid pathways and from bone morphogenetic proteins or BMP, Notch, Cdx and Hox pathways) or in placental angiogenesis, sclerotome development or CMI-associated syndromes. Single-marker analysis identified nominal associations with 18 SNPs in 14 genes (CDX1, FLT1, RARG, NKD2, MSGN1, RBPJ1, FGFR1, RDH10, NOG, RARA, LFNG, KDR, ALDH1A2, BMPR1A) considering the whole CMI sample. None of these overcame corrections for multiple comparisons, in contrast with four SNPs in CDX1, FLT1 and ALDH1A2 in the classical CMI group. Multiple marker analysis identified a risk haplotype for classical CMI in ALDH1A2 and CDX1. Furthermore, we analyzed the possible contributions of the most significantly associated SNPs to different PCF morphometric traits. These findings suggest that common variants in genes involved in somitogenesis and fetal vascular development may confer susceptibility to CMI.
Introduction
The Chiari malformations, first described in 1895 [1], are a group of disorders sharing the common feature of ectopia of the cerebellar tonsils, which are downwardly displaced through the foramen magnum. Chiari malformation type I (CMI) is currently defined by the observation, on cranial midsagittal magnetic resonance imaging (MRI), of a descent of the cerebellar tonsils across the foramen magnum of at least 3 mm [2]. In contrast with Chiari II and III malformations, in CMI the rest of hindbrain structures remain within the posterior fossa.
CMI causes neurological dysfunction by direct compression of the neural tissue at the craniovertebral junction or cerebrospinal fluid disturbances that give rise to syringomyelia or hydrocephalus. The most frequent symptoms are headache, ocular disturbances, vertigo, sleep apnea and lower cranial nerve signs, including tongue fasciculation, dysphagia or dysarthria. Motor and sensory symptoms derived from spinal cord disturbances associated with syringomyelia or scoliosis, [3], [4] as well as signs and symptoms mimicking pseudotumor cerebri are also often encountered. Although presentation typically occurs in middle age, it may start in childhood or infancy, where it is a cause of sudden infantile death syndrome [5], [6]. Conversely, cases of CMI can often be asymptomatic and tonsillar descent is often regarded as an incidental neuroradiological finding [7], [8]. The prevalence of CMI is estimated to be in the 1/1,000 to 1/5,000 range [9]. A retrospective analysis of 22,591 serial cranial MRI studies, using a conservative definition of cerebellar tonsillar herniation of ≥5 mm, showed the presence of CMI in 0.77% of the subjects and asymptomatic CMI in 0.11%, with an estimated prevalence of 1/1,280 [10].
Regarding pathogenesis, different mechanisms are to be distinguished. In the classical form, a shallow posterior cranial fossa (PCF) is unable to house a normal hindbrain and hence the tonsillar herniation occurs (Figure 1A). Seminal experimental work by Marin-Padilla and Marin-Padilla [11] demonstrated that underdevelopment of the occipital bone and overcrowding of the cerebellum in a small posterior fossa are the fundamental defects in CMI [12], [13], [14]. This has been underscored by several radiological morphometric studies [3], [15], [16], [17], [18], [19]. However, cerebellar herniation may also occur in conditions such as cranial settling associated with connective tissue disorders, intraspinal hypotension following CSF leaks or lumboperitonial shunting, cord traction in tethered cord syndrome and intracranial hypertension caused by space-occupying lesions or hydrocephalus [20], where the PCF has a normal size and the term “secondary” CMI is preferred. In the classical form of CMI there is shortness of the basichondrocranium [3], [15], [16], [17], [19], [20], [21], [22], suggesting underdevelopment of occipital somites and pointing at insufficiency of the paraxial mesoderm as the central event in CMI pathogenesis [23].
Figure 1. Typical neuroradiological findings in CMI.
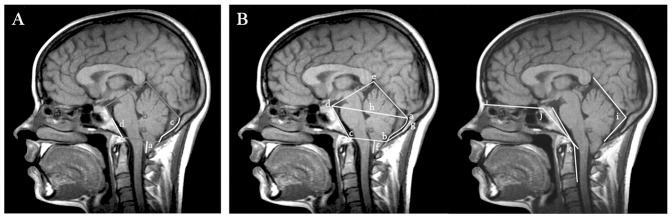
A) T1-weighted mid-sagittal MR image showing downward herniation of the cerebellar tonsils (a) and hypoplasia of the occipital components of the posterior cranial fossa (b): the supraocciput (c) and the basiocciput (d). B) Morphological measurements performed: length of tonsillar descent (f), supraocciput (g-b) and clivus (c–d) and foramen magnum diameter (b–c). The antero-posterior diameter of PCF (h) was inferred from a line running from the internal occipital protuberance (a) to top of the dorsum sellae (d). The PCF area was estimated as the polygon delimited by (a) (b) (c) (d) and (e) and the osseus PCF area as the one delimited by (a) (b) (c) and (d). Angular measurements: tentorium angle (i), basal angulation (j) and Wackenheim angle (k).
Whether CMI mesodermal insufficiency is a primary (genetic) condition or occurs as the result of early gestational injuries, or even post-natal osseous growth disturbances [16] is unknown. However, a number of evidences suggest that, at least in a subset of CMI cases, genetic factors may play an important role, including CMI familial aggregation [9], [24], higher degree of concordance in monozygotic twins and CMI occurrence as part of Mendelian syndromes [25], [26], [27]. In the instance of Crouzon syndrome, a specific FGFR2 mutation has been associated with CMI and syringomyelia in a small series of cases [28] and a PAX2 mutation was found in two siblings with CMI and coloboma [29]. The only reported genome-wide linkage screen, performed in 23 CMI multiplex families, uncovered two loci on chromosomes 9 and 15, with maximal two-point LOD (logarithm of the odds) score of 3.3 on 15q [30]. To date, no single gene has been identified as causing CMI.
If heredity does indeed play a role in CMI, it is also possible that this may occur via a polygenic mechanism. Common genetic variants in genes involved in growth and shaping of the PCF should be among the first to be explored. A similar approach has been used in association studies performed in patients with Chiari malformation type II, where screening of genes critical for neurulation and neural tube closure disclosed some risk factors but no major causative gene (reviewed in [31]). In contrast, no previous association study has been carried out in CMI. We here sought to identify common variants associated with development of CMI among critical genes expressed during occipital somite, sclerotome and placental development.
Subjects and Methods
Ethics Statement
The present investigation was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Ethics Committee of the Vall d'Hebron University Hospital. All participants signed an informed consent prior to their participation in the study.
Subjects
A total of 415 patients (42.2±5.9 years) with symptomatic CMI were recruited between 2004 and 2010. Three hundred and thirty seven patients were diagnosed and treated at the Division of Neurosurgery and the Pediatric Neurology Service of the Vall d'Hebron University Hospital in Barcelona. In all of them evaluation included direct anamnesis, physical exam, neurophysiological studies (somatosensory evoked potentials, brain auditory evoked potentials and polysomnography) and a cranial and spinal MRI study. For an additional 78 cases that were referred from two Spanish CMI patient associations, review of clinical summaries, radiological reports and telephone interviews were used to confirm the diagnosis, but direct review of the MRI studies was not always possible. Diagnosis of CMI was based on MRI demonstration of downward herniation ≥3 mm of the cerebellar tonsils on a midsagittal T1-weighted image in the presence of signs or symptoms indicating neural compression at the craniovertebral junction, syringohydromyelia, cerebellar dysfunction or intracranial hypertension
Since recent studies have clearly demonstrated that various mechanisms may lead to tonsillar herniation, we tried to identify the subset of patients with small PCF in an attempt to increase sample homogeneity. To this end we performed a morphometric PCF analysis in 211 cases with available digital MRI studies (Figure 1B). After applying a previously described logistic regression model based on several PCF measurements performed in CMI cases and a control population [32] we identified a subgroup of 186 patients representative of classical CMI, i.e., with underdeveloped PCF.
Clinical data was retrieved from an in-house patient database devoted to Chiari malformations and related disorders, a clinical research repository that included detailed information about the medical history, family history, symptoms and signs, treatment and any relevant associated conditions.
The control sample was composed of 524 healthy blood donors (42.6±14.4 years), sex-matched with the case sample and recruited at Vall d'Hebron University Hospital in Barcelona. To minimize ethnic heterogeneity only Caucasian subjects of Spanish origin were included in this study.
DNA isolation and Quantification
Genomic DNA was isolated from peripheral blood lymphocytes by the salting-out method [33] and from saliva with Oragen DNA Self-Collection kit according to the manufacturer's recommendations (Genotek Inc. Ottawa, Ontario, Canada). DNA concentrations of all samples were determined on a NanoDrop spectrophotometer (NanoDrop Technologies, LLC, Wilmington, DE).
Selection of genes and SNPs
We selected 58 functional candidate genes. Most belonged to gene pathways involved in the early paraxial mesoderm development, leading to formation of the occipital somites, such as retinoic acid (RBP4, RBP1, STRA6, ADH4, RDH10, CRABP1, CYP26A1, CYP26C1, RARG, RARA, RXRA, ALDH1A2) reviewed in [34], FGF8 (FGF8, FGFR1, DUSP4, HES7, SNAI1, SNAI2, CDH1) [35], [36], [37], Wnt (WNT3A, CTNNB1, Axin2, T, TBX6, MSGN1, NKD1, NKD2) [38], [39], [40], Notch (DLL1, RBPJ, LFNG, NOTCH1, MESP2, ID1, ID2) [37], [40], bone morphogenetic proteins (BMPR1A, BMP2) [41], Cdx (CDX1) [39], [42] and Hox pathways (HOXA1, HOXA2, HOXA3, HOXA4, HOXB1, HOXB2, HOXB3, HOXB4, HOXC4, HOXD1, HOXD3, HOXD4) reviewed in [42]. In addition, genes involved in sclerotome development, chondrogenesis and osteogenesis (NOG, SHH, NKX3-2, PRRX1) [43], [44], [45], [46], genes from the VEFG family crucial for placental angiogenesis (VEGFA, KDR, FLT1) [47], [48] and genes responsible for two conditions frequently associated with CMI, NF1 (neurofibromatosis) and STAT3 (Hyper-IgE Syndrome), were also included [49], [50].
The SNP selection was based on genetic coverage criteria, by considering linkage disequilibrium (LD) patterns within the candidate genes [51]. SNPs covering each gene plus 5 Kb flanking sequences were picked from the CEU panel of the HapMap database (www.hapmap.org, phases 1+2+3, release 27) [52], [53]. We used the LD-select software (droog.gs.washington.edu/ldSelect.html) to evaluate LD of the genomic regions in order to minimize redundancy between the selected SNPs [54], [55]. TagSNPs were selected with the following criteria: r2<0.85 from any other SNP according to CEU HapMap data and a minor allele frequency (MAF)>0.10 for genes with less than 20 tag SNPs and MAF>0.25 for those genes with more than 20 tagSNPs (FLT1, KDR, NOTCH1, RXRA).
Plex design, genotyping and quality control
A total of 384 tagSNPs were initially selected in our study (Table S1): 77 SNPs for the retinoic acid pathway, 44 SNPs for the FGF8 pathway, 55 SNPs for the Wnt pathway, 44 SNPs for the Notch pathway, 24 SNPs for BMP, 6 SNPs for CDX, 38 SNPs for Hox pathways, 30 SNPs for genes involved in sclerotome development, 51 SNPs for the VEGF pathway and 15 SNPs for NF1 and STAT3.
Genotyping was performed at the Barcelona node of the National Genotyping Center (CeGen, www.cegen.org) using the VeraCode technology (Illumina, San Diego, CA, USA) [56]. A total of 9 HapMap individuals were included as controls in the genotyping assay, obtaining a 100% concordance rate with HapMap genotype data. In addition, no differences were found in the genotypes of thirteen replicates.
Statistical analysis
The analysis of minimal statistical power was performed a priori with the CaTS Power Calculator software (sph.umich.edu/csg/abecasis/CaTS) [57], assuming an odds ratio (OR) of 1.5, estimated disease prevalence of 0.008 [7], [10], [24], significance level of 0.05 (α) and MAF of 0.10, under the additive model.
All individuals with genotyping rates under 80% were excluded from the study. From 384 SNPs genotyped in our study, 303 passed quality controls filters and were included in the analysis, whereas 81 (21%) were excluded for the following reasons: more than 15% missing genotypes, deviation from Hardy-Weinberg equilibrium (HWE threshold set at p = 0.01 in our control population), monomorphism, MAF<0.10 or MAF<0.25 and r2>0.85 from any other studied SNP in our control sample. The evaluation of LD patterns and r2 were performed from the genotype data of controls using Haploview v4.2 [58].
Single-marker analysis
The analysis of HWE and the case-control association study were performed with the SNPassoc R package [59]. For the case-control study we analysed all single markers under the additive model using the Cochran-Armitage Trend Test (ATT) (Table S1). A quantile-quantile plot (Q-Q plot) was generated with the ggplot2 R library [60]. The correction for multiple testing was performed using the Q-value R library assuming a 10% False Discovery Rate (FDR) [61] and the Bonferroni correction.
Multiple-marker analysis
To minimize multiple testing and type I (α) errors, we restricted the haplotype-based association study to only those LD blocks that included the SNPs overcoming the 10% FDR correction.
The linkage disequilibrium patterns for CDX1, ALDH1A2 and FLT1 were constructed from the genotyping data of our control individuals, using Haploview v4.2 software [58]. The haplotype-based analysis was performed using the same software [58]. To obtain a measure of significance for multiple testing in haplotype analysis, a total of 10,000 permutations were performed. Haplotypes with frequencies <0.05 were excluded.
PCF morphometric traits and risk genotypes
We used the Scheffé's post hoc test to evaluate whether common variants contributed to variation in any specific parameters as measured in PCF morphometric analyses, including cerebellar tonsillar descent, length of supraocciput, length of the clivus, foramen magnum diameter, PCF area, osseous PCF area, antero-posterior diameter of the PCF (PCF length), tentorium angle, basal angulation and Wackenheim angle, as previously described [32]. The analysis was carried out only in the 186 patients where the PCF morphometry confirmed the diagnosis of classical CMI and was limited to markers overcoming 10% FDR corrections for multiple testing in the single-marker analysis. The Kruskal-Wallis nonparametric test was used for variables not normally distributed (Kolmogorov-Smirnov test P<0.05) or showing no homogeneity of variances (Levene's test P<0.05)).
Results
The most common signs and symptoms in the 415 cases included in the study are listed in Table 1. Headache was the most prevalent symptom (69.1%), often featuring neck irradiation (43.3% of those with headache), followed by dizziness (35.1%), motor weakness (33.6%), instability (32.1%), upper limb paresthesias (30.7%), sensory loss (29.7%) and fatigue (28.1%). Syringomyelia was present in 35.1% of cases. Hydrocephalus (15.1%) required specific management. Other, less common manifestations, as well as co-morbid conditions, were typical for CMI (Table 1). Positive CMI family history, defined as at least one MRI-confirmed CMI first relative, was present in only 6% of the cases. Surgical PCF reconstruction was performed in 55.8% of patients.
Table 1. Clinical findings in 415 patients with Chiari malformation type I.
| CMI | |
| Number of patients | 415 |
| Sex (M/F) | 122/293 |
| Age (y) | 42.2±50.9 |
| Age at diagnosis | 30.6±58.7/323 |
| Hydrocephalus | 57/377 (15.1%) |
| Syringomyelia | 133/379 (35.1%) |
| Intracranial Hypertension | 20/376 (5.3%) |
| Retrocurved Odontoid | 40/352 (11.4%) |
| Basilar impression | 11/382 (2.3%) |
| Platibasia | 8/382 (2.1%) |
| Klippel-Feil Malformation | 3/377 (0.8%) |
| Other Craniocervical Malformations | 24/377 (6.4%) |
| SIGNS AND SYMPTOMS | |
| Time elapsed from onset (mo) | 113.3±59.4/323 |
| Headache | 246/356 (69.1%) |
| Occipital Headache/Cervicalgia | 97/224 (43.3%) |
| Cough Headache | 87/358 (24.3%) |
| Dizziness | 128/365 (35.1%) |
| Vertigo | 82/365 (22.5%) |
| Visual Alterations | 71/364 (19.5%) |
| Nystagmus | 41/276 (14.8%) |
| Kyphoscoliosis | 30/160 (18.7%) |
| Fatigue | 102/363 (28.1%) |
| Instability | 117/364 (32.1%) |
| Sensory Loss | 108/364 (29.7%) |
| Motor Weakness | 123/366 (33.6%) |
| Dysphagia | 67/365 (18.3%) |
| Dysphonia | 16/365 (4.4%) |
| Gait Disturbances | 73/364 (20.0%) |
| Paresthesia Upper Limbs | 112/365 (30.7%) |
| Paresthesia Lower Limbs | 47/366 (12.9%) |
| Upper Limb Pain | 47/365 (12.9%) |
| Lower Limb Pain | 16/365 (4.4%) |
| Neck Pain | 169/366 (46.2%) |
| Difficulty Swallowing | 66/365 (18.1%) |
| Anxiety | 38/360 (10.5%) |
| Depression | 33/361 (9.1%) |
| Neurofibromatosis type 1 | 45/382 (11.8%) |
| TREATMENT | |
| Posterior fossa reconstruction | 210/376 (55.8%) |
Criteria for surgery were a >3 mm descent of the cerebellar tonsils in i) symptomatic patients, ii) oligosymptomatic patients with sleep apnea syndrome predominantly of the central type, and iii) patients with syringomyelia. Posterior fossa reconstruction was carried out in all patients to restore the volumetric capacity of the posterior fossa and to restore normal CSF dynamics at the craniovertebral junction. In summary, the technique consisted of the removal of a large amount of occipital bone and the posterior arch of the atlas. The dura mater was opened and in most cases an extra-arachnoidal technique was used. An extensive dural graft secured with tenting sutures to the cervical fascia was used to avoid arachnoid scarring and to allow for the expansion of the compressed cisterna magna or the creation of a pseudocisterna magna
Initially, 384 SNPs from 58 candidate genes were genotyped (Table S1). Eighty-one SNPs were excluded from statistical analysis after data filtering: 40 SNPs had a genotyping rate below 85%, 4 SNPs showed deviation from HWE, 20 SNPs were monomorphic or had MAF<0.10 (or MAF<0.25) and 17 had a r2>0.85 with other SNPs in the same candidate gene (Table S1).
All individuals with genotyping rates below 80% were excluded from the study, resulting in a range of genotyping efficiency of 94–97%. Thus, 303 SNPs were finally assessed in the association study in a total of 404 patients (286 female) and 519 sex-matched controls. The minimal statistical power of our sample, under the additive model, was 78% for the whole sample and 53% after subgrouping into classical CMI.
Single Marker Analysis
We used a two-tiered single marker analysis. The first step comprised the whole patient sample while the second one included the more homogeneous subset of morphometry-proven classical CMI patients. In both we used the same control population.
The case-control association study with the whole CMI sample (404 patients and 519 controls) found nominal associations (P-values<0.05) for 18 SNPs located in 14 genes (Table S1). Only 5 SNPs, located in three different genes, displayed P-values<0.01: CDX1 (rs887343, rs2282809), FLT1 (rs17086609) and RARG (rs1554753, rs6580936) (P-values and genotype counts are shown in Table 2 and Figure 2A, and P-values for the remaining SNPs are shown in Table S1). However, no marker remained significant after applying 10% FDR or the Bonferroni correction (P value = 1.6E-04 ( = 0.05/303 SNPs)).
Table 2. Case-control association study of 404 Spanish CMI patients and 519 sex-matched controls.
| Gene | Control Genotypes N (%) | All CMI Genotypes N (%) | Classical CMI Genotypes N (%) | |||||||||
| SNP (P<0.01) | 11 | 12 | 22 | 11 | 12 | 22 | P-val | 11 | 12 | 22 | P-val | |
| ALDH1A2 | rs2899611 | 110 (21) | 264 (51) | 145 (28) | NS | 59 (32) | 90 (48) | 37 (20) | 0.00201 * | |||
| rs6493979 | 78 (15) | 273 (53) | 168 (32) | 84 (21) | 207 (51) | 113 (28) | 0.02436 | 46 (25) | 95 (51) | 45 (24) | 0.00209 * | |
| rs2067062 | 148 (29) | 258(50) | 105 (21) | NS | 35 (20) | 93 (52) | 51 (28) | 0.00425 | ||||
| BMPR1A | rs12415784 | 5 (1) | 135 (26) | 379 (73) | 5 (1) | 76 (19) | 323 (80) | 0.02744 | 2 (1) | 29 (16) | 155 (83) | 0.00704 |
| CDX1 | rs887343 | 85 (16) | 252 (49) | 180 (35) | 43 (11) | 187 (46) | 174 (43) | 0.00181 | 16 (9) | 82 (44) | 88 (47) | 0.00044 * |
| rs2282809 | 71 (14) | 240 (46) | 206 (40) | 37 (9) | 174 (43) | 193 (48) | 0.00489 | 15 (8) | 76 (41) | 95 (51) | 0.00308 | |
| FLT1 | rs17086609 | 242 (47) | 222 (43) | 50 (10) | 149 (37) | 196 (49) | 55 (14) | 0.00183 | 67 (37) | 83 (45) | 33 (18) | 0.00137 * |
| rs9554320 | 104 (20) | 261 (50) | 153 (30) | NS | 25 (14) | 88 (47) | 73 (39) | 0.00575 | ||||
| MSGN1 | rs11689375 | 117 (23) | 245 (47) | 156 (30) | 60 (15) | 203 (51) | 137 (34) | 0.01225 | 21 (11) | 98 (53) | 64 (36) | 0.00810 |
| RARG | rs1554753 | 310 (60) | 182 (35) | 27 (5) | 272 (67) | 121 (30) | 11 (3) | 0.00732 | 146 (79) | 36 (19) | 4 (2) | 0.01752 |
| rs6580936 | 358 (69) | 146 (28) | 15 (3) | 310 (77) | 87 (21) | 7 (2) | 0.00767 | NS | ||||
| RDH10 | rs16938610 | 305 (59) | 178 (34) | 36 (7) | 262 (64) | 126 (31) | 16 (4) | 0.02209 | 127 (68) | 53 (29) | 6 (3) | 0.00903 |
An additional study analyzed 186 classical CMI patients and the same control population. Only markers with P-value<0.01 (Cochran-Armitage trend test) are shown.
Significant associations after applying 10% FDR.
Figure 2. Quantile-quantile plot of the 303 P-values obtained in the association study under the additive model.

404 CMI patients versus 519 controls (A), and 186 small-PCF CMI versus 519 controls (B). SNPs with P-value<0.01 are indicated. Asterisks denote SNPs displaying association after 10% FDR correction for multiple testing (P<2.09E-03).
The second analysis considered 186 classical CMI patients (patients with MRI morphometry-confirmed small PCF) and the 519 controls. Nominal associations (P-values<0.05) were found under the additive model for 26 SNPs located in 13 genes (Table S1). The following 10 SNPs, located in 6 different genes, displayed P-values<0.01: ALDH1A2 (rs2899611, rs6493979, rs2067062), BMPR1A (rs12415784), CDX1 (rs887343, rs2282809), FLT1 (rs17086609, rs9554320), MSGN1 (rs11689375) and RDH10 (rs16938610) (Table 2 and Figure 2B). P-values for the remaining SNPs are shown in Table S1.
After applying a 10% FDR correction (P<2.09E-03) four associations did remain significant: rs887343 (P = 4.4E-04) in CDX1, rs17086609 (P = 1.37E-03) in FLT1, rs2899611 (P = 2.01E-03) and rs6493979 (P = 2.09E-03) in the ALDH1A2 gene. Under the more strict Bonferroni correction no SNP reached significance.
Multiple Marker Analysis
The haplotype analysis was addressed only for those genes containing SNPs that passed 10% FDR in the single marker analysis. The pairwise linkage disequilibrium map of ALDH1A2, CDX1 and FLT1 genes was calculated in the sample under study (Figure 3). LD patterns across the FLT1 gene defined five blocks (block 1 of <1 kb: rs9551462, rs9554325; block 2 of 6 kb: rs17086617, rs9508021 and rs2104330; block 3 of 1 kb: rs11149523 and rs8002446; block 4 of 22 kb: rs9513112, rs9513114 and rs7330109; block5 of 5 kb: rs12858139 and rs677471); two blocks were defined in ALDH1A2 (block 1 of 11 kb: rs3784264, rs3784260, rs4646615; block 2 of 17 kb: rs4238326, rs6493979, rs4238328, rs2067062 and rs7169439) and only one block in CDX1 (block 1 of 2 kb: rs887343 and rs2282809). The haplotype analysis was assessed only for those blocks containing SNPs significantly associated (block2 of ALDH1A2 and the only block of CDX1) (Figure 3). The SNPs rs2899611 (in ALDH1A2) and rs17086609 (in FLT1) were singletons (Figure 3). The results in the CDX1 gene identified a risk haplotype constituted of G-G allelic combination (Permutated P-value = 9.4 E-03; OR = 1.54, 95% CI = 1.19–2.01) (Table 3). The results in the ALDH1A2 gene identified an over-representation of the C-A-G-C-G haplotype in cases (Permuted P-value = 0.0179; OR = 1.53, 95% CI = 1.16–2.02).
Figure 3. Haploview graphs showing the markers tested and haplotype blocks constructed for ALDH1A2, CDX1 and FLT1.

D′ values are indicated (tones from white to dark grey indicate D′ values from 0 to 1, respectively). The genomic structures of ALDH1A2, CDX1 and FLT1 genes are drawn with coding exons indicated as black boxes. The SNPs with P-value<0.01 are indicated with an asterisk (*); the SNPs showing association after 10% FDR correction for multiple testing (P<2.09E-03) are indicated with a double asterisk (**).
Table 3. Haplotype analysis for the linkage disequilibrium blocks containing SNPs that showed significant associations in the single marker analysis (Haploview v4.2 software).
| Population | Gene | LD block: SNPs | Haplotype | Cases % | Controls % | P-value (permP)a | Odds Ratio [95% CI] |
| Classical CMI | CDX1 | Block 1: rs887343-rs2282809 | GG | 255 (71) | 614 (62) | 0.0014 (0.0094) | 1.54 [1.19–2.01] |
| CA | 103 (29) | 384 (38) | 0.0012 (0.0092) | 0.65 [0.50–0.84] | |||
| Classical CMI | ALDH1A2 | Block 2: rs4238326-rs6493979-rs4238328-rs2067062-rs7169439 | TGGAG | 99 (28) | 328 (33) | 0.0732 (0.3975) | 0.79 [0.60–1.02] |
| CAGCG | 100 (28) | 201 (20) | 0.0025 (0.0179) | 1.53 [1.16–2.02] | |||
| TGAAG | 66 (19) | 233 (24) | 0.0563 (0.3233) | 0.75 [0.55–1.01] | |||
| CAGCA | 49 (14) | 140 (14) | 0.882 (1.0000) | 0.97 [0.69–1.38] | |||
| TAGCG | 38 (11) | 87 (9) | 0.2624 (0.8997) | 1.24 [0.83–1.86] |
A total of 10,000 permutations were performed using Haploview v4.2 to obtain a measure of significance corrected for multiple testing.
PCF morphometric traits and risk genotypes
We sought to identify a possible relationship between genotypes of the four SNPs in three genes that passed 10% FDR correction (rs289961, rs6493979 (ALDH1A2), rs887343 (CDX1), rs17086609 (FLT1)) and any of the PCF morphological measurements performed on the MRI studies of 211 patients (Table 4). A modest association was found between the two SNPs in ALDH1A2 and the Wackenheim angle and the degree of basal angulation (P-value<0.05).
Table 4. Association between risk genotypes and PCF morphometric traits in 211 patients with CMI.
| ALDH1A2 | ALDH1A2 | CDX1 | FLT1 | |
| Measures | rs2899611 | rs6493979 | rs887343 | rs17086609 |
| TD | 0.105 | 0.684 | 0.226 | 0.328 |
| Supraocciput | 0.477 | 0.352 | 0.777 | 0.829 |
| Clivus | 0.922 | 0.976 | 0.593 | 0.769 |
| FM | 0.080 | 0.595 | 0.227 | 0.933 |
| PCF area | 0.248 | 0.560 | 0.556 | 0.459 |
| OPCF area | 0.367 | 0.636 | 0.532 | 0.356 |
| PCF length * | 0.750 | 0.300 | 0.094 | 0.983 |
| Tentorium angle | 0.456 | 0.836 | 0.410 | 0.118 |
| Basal angulation | 0.016 | 0.067 | 0.800 | 0.228 |
| Wackenheim angle | 0.006 | 0.039 | 0.824 | 0.146 |
Data presented are P-values (Scheffé's post-hoc and Kruskal-Wallis tests).
Assessed as the distance from the dorsum sellae to the internal occipital protuberance; in bold, P<0.05.TD: tonsillar descent; OPCF: osseous PCF, FM: foramen magnum.
Discussion
In the present association study performed in a sample of CMI patients from the Spanish population we found nominal association for SNPs in several genes involved in the early development of paraxial mesoderm and vascular development of the placenta. After corrections for multiple testing, only associations with four SNPs within CDX1, FLT1 and ALDH1A2 remained significant in the subsample where MRI morphometric analysis documented a reduced PCF. Considering these four SNPs, we also found a modest correlation between the two in ALDH1A2 and specific morphometric data of the PCF.
The Chiari malformations have long been considered sporadic conditions, without a heritable etiology. However, there have been a number of case reports identifying familial aggregation and clustering of CMI, suggesting a genetic basis [9], [25] although the involved genes and even type of inheritance are presently unknown. Given the predominance of apparently sporadic patients and the relatively high prevalence of the disorder, a polygenic model of inheritance seems plausible in a majority of CMI cases. To our knowledge, however, this is the first genetic association study ever to be carried out in CMI.
The contribution of common genetic variants has been examined in Chiari malformation type II (CMII), where several studies failed to demonstrate consistent associations with developmental genes such as CRABP1, CRABP2, ALDH1A2, RALDH2, CYP26, HOX, NOG, SHH and TBX6 (reviewed in [62], [63], [64]). Mutational screens of some of these genes did not find definitive evidence for disease-causing mutations [63]. Since the most accepted pathogenic hypothesis in CMI is that of insufficiency of paraxial mesoderm in the third and fourth week of gestation, we decided to explore whether variants in genes expressed during somitogenesis could be associated with the milder and more frequent CMI phenotype.
Association study results
A number of nominal associations were found in the whole CMI sample. Despite none of them overcoming corrections for multiple testing, three of the selected candidate genes, CDX1, FLT1 and RARG, contained SNPs that showed association with P-values<0.01, thus suggesting that these genes are worth of scrutiny in further samples. However, our cohort was selected according to the radiographic standard criterion for diagnosis of CMI, a cerebellar tonsillar descent exceeding 3 mm on midsagittal MRI. Application of this single criterion may have resulted in clinical (and presumably genetic) heterogeneity, as in fact is suggested by the PCF morphometric data we obtained in the subgroup of cases where digitized MRI studies were available. Thus, it is not surprising that statistical significance of the association was higher in the group of 186 patients with underdeveloped PCF. Among the 26 nominally associated SNPs within 13 genes, in this “classical CMI” subsample, association with P-values<0.01 were found for SNPs within six genes: ALDH1A2, RDH10, CDX1, BMPR1A, MSGN1 and FLT1. The association remained significant after a 10% FDR correction for SNPs within three genes: rs887343 in CDX1, rs17086609 in FLT1 and rs2899611 plus rs6493979 in the ALDH1A2 gene.
Regarding the potential functional relevance of the identified variants, four of the genes with the best association signals (P<0.01 in the single marker analysis of either CMI or classical CMI), ALDH1A2, RDH10, RARG and CDX1, are directly or indirectly related with retinoic acid (RA) signalling during somitogenesis. RA is a transcription factor needed for elongation of the embryonic body axis, acting as a molecular oscillator in the somitic precursors of the paraxial mesoderm [39] and promoting somite differentiation through regulation of the Hox genes [42]. Expression of RA synthetic enzyme, RALDH2, encoded by ALDH1A2, is crucial at these presomitic or early somatic stages. Avian or mouse RA-deficient embryos develop abnormally small somites (reviewed in [34]), while exposure to teratogenic RA levels give rise to congenital dysraphism in humans [65]. RDH10 encodes a retinol dehydrogenase critical for embryonic vitamin A metabolism. Murine Rdh10 mutants display lethal abnormalities characteristic of a RA-deficiency phenotype [66]. RARG binds to DNA after heterodimerization with any of the retionid X receptors, leading to activation of RA response elements (RAREs), which displace transcriptional corepressors and recruits coactivators [67]. Through this pathway, RA regulates specific Hox expression in paraxial mesoderm, determining embryonic anterior-posterior patterning throughout somitogenesis (reviewed in [34], [67]). This may origin spinal column defects such as scoliosis [68], present in nearly 20% of our CMI patients. A third RA-related gene showing association is CDX1. Cdx genes encode homeodomain transcription factors containing atypical RARE [69], [70]. In the mouse embryo primitive streak, expression of Cdx1 begins at postconceptional day 7.5 in concurrence with onset of RA signalling and Hox expression. Of note, Cdx1-null mutants display alterations in the basioccipital bone [69].
The other three listed genes, BMPR1A, MSGN1 and FLT1, belong to less interrelated pathways. BMPR1A has been found to regulate the recruitment of prospective paraxial mesoderm cells in the mouse epiblast. Mutant embryos have a shorter primitive streak, a more proximal node and a defective extension of the primitive streak [41]. MSGN1 is a direct target of Wnt and TBX6 during the specification, maturation and segmentation of the paraxial mesoderm in the mouse [38]. FLT-1 encodes VEGFR-1, a vascular endothelial growth factor receptor expressed in the endothelium of blood vessels [71]. Vasculogenesis at the chorionic villi tree is evident by post conceptional day 21, during the four-somite embryonic stage (reviewed in [72]). A FLT-1 mutation resulted in endothelial disorganization and abnormal vessel formation during early embryo development [73]. During the fourth week of life the human embryo switches from the exchange of nutrients and wastes by simple diffusion to establishment of utero-placental circulation. It is conceivable that failure to properly transition through these two stages could impair critical processes as mesodermal proliferation or neurulation.
Finally, the four variants associated with classical CMI (rs887343 in CDX1, rs17086609 in FLT1 and rs2899611 plus rs6493979 in ALDH1A2) are intronic, with no direct effect on the protein sequence. It could be that the identified risk variants may produce by themselves functional alterations or they might lie in LD with other susceptibility loci, in the case of an indirect association. Since all these SNPs are tagSNPs, we analyzed these variants and those in LD with them by means of the web utility SNP Function Prediction [74]. The predictions of functional effects for transcription factor binding sites, exonic splicing enhancers or silencers, changing of splicing pattern or efficiency by disrupting splice sites, regulation of protein translation by affecting microRNA binding sites and conservation scores were all negative.
Genetic-morphometric analysis
We also investigated whether any specific PCF morphological feature might relate to any of the gene variants showing association with CMI. A short anteroposterior diameter of the PCF and an increased basal angle have been described in some CMI morphometric studies [17], [19]. However, no correlation was found among genotypes and any of the previously considered more typical features of CMI, such as clivus or supraocciput length, PCF area or tentorial angle [15]. In our study, two SNPs in ALDH1A2 displayed a correlation with the Wackenheim angle and one of them with the basal angulation, in keeping with previous evidence pointing at basiocciput, rather than supraocciput, abnormalities in CMI. Whether an abnormal clivus slope does constitute a marker of genetically-determined CMI deserves further investigation.
Methodological issues
The present case–control association study raises several methodological questions. First, the modest sample size (404 patients and 519 controls) may have prevented from detecting subtle phenotypic effects, as the statistical power calculated for this cohort is close to 80% when an OR of 1.5 is assumed. Second, we do not present a replication study of our findings in an independent cohort. This notwithstanding, ours was a relatively large sample considering the low prevalence of CMI (1∶1000 to 1∶5000), bordering that of rare diseases. It was also the first CMI sample being explored for association with a large set of candidate genes. Third, although the SNP selection was designed to cover the 58 candidate genes on the basis of existing linkage disequilibrium patterns, gaps still exist in 12 genes due to experimental constraints, and 21% of the 384 SNPs could not be studied. Last, lack of MRI studiy of the control population may have resulted in inclusion of some asymptomatic CMI cases, although given the estimated low prevalence of tonsillar descent, this should not be expected to influence the validity of our findings
In conclusion our genetic data give support to the hypothesis that variants in genes involved in paraxial mesoderm may determine PCF size to the extent of the development of the CMI. Specifically, our results point to a putative role for defective RA signalling and fetal vasculogenesis in causing hypoplasia of the basichondrocranium. Although confirmatory studies are needed, these are among the first genetic susceptibility factors to be defined in CMI. Given the limitations of the current radiographic diagnostic criteria, understanding of the genetic underpinnings of CMI should have important clinical implications, including earlier recognition of subjects at risk of becoming symptomatic and the timely indication of the surgical reconstruction of the PCF.
Supporting Information
Description of the 384 SNPs initially selected for the genotyping assay using the VeraCode technology.
(DOCX)
Acknowledgments
We thank the Spanish Chiari Malformation associations, Asenchi and ANAC, and all the patients for their valuable contribution to this study. We thank Elizabeth Solana and Olga Mestre for assistance in recruiting the patient cohort and obtaining samples.
Funding Statement
Work supported by Instituto de Salud Carlos III, Spain,grant PI061606; Fundació La Marató TV3, Spain, grant 062710; and Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), Spain, grant 2009SGR -0078. A.U. is the recipient of a scholarship from AGAUR (Spain). C.T. was supported by the European Union (Marie Curie, PIEF-GA-2009-254930). Funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chiari H (1987) Concerning alterations in the cerebellum resulting from cerebral hydrocephalus. 1891. Pediatr Neurosci 13: 3–8. [DOI] [PubMed] [Google Scholar]
- 2. Barkovich AJ, Wippold FJ, Sherman JL, Citrin CM (1986) Significance of cerebellar tonsillar position on MR. AJNR Am J Neuroradiol 7: 795–799. [PMC free article] [PubMed] [Google Scholar]
- 3. Milhorat TH, Chou MW, Trinidad EM, Kula RW, Mandell M, et al. (1999) Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. Neurosurgery 44: 1005–1017. [DOI] [PubMed] [Google Scholar]
- 4. Tubbs RS, Beckman J, Naftel RP, Chern JJ, Wellons JC 3rd, et al. (2011) Institutional experience with 500 cases of surgically treated pediatric Chiari malformation Type I. J Neurosurg Pediatr 7: 248–256. [DOI] [PubMed] [Google Scholar]
- 5. Greenlee JD, Donovan KA, Hasan DM, Menezes AH (2002) Chiari I malformation in the very young child: the spectrum of presentations and experience in 31 children under age 6 years. Pediatrics 110: 1212–1219. [DOI] [PubMed] [Google Scholar]
- 6. Martinot A, Hue V, Leclerc F, Vallee L, Closset M, et al. (1993) Sudden death revealing Chiari type 1 malformation in two children. Intensive care medicine 19: 73–74. [DOI] [PubMed] [Google Scholar]
- 7. Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, et al. (2007) Incidental findings on brain MRI in the general population. N Engl J Med 357: 1821–1828. [DOI] [PubMed] [Google Scholar]
- 8. Morris Z, Whiteley WN, Longstreth WT Jr, Weber F, Lee YC, et al. (2009) Incidental findings on brain magnetic resonance imaging: systematic review and meta-analysis. Bmj 339: b3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Speer MC, George TM, Enterline DS, Franklin A, Wolpert CM, et al. (2000) A genetic hypothesis for Chiari I malformation with or without syringomyelia. Neurosurg Focus 8: E12. [DOI] [PubMed] [Google Scholar]
- 10. Meadows J, Kraut M, Guarnieri M, Haroun RI, Carson BS (2000) Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg 92: 920–926. [DOI] [PubMed] [Google Scholar]
- 11. Marin-Padilla M, Marin-Padilla TM (1981) Morphogenesis of experimentally induced Arnold–Chiari malformation. J Neurol Sci 50: 29–55. [DOI] [PubMed] [Google Scholar]
- 12. Barry A, Patten BM, Stewart BH (1957) Possible factors in the development of the Arnold-Chiari malformation. J Neurosurg 14: 285–301. [DOI] [PubMed] [Google Scholar]
- 13. Gardner WJ, Goodall RJ (1950) The surgical treatment of Arnold-Chiari malformation in adults; an explanation of its mechanism and importance of encephalography in diagnosis. J Neurosurg 7: 199–206. [DOI] [PubMed] [Google Scholar]
- 14. McLone DG, Knepper PA (1989) The cause of Chiari II malformation: a unified theory. Pediatr Neurosci 15: 1–12. [DOI] [PubMed] [Google Scholar]
- 15. Nishikawa M, Sakamoto H, Hakuba A, Nakanishi N, Inoue Y (1997) Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg 86: 40–47. [DOI] [PubMed] [Google Scholar]
- 16. Noudel R, Jovenin N, Eap C, Scherpereel B, Pierot L, et al. (2009) Incidence of basioccipital hypoplasia in Chiari malformation type I: comparative morphometric study of the posterior cranial fossa. Clinical article. J Neurosurg 111: 1046–1052. [DOI] [PubMed] [Google Scholar]
- 17. Aydin S, Hanimoglu H, Tanriverdi T, Yentur E, Kaynar MY (2005) Chiari type I malformations in adults: a morphometric analysis of the posterior cranial fossa. Surg Neurol 64: 237–241; discussion 241. [DOI] [PubMed] [Google Scholar]
- 18. Schady W, Metcalfe RA, Butler P (1987) The incidence of craniocervical bony anomalies in the adult Chiari malformation. J Neurol Sci 82: 193–203. [DOI] [PubMed] [Google Scholar]
- 19. Karagoz F, Izgi N, Kapijcijoglu Sencer S (2002) Morphometric measurements of the cranium in patients with Chiari type I malformation and comparison with the normal population. Acta Neurochir (Wien) 144: 165–171; discussion 171. [DOI] [PubMed] [Google Scholar]
- 20. Milhorat TH, Nishikawa M, Kula RW, Dlugacz YD (2010) Mechanisms of cerebellar tonsil herniation in patients with Chiari malformations as guide to clinical management. Acta Neurochir (Wien) 152: 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stovner LJ, Bergan U, Nilsen G, Sjaastad O (1993) Posterior cranial fossa dimensions in the Chiari I malformation: relation to pathogenesis and clinical presentation. Neuroradiology 35: 113–118. [DOI] [PubMed] [Google Scholar]
- 22. Vega A, Quintana F, Berciano J (1990) Basichondrocranium anomalies in adult Chiari type I malformation: a morphometric study. J Neurol Sci 99: 137–145. [DOI] [PubMed] [Google Scholar]
- 23. Marin-Padilla M (1991) Cephalic axial skeletal-neural dysraphic disorders: embryology and pathology. Can J Neurol Sci 18: 153–169. [DOI] [PubMed] [Google Scholar]
- 24. Schanker BD, Walcott BP, Nahed BV, Kahle KT, Li YM, et al. (2011) Familial Chiari malformation: case series. Neurosurg Focus 31: E1. [DOI] [PubMed] [Google Scholar]
- 25. Speer MC, Enterline DS, Mehltrette L, Hammock P, Judith Joseph MD, et al. (2003) Chiari Type I Malformation With or Without Syringomyelia: Prevalence and Genetics. Journal of Genetic Counseling 12: 297–311. [DOI] [PubMed] [Google Scholar]
- 26. Fujita K, Aida N, Asakura Y, Kurosawa K, Niwa T, et al. (2009) Abnormal basiocciput development in CHARGE syndrome. AJNR Am J Neuroradiol 30: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wojcik C, Volz K, Ranola M, Kitch K, Karim T, et al. (2010) Rubinstein-Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome? American journal of medical genetics Part A 152A: 479–483. [DOI] [PubMed] [Google Scholar]
- 28. Fujisawa H, Hasegawa M, Kida S, Yamashita J (2002) A novel fibroblast growth factor receptor 2 mutation in Crouzon syndrome associated with Chiari type I malformation and syringomyelia. J Neurosurg 97: 396–400. [DOI] [PubMed] [Google Scholar]
- 29. Schimmenti LA, Shim HH, Wirtschafter JD, Panzarino VA, Kashtan CE, et al. (1999) Homonucleotide expansion and contraction mutations of PAX2 and inclusion of Chiari 1 malformation as part of renal-coloboma syndrome. Hum Mutat 14: 369–376. [DOI] [PubMed] [Google Scholar]
- 30. Boyles AL, Enterline DS, Hammock PH, Siegel DG, Slifer SH, et al. (2006) Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet A 140: 2776–2785. [DOI] [PubMed] [Google Scholar]
- 31. Kibar Z, Capra V, Gros P (2007) Toward understanding the genetic basis of neural tube defects. Clinical genetics 71: 295–310. [DOI] [PubMed] [Google Scholar]
- 32. Urbizu A, Poca M, Vidal X, Rovira A, Sahuquillo J, et al. (2012) MRI-based morphometric analysis of posterior cranial fossa: a probability model for diagnosis of Chiari malformation type I. Journal of Neuroimaging In press. [DOI] [PubMed] [Google Scholar]
- 33. Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niederreither K, Dolle P (2008) Retinoic acid in development: towards an integrated view. Nat Rev Genet 9: 541–553. [DOI] [PubMed] [Google Scholar]
- 35. Bottcher RT, Niehrs C (2005) Fibroblast growth factor signaling during early vertebrate development. Endocrine reviews 26: 63–77. [DOI] [PubMed] [Google Scholar]
- 36. Shapiro R, Robinson F (1976) Embryogenesis of the human occipital bone. AJR Am J Roentgenol 126: 1063–1068. [DOI] [PubMed] [Google Scholar]
- 37. Kageyama R, Niwa Y, Shimojo H (2009) Rhythmic gene expression in somite formation and neural development. Molecules and cells 27: 497–502. [DOI] [PubMed] [Google Scholar]
- 38. Wittler L, Shin EH, Grote P, Kispert A, Beckers A, et al. (2007) Expression of Msgn1 in the presomitic mesoderm is controlled by synergism of WNT signalling and Tbx6. EMBO reports 8: 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aulehla A, Pourquie O (2009) Signaling gradients during paraxial mesoderm development. Cold Spring Harb Perspect Biol 2: a000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. William DA, Saitta B, Gibson JD, Traas J, Markov V, et al. (2007) Identification of oscillatory genes in somitogenesis from functional genomic analysis of a human mesenchymal stem cell model. Developmental biology 305: 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miura S, Davis S, Klingensmith J, Mishina Y (2006) BMP signaling in the epiblast is required for proper recruitment of the prospective paraxial mesoderm and development of the somites. Development 133: 3767–3775. [DOI] [PubMed] [Google Scholar]
- 42. Alexander T, Nolte C, Krumlauf R (2009) Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol 25: 431–456. [DOI] [PubMed] [Google Scholar]
- 43. Olsen BR, Reginato AM, Wang W (2000) Bone development. Annual review of cell and developmental biology 16: 191–220. [DOI] [PubMed] [Google Scholar]
- 44. Monsoro-Burq AH (2005) Sclerotome development and morphogenesis: when experimental embryology meets genetics. The International journal of developmental biology 49: 301–308. [DOI] [PubMed] [Google Scholar]
- 45. Tribioli C, Lufkin T (1999) The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development 126: 5699–5711. [DOI] [PubMed] [Google Scholar]
- 46. Martin JF, Bradley A, Olson EN (1995) The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes & development 9: 1237–1249. [DOI] [PubMed] [Google Scholar]
- 47. Kaufmann P, Mayhew TM, Charnock-Jones DS (2004) Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 25: 114–126. [DOI] [PubMed] [Google Scholar]
- 48. Demir R, Kayisli UA, Seval Y, Celik-Ozenci C, Korgun ET, et al. (2004) Sequential expression of VEGF and its receptors in human placental villi during very early pregnancy: differences between placental vasculogenesis and angiogenesis. Placenta 25: 560–572. [DOI] [PubMed] [Google Scholar]
- 49. Tubbs RS, Rutledge SL, Kosentka A, Bartolucci AA, Oakes WJ (2004) Chiari I malformation and neurofibromatosis type 1. Pediatric neurology 30: 278–280. [DOI] [PubMed] [Google Scholar]
- 50.Freeman AF, Davis J, Hsu AP, Holland SM, Puck JM (1993) Autosomal Dominant Hyper IgE Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA). [Google Scholar]
- 51. Gu CC, Yu K, Rao DC (2008) Characterization of LD structures and the utility of HapMap in genetic association studies. Adv Genet 60: 407–435. [DOI] [PubMed] [Google Scholar]
- 52. Thorisson GA, Smith AV, Krishnan L, Stein LD (2005) The International HapMap Project Web site. Genome Res 15: 1592–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barnes MR (2006) Navigating the HapMap. Brief Bioinform 7: 211–224. [DOI] [PubMed] [Google Scholar]
- 54. Mueller JC (2004) Linkage disequilibrium for different scales and applications. Brief Bioinform 5: 355–364. [DOI] [PubMed] [Google Scholar]
- 55. Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, et al. (2004) Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74: 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin CH, Yeakley JM, McDaniel TK, Shen R (2009) Medium- to high-throughput SNP genotyping using VeraCode microbeads. Methods Mol Biol 496: 129–142. [DOI] [PubMed] [Google Scholar]
- 57. Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38: 209–213. [DOI] [PubMed] [Google Scholar]
- 58. Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21: 263–265. [DOI] [PubMed] [Google Scholar]
- 59. Gonzalez JR, Armengol L, Sole X, Guino E, Mercader JM, et al. (2007) SNPassoc: an R package to perform whole genome association studies. Bioinformatics 23: 644–645. [DOI] [PubMed] [Google Scholar]
- 60.Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer, New York. [Google Scholar]
- 61. Storey JD (2002) A direct approach to false discovery rates. Journal of the Royal Statistical Society, Series B 64 64: 479–498. [Google Scholar]
- 62. Boyles AL, Hammock P, Speer MC (2005) Candidate gene analysis in human neural tube defects. American journal of medical genetics Part C, Seminars in medical genetics 135C: 9–23. [DOI] [PubMed] [Google Scholar]
- 63. Kibar Z, Capra V, Gros P (2007) Toward understanding the genetic basis of neural tube defects. Clin Genet 71: 295–310. [DOI] [PubMed] [Google Scholar]
- 64. Greene ND, Stanier P, Copp AJ (2009) Genetics of human neural tube defects. Human molecular genetics 18: R113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, et al. (1985) Retinoic acid embryopathy. The New England journal of medicine 313: 837–841. [DOI] [PubMed] [Google Scholar]
- 66. Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, et al. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes & development 21: 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Duester G (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell 134: 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kane WJ (1977) Scoliosis prevalence: a call for a statement of terms. Clinical orthopaedics and related research 43–46. [PubMed] [Google Scholar]
- 69. Houle M, Sylvestre JR, Lohnes D (2003) Retinoic acid regulates a subset of Cdx1 function in vivo. Development 130: 6555–6567. [DOI] [PubMed] [Google Scholar]
- 70. Houle M, Prinos P, Iulianella A, Bouchard N, Lohnes D (2000) Retinoic acid regulation of Cdx1: an indirect mechanism for retinoids and vertebral specification. Molecular and cellular biology 20: 6579–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, et al. (1992) Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochemical and biophysical research communications 187: 1579–1586. [DOI] [PubMed] [Google Scholar]
- 72. Demir R, Seval Y, Huppertz B (2007) Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem 109: 257–265. [DOI] [PubMed] [Google Scholar]
- 73. Fong GH, Rossant J, Gertsenstein M, Breitman ML (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376: 66–70. [DOI] [PubMed] [Google Scholar]
- 74. Xu Z, Taylor JA (2009) SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic acids research 37: W600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of the 384 SNPs initially selected for the genotyping assay using the VeraCode technology.
(DOCX)