

Abstract
Millions of years of coevolution between plants and pathogens can leave footprints on their genomes and genes involved on this interaction are expected to show patterns of positive selection in which novel, beneficial alleles are rapidly fixed within the population. Using information about upregulated genes in maize during Colletotrichum graminicola infection and resources available in the Phytozome database, we looked for evidence of positive selection in the Poaceae lineage, acting on protein coding sequences related with plant defense. We found six genes with evidence of positive selection and another eight with sites showing episodic selection. Some of them have already been described as evolving under positive selection, but others are reported here for the first time including genes encoding isocitrate lyase, dehydrogenases, a multidrug transporter, a protein containing a putative leucine-rich repeat and other proteins with unknown functions. Mapping positively selected residues onto the predicted 3-D structure of proteins showed that most of them are located on the surface, where proteins are in contact with other molecules. We present here a set of Poaceae genes that are likely to be involved in plant defense mechanisms and have evidence of positive selection. These genes are excellent candidates for future functional validation.
Keywords: positive selection, Poaceae, Zea mays, defense related genes, plant-pathogen interaction, molecular evolution, adaptation, resistance, disease
Introduction
Antagonistic coevolution between plants and pathogens triggers an evolutionary arms race in which the host evolves to escape pathogen infections, and pathogens evolve to escape host defenses. Genes involved in this interaction are expected to show signatures of adaptive molecular evolution through positive selection1 where advantageous mutations are retained in the population. For instance, several plant genes related to defense mechanisms have been reported to evolve under positive selection.2-4
The maize genome contains more than 32,500 protein coding genes. 22,874 of the proteins were assigned to a least one InterPro term.5 Domains associated with defense related genes (DRGs) are well represented, including 240 glycoside hydrolases, 213 pathogenesis-related transcriptional factors, and 211 with N-terminal leucine-rich repeats. Identifying those genes that play a fundamental role during pathogen attack is a difficult but important task. In a previous study, we performed suppression subtractive hybridization experiments in the maize-Colletotrichum graminicola pathosystem during early stages of anthracnose leaf blight development.6 We found more than 200 differentially expressed genes from maize, including 36 DRGs and 34 genes encoding hypothetical or unknown proteins (HUPGs) identified as upregulated during infection. Moreover, the recent release of Phytozome v8.07 offers the opportunity for fast and accurate access to families of orthologous and paralogous genes in the Poaceae lineage. Using the information available in Phytozome, we further analyzed the previously identified genes,6 to investigate genomic patterns of positive selection on HUPGs and DRGs across the Poaceae lineage.
Results and Discussion
A total of 74 clusters were identified in Phytozome across the Poaceae (grass) node, which contain the 70 upregulated maize genes (36 DRGs and 34 HUPGs). Seven sequences were classified in at least two different clusters and six out the 74 clusters contain the maize sequence as the only member. Three of them GRMZM2G040493, GRMZM2G080466 and GRMZM2G078124, encode proteins with unknown function and without any known functional domains. The remaining are genes GRMZM2G016922 (a putative kaurene synthase), GRMZM2G001084 (putative ATP-dependent Clp protease) and GRMZM2G088088 (a putative importin subunit α protein). It is interesting to highlight that sequence GRMZM2G001084 (cluster 32722009) shows high similarity (99.7%) with GRMZM2G123922 (cluster 31448528), however they are grouped into different clusters. While cluster 31448528 contains one member for each grass species, GRMZM2G001084 is the only member of cluster 32722009. All sequences in cluster 31448528 show high similarity along the full length of the protein and GRMZM2G001084 differs only in the N-terminal region, outside the highly conserved functional domain, which may be responsible for the separation into two different clusters. There is no supporting EST or protein evidence for the N-terminal region of the GRMZM2G001084 gene model so we cannot exclude the possibility that the gene is incorrectly annotated. Subcellular localization analysis using TargetP v1.18 suggests that the GRMZM2G123922 protein is targeted to the chloroplasts while that signal is absent in GRMZM2G001084. These results suggest that either GRMZM2G001084 is not correctly annotated, or that GRMZM2G001084 represents an ancient gene copy of GRMZM2G123922 that has gained a novel function after the gene duplication event.
In addition, genes GRMZM2G080466 and GRMZM2G016922, unique members of specific clusters, are upregulated during infection with both, C. graminicola and Ustilago maydis.9,10,6 These genes induced during infection with both fungal species may represent evolutionary innovations in the maize genome directly related with defense mechanisms and are very interesting candidates for future functional characterization.
The remaining 68 clusters were analyzed in order to obtain a set of orthologous genes and filtered to avoid highly divergent sequences that may cause false-positive results in the positive selection tests. A total of 43 clusters outperformed the filtering and were tested for signatures of positive selection. CODEML implemented in the PAML v4 software package11 was used to fit two kinds of models to the data: models that allow positive selection (M3 and M8) and models that do not (M0 and M7). For each model, the log likelihood (lnL) value were obtained and two Likelihood Ratio Test (LRTs) were performed (M0vsM3 and M7vsM8) as 2*(lnL1-lnL0) = 2ΔL, which were compared with a χ2 distribution to test whether ω was statistically different from one. Fourteen clusters showed statistically significant differences at any of the two LRTs, indicating that either there was heterogeneity in ω values among sites, or evidence of positive selection in the clusters. Next, we used BLAST to search for additional Poaceae orthologous sequences in public databases for each cluster and the positive selection tests were performed again.
Results and characteristics for the 14 clusters are summarized in Table 1. The comparison M0vsM3 shows that all 14 clusters have variations for dN/dS ratio (ω) among their codons, indicating that selective constraints are heterogeneous between sites as expected. A total of 6 clusters (31469106-se, 31456723-e, 31466309-se, 31477240-se, 31452004-e and 31443992-se) showed evidence for positive selection through M7vsM8 comparison. Using the Bayes empirical Bayes method implemented under M8 (M8+BEB),12 we identified several sites in the alignment for each cluster in which the approximate mean of the posterior distribution for ω was > 1.
Table 1. Results for positive selection tests and characteristics for the 14 clusters.
| |
|
|
|
M0 vs M3 |
M7 vs M8 |
MEME |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Maize Gene | Genome Annotation | Cluster | Species | ω | Δ2L | ω | Δ2L | PSS (BEB) | PSS | |
| GRMZM2G149800 |
Hypothetical protein LOC100277913 |
31469106-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, Pe |
24.09 |
245.65 |
2.59 |
21.2360 |
None |
71T, 227R, 232S, 342Y |
|
| GRMZM2G324297 |
Unknown (similar to arogenate dehydrogenase) |
31456723-e |
Zm, Si, Bd, Os, Sb, Ta, Hv |
1.10 |
470.95 |
1.30 |
17.4570 |
151Q**, 158R, 240K, 253D, 257A, 306S, 323Q, 349R, 368F |
147A, 182S, 211L, 301S, 358R |
|
| GRMZM2G453805 |
Chitinase class III |
31466309-se |
Zm, Si, Bd, Os, Sb, Ta, Pe |
6.83 |
216.60 |
7.31 |
14.2870 |
4A**, 9A, 260P, 285I* |
87G, 138G, 160S, 169A, 184L, 222G, 224A, 269I, 282I, 293V |
|
| GRMZM2G168502 |
Hypothetical protein LOC100217285 |
31477240-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, Pe |
0.83 |
222.73 |
3.85 |
10.2076 |
130V, 347S*, 356T |
32V, 99N, 120N, 144A, 184V, 203C, 206A, 235E, 293S, 296C |
|
| GRMZM2G056369 |
Putative Isocitrate lyase |
31452004-e |
Zm, Si, Bd, Os, Sb, Ta, Hv |
0.40 |
145.16 |
1.10 |
8.5392 |
30G, 76G, 334K, 345R, 371T, 560P, 561R, 564T, 572M |
142L, 293S, 320C, 330G, 331V, 344D, 345R |
|
| GRMZM2G402631 |
Pathogenesis-related protein 5 |
31443992-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, As, Pe, Sc, Or, Tm |
1.12 |
201.12 |
1.31 |
8.3213 |
57P, 91Q, 148V, 166M |
39G, 53T, 56N, 58G, 84G, 98A, 109L, 146R, 182T, 185P |
|
| GRMZM2G465226 |
Pathogenesis-related protein 1 |
31458297-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, Pe |
0.79 |
219.62 |
7.57 |
1.8310 |
None |
99S, 108D, 128A, 134V |
|
| GRMZM2G338809 |
Hypothetical protein LOC100382111 |
31445034-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, Pe |
0.33 |
272.13 |
1.63 |
0.5130 |
None |
35T, 37A, 121F, 131A, 266V, 271S, 278C, 342I, 393K, 413A |
|
| GRMZM2G011085 |
Putative uncharacterized protein |
31447249-e |
Zm, Si, Bd, Os, Sb, Hv, Pe |
0.10 |
20.92 |
1.00 |
0.0032 |
None |
95L, 160T, 195V |
|
| GRMZM2G039639 |
Protein P21 |
31461924-se |
Zm, Si, Bd, Os, Sb, Ta, Hv, Pe |
0.80 |
179.11 |
1.00 |
0.0002 |
None |
23L, 34V, 98V, 107A, 145G, 161S, 165A, 202K, 209P |
|
| GRMZM2G080499 |
Hypothetical protein |
31455526-e |
Zm, Si, Bd, Os, Sb, Hv |
0.52 |
82.64 |
1.00 |
0.0001 |
None |
67T, 90T, 93T, 161P, 400S, 452T, 803V |
|
| GRMZM2G117971 |
Hypothetical protein LOC100191593 |
31462333-se |
Zm, Si, Bd, Os, Sb, Ta, Hv |
0.45 |
133.46 |
2.57 |
0.0001 |
None |
33Q, 36G, 50N, 103T, 108S, 118S, 131K, 138Q |
|
| GRMZM2G322129 |
Putative uncharacterized protein |
31457048 |
Zm, Si, Bd, Os, Sb |
0.85 |
189.35 |
1.00 |
1.6598 |
None |
28V, 37P, 38H, 119R, 135L, 172G, 173S, 203S, 228R, 250K, 255D, 263M, 284H, 312D, 434L, 595S |
|
| GRMZM2G057093 | Chitinase | 31480783 sec | Zm, Si, Bd, Os, Sb | 0.74 | 80.97 | 4.38 | 0.0614 | None | 177T, 277S | |
Cluster: clusters are named according to Phytozome v8.0 (Grass node), adding “e” when the cluster was expanded to other Poaceae species and “s” when a subtree was extracted from original Phytozome cluster. Species: one sequence for each of the following species was included in the cluster: Zm: Zea mays, Sb: Sorghum bicolor, Si: Setaria italica, Os: Oryza sativa, Bd: Brachypodium distachyon, Ta: Triticum aestivum, Hv: Hordeum vulgare, Pe: Phyllostachys edulis, As: Aegilops speltoides, Sc: Secale cereale, Or: Oryza rufipogon, Tm: Triticum monococcum. ω = dN/dS estimated under M3 and M8. Δ2L: likelihood ratio estimated as 2*(lnL1-lnL0) between M0vsM3 and M7vsM8, in bold those are statically significant at 0.05. PSS: Positively selected sites indentified by BEB when the approximate mean of the posterior distribution for w is > 1. Clusters with posterior probability > 0.8, * > 0.95 and ** > 0.99 are in bold type. PSS (MEME) show sites identified by MEME as under episodic selection (p-value < 0.1). In both cases amino acids refer to maize sequence.
Since the M7vsM8 LRT is a very stringent test13 and lacks power to identify sites where episodes of positive selection are confined to a small subset of branches in a phylogenetic tree, we also used the Mixed Effects Model of Evolution(MEME), which is able14 to identify lineage-specific events of positive selection (episodic selection) even though the same site is neutrally or negatively evolving in the rest of the lineages. As expected, MEME showed a greater power to detect sites under positive selection, finding episodic selection in all 14 clusters.
Two of six clusters identified by LRT (M7vsM8) have already been described as evolving under positive selection in plants. One of them is cluster 31466309-se, in which four sites showed ω > 1 in the approximate mean of the posterior distribution using M8+BEB, two of them with posterior probabilities (Pr ω > 1) > 0.95 (sites 4A and 285I). Additionally, 10 sites were identified by MEME showing episodic selection. This cluster contains members of the chitinase class III, a well-known defense related protein involved in fungal cell-wall degradation, and described by Bishop et al.3 as evolving under positive selection. Furthermore, cluster 31443992-se showed 4 sites with ω > 1 using M8+BEB, two of them with (Pr ω > 1) > 0.8 (148V and 166M) and 10 sites under episodic selection identified by MEME. Three-D structure of maize protein (GRMZM2G402631_P01) was predicted based on template PDB ID: 1AUN with confidence = 100% and coverage = 86%. Sites predicted as evolving under positive selection were then mapped onto the tertiary structure (Fig. 1D), and sites under positive selection are situated on the surface of the protein, which is consistent with the results already described by Zamora et al.4 This cluster contains members of the PR5 family, thaumatin-like proteins with known antifungal and anti-insect activity.

Figure 1. Modeled 3-D structure for six maize proteins based on fold recognition using Phyre2. A) GRMZM2G149800_P02 (cluster: 31469106-se, PDB ID: 3OOX). B) GRMZM2G324297_P02 (cluster: 31456723-e, PDB ID: 3KTD). C) Tetrameric structure of isocitrate lyase from A. nidulans (PDB ID: 1DQU). Domain II highlighted in yellow. D) GRMZM2G402631_P01 (cluster: 31443992-se, PDB ID: 1AUN). E) GRMZM2G039639_P01 (cluster: 31461924-se, PDB ID: 1DU5). F) GRMZM2G117971_P01 (cluster: 31462333-se, PDB ID: 1BW3). Amino acids inferred as evolving under positive selection are colored. PSS (M8+BEB) when the approximate mean of the posterior distribution for ω is > 1 and posterior probability > 0.5 (yellow) and > 0.8 (red). PSS (MEME), p-value < 0.1 (cyan).
Among clusters without previous evidence of positive selection we found 31452004-e. This cluster contains members of isocitrate lyase enzyme, which in plants is an enzyme exclusively found in the glyoxylate cycle. Even though its specific role in defense mechanisms has not been elucidated yet, glyoxylate metabolism seems to be an important part of the defense mechanisms activated by plants during infection by pathogens.15
Sequence analysis revealed that nine sites showed ω > 1 using M8+BEB, four of them with (Pr ω > 1) > 0.8 (334K, 345R, 371T and 560P), and seven sites displayed episodic selection after analysis using MEME. The 3-D structure for protein GRMZM2G056369_P01 was modeled based on PDB template 1DQU (confidence = 100% and coverage = 90%), the crystal structure of the tetrameric isocitrate lyase (ICL) from Aspergillus nidulans. Each subunit defines two domains. Domain I is associated with the center of the tetramer and shows high similarity to triose phosphate isomerase (TIM) barrel. Domain II forms a peripheral head to the subunit.16 Most sites identified as evolving under positive selection were located between sites 293 and 371 in the protein alignment (Fig. 2), which match with residues belonging to domain II (Fig. 1C). This domain appears to be unique to eukaryotic ICLs and has been proposed to be important for the association of ICL with peroxisomes.17 Further functional studies will contribute to a better understanding of domain II and its functional relevance during plant-pathogen interactions.
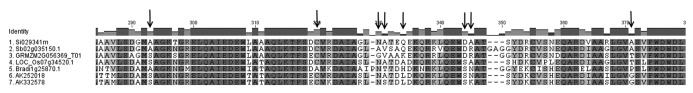
Figure 2. Multiple sequence alignment of protein cluster 31452004-e. Arrows indicate sites identified under positive selection by both methods, M8+BEB and MEME.
Clusters 31469106-se, 31456723-e and 31477240-se contain proteins annotated as hypothetical or with unknown function. In the case of 31469106-se, sites under positive selection could not be identified by M8+BEB, but MEME identified four sites with evidence of episodic selection. Even though the protein GRMZM2G149800_P02 (cluster 31469106-se) has been annotated as a hypothetical protein, its 3-D structure was modeled using as template the crystal structure of a putative 2OG-Fe(II) oxygenase (PDB ID: 3OOX, confidence = 100.0% and coverage = 84%) (Fig. 1A). In addition, using the Superfamily database18 we found that this maize protein belongs to a superfamily of clavaminate synthase-like proteins, an oxidoreductase involved in the biosynthesis of clavulanic acid and other 5S clavams.19 Clavulanic acid is a well-known antibiotic, with metabolites of the clavam family having shown antibacterial and antifungal activities.20 In the case of cluster 31456723-e, nine sites showed ω > 1 using M8+BEB, six of them with (Pr ω > 1) > 0.8. In addition, MEME identified five sites under episodic positive selection. The 3-D structure of maize protein belonging to this cluster (GRMZM2G324297_P02) was predicted based on template PDB ID: 3KTD (confidence = 100% and coverage = 65%) and sites identified as evolving under positive selection were located throughout the protein surface (Fig. 1B). Proteins in this cluster show similarities with arogenate/prephenate dehydrogenases. Members of this group of enzymes catalyze a step during tyrosine biosynthesis in the shikimate pathway, which is involved in the biosynthesis of aromatic amino acids and a wide range of secondary metabolites. Although the regulation and coordination of the synthesis of these amino acids are not well understood, many secondary metabolites have shown to be important during plant defense against herbivores, pests and pathogens.21,22 In fact, the accumulation of phenolic compounds and increased levels of hydroxycinnamic acid derivatives have been detected in maize leaves during C. graminicola infection.6 Finally, cluster 31477240-se showed three sites with ω > 1 using M8+BEB, one of them with (Pr ω > 1) > 0.95 and 10 sites with evidences of episodic selection. While annotated as hypothetical proteins, all sequences in this cluster contain two copies of a domain associated with drug/metabolite transport. Simmons et al.23 have also described a putative multidrug transporter (Zm-mfs1) upregulated in maize during Cochliobolus heterostrophus and Cochliobolus carbonum infection. Multidrug transporters play critical roles in plant defense, such as decreasing the accumulation of toxins secreted by the pathogen and exporting secondary metabolites out of the cell.24 Ten transmembrane regions were predicted using TMHMM v.0.92b25 for all sequences in this cluster and eight out of ten sites identified by MEME were found in the transmembrane regions (Fig. 3). Interestingly, substitutions located at transmembrane segments cause modifications in the activity and substrate specificity of some transporters belonging to this family.26,27 Unlike most transporters, the relationship between multidrug transporters and their substrates is not highly specific, and they have the ability to recognize and transport a wide variety of structurally different organic compounds.28 However, it is possible that the sites under positive selection might be crucial to determine the kinetic capabilities of the transporter. In this sense, rapid evolution would be acting on the selection of a more efficient transporter protein to grant a better metabolic fitness of the host cells during the pathogenic attack.
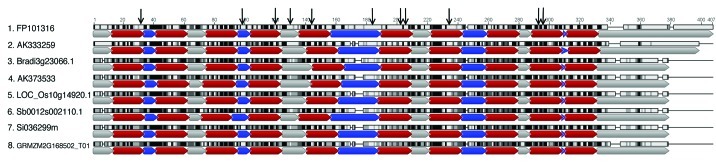
Figure 3. Multiple sequence alignment of protein cluster 31477240-se. Vertical black arrows indicate sites identified under positive selection, horizontal arrows indicate: potential transmembrane regions (red), potential cytoplasmic regions (gray) and potential extracellular regions (blue). Most sites under positive selection appear at the potential transmembrane regions.
The clusters of proteins annotated as hypothetical proteins deserve attention since they may represent genes that have not been previously described as important during plant-pathogen interactions. One of them is cluster 31462333-se, which showed eight sites under episodic selection. Maize protein (GRMZM2G117971_P01) shows high similarity with Barwin proteins and the 3-D structure was predicted based on template PDB ID: 1BW3 (confidence = 100% and coverage = 83%, Figure 1F). Barwin domains are common to pathogenicity related proteins IV (PR4), which have been described with antifungal activity against a wide variety of pathogens, such as LrPR4.29 In the case of cluster 31455526-e, seven sites showed signatures of episodic selection. Maize protein in this cluster (GRMZM2G080499_P01) is annotated as unknown function protein, but presents a serine/threonine protein kinases domain. Several serine/threonine protein kinases have shown to be involved in the recognition of pathogen-derived signal molecules30-32 and some have already been identified as evolving under positive selection in plants.3
Another cluster with several sites presenting episodic selection is cluster 31457048. A total of 16 sites were identified by MEME, the highest number of sites between all the clusters analyzed in this study. The maize protein in this cluster (GRMZM2G322129_P01) shows similarity to a transducin-like protein from sorghum. Ontologies associated with this cluster in Phytozome are mainly related to nucleotide/protein binding activities. In addition, the C-terminal region shows similarity to the armadillo (Arm) repeat, which is present in proteins acting in intracellular signaling events and in proteins with other essential functions such as cytoskeletal regulation.33 Interestingly, 15 out of 16 sites under episodic selection appear outside the armadillo domain, at the N-terminal region of the protein. Four motifs with the conserved pattern LxxLxL in the C-terminal region were also identified in all sequences in the cluster with the exemption of GRMZM2G322129_P01 (containing only three motifs). The LxxLxL pattern has been proposed by molecular modeling as being sufficient to provide the characteristic horseshoe curvature present in leucine-rich repeat (LRRs) proteins.34 Among the most important LRR proteins involved in plant defense are the nucleotide-binding site-leucine-rich repeat (NBS-LRR), pivotal players for pathogen detection. However, no NBSs were detected in any of the proteins of the cluster. Furthermore, most sites under positive selection were detected outside of the putative LRR region, where the recognition of the pathogen would be expected to occur. However, Mondragón-Palomino et al.35 have already described sites under positive selection outside of the LRR region in this family, concluding that these sites could also be important for the detection and signaling transduction during plant pathogen interactions.
We also found evidence of episodic selection in two additional clusters containing hypothetical or uncharacterized proteins. The first is cluster 31445034–se that includes proteins similar to ammonium transporters. These transporters may play important roles in plant defense against alkalinizing pathogens as Colletotrichum spp, which secrete ammonia to increase the pH of the host tissue and infect the host.36,37 The second cluster is 31447249-e, which contains proteins with similarity to the PRELI/MSF1 domain, and has three sites that have undergone episodic selection. The function of the PRELI/MSF1 domain is unknown, although it is conserved in lipid-binding proteins and proteins involved in vesicle transport and secretion mechanisms.38
Finally, we obtained three clusters of proteins with known function where MEME detected sites under episodic selection. Two of them, 31458297-se and 31480783 sec contain well-known pathogenicity related proteins as PR1 and chitinases displaying just four and two sites evolving rapidly, respectively. The third cluster is 31461924-se and showed nine sites under episodic selection. The maize protein from this cluster (GRMZM2G039639_P01) is annotated as protein P21, a family that belongs to the thaumatin-like proteins and with highly similarity to osmotins PR5. Three-D structure for GRMZM2G039639_P01 was modeled based on template PDB ID: 1DU5 (confidence = 100% and coverage = 89%), the crystal structure of zeamatin, an antifungal protein from maize with membrane-permeabilizing activity.39,40 Sites predicted as evolving under episodic selection were mapped along the whole surface of the protein (Fig. 1E), showing a similar pattern to the PR5 previously described in this work and by Zamora et al.4 Although the mechanism of action of osmotins has not been elucidated yet, it has been proposed that these proteins may target cell-wall and membranes of the fungal cell.41
We present here a total of 14 gene clusters with evidence for positive selection in the Poaceae lineage. Maize genes belonging to these clusters have been shown to be upregulated in maize during development of anthracnose caused by the hemibiotrophic fungus C. graminicola. Some of them have already been identified as evolving under positive selection while others are reported here for the first time. In general, most of the residues under positive selection detected in this study are exposed to the surface of the proteins (Fig. 1). These results are expected for proteins with important functions such as ligand-protein interactions, allosteric regulations and signal perception. Six clusters showed positive selection even using one of the most stringent tests, the LRT between models M7 and M8 implemented in CODEML. According to Phytozome v8.0, five out the 14 clusters contain a single-copy gene at each of the analyzed Poaceae species (31456723-e, 31452004-e, 31447249-e, 31455526-e and 31457048), therefore these genes are excellent candidates for further functional analysis.
Conclusions and Perspectives
We identified a set of genes that probably have been playing important roles during plant-microbe interactions for millions of years of antagonist coevolution between grasses and their pathogens. This information may aid the understanding of molecular mechanisms involved in plant defense since products of these genes may be interacting with effectors produced by pathogens or involved in metabolic pathways that are important for defense. Accordingly, these genes represent a set of important candidates for functional validation through biochemical and genetic studies in order to identify targets to take into account in plant breeding programs as well as in pursuit of environmentally friendly plant protection compounds.
Materials and Methods
Tests for positive selection
Figure 4 shows the workflow designed to analyze positive selection in the 36 DRGs and the 34 HUPGs. It consists of three main steps: Step 1) Identification of orthologs and paralogs. Using a custom Python script we systematically identified Phytozome gene clusters in the Poaceae (Grass) node looking for all clusters containing at least one of the 70 genes IDs under study. In some cases, maize genes appear in more than one cluster, so all of them were taken into account for the analysis. Grass gene clusters in Phytozome v8.0 contain sequences from Zea mays (Zm), Sorghum bicolor (Sb), Setaria italica (Si), Oryza sativa (Os) and Brachypodium distachyon (Bd). Gene families with more than 1 sequence for each species were manually curated by analyzing the phylogenetic tree and selecting those sequences clustered together in a subtree resembling the species phylogeny, an indication of orthology.4 We additionally analyzed gene clusters containing more than 4 sequences of maize in order to look patterns of positive selection in groups of paralogs. Step 2) Remove highly divergent gene clusters. We first aligned protein clusters using MUSCLE v3.842 with the default options and then filtered the multiple sequence alignments (MSA) to discard clusters with highly divergent sequences. MSAs were retained for further analysis if the average pairwise identity within the cluster was greater than 70%, the percentage of gapped residues was less than 25%35 and the percentage of sites conserved by Gblocks (using the default parameters)43 was greater than 70%. Clusters that did not meet these requirements were discarded (as in the case of both paralogous sets) and the ones that did it were then tested for positive selection. Step 3) Positive selection tests. CODEML implemented in the PAML v4 software package11 was used to fit two kinds of models to the data, models that allow positive selection (M3 and M8) and models that do not (M0 and M7). For each model, the log likelihood (lnL) values were obtained and two likelihood ratio tests (LRTs) were performed (M0vsM3 and M7vsM8) as 2*(lnL1-lnL0) = 2ΔL, which was compared with a χ2 distribution to test whether ω was statistically different from one (critical value 5.99 at 5% significance level).

Figure 4. Flowchart of methods used to identify families of defense-related genes (DRGs) and hypothetical or unknown protein-coding genes (HUPGs) under positive selection in the Poaceae lineage.
Clusters showing statistically significant differences at any of the two LRTs were “expanded” by adding more orthologous sequences from other Poaceae species and then conducting the positive selection tests again. For each of the 14 clusters, we used the protein sequence from maize as query in TBLASTN searches against the “nr” and “est” DNA databases and filtering results for Poaceae species. An e-value < 10−20, and a coverage > 90% of the complete ORF were required to be considered as putative orthologs. The new “expanded” orthologous clusters were then tested again using CODEML. When the LRT indicated positive selection, the Bayes empirical Bayes (BEB) method was used to calculate the posterior probabilities that each codon is from the site class of positive selection under model M8.12
We used the MEME algorithm,14 part of the HyPhy package44 implemented in the Datamonkey webserver,45 to test for evidence of episodic selection. The LRT is reliable even if only a few similar sequences are analyzed,13 but the power of prediction of positive selection sites by M8+BEB is low when few closely related sequences are used.46 Sizes of clusters analyzed here were between 5 and 12 members and the relative sequence divergence was between 0.11 (31452004-e) and 0.56 (31456723-e) nucleotide changes per codon per branch (estimated as S/(2T-3) according to Anisimova et al.,13 so the low numbers of taxa as well as the low sequence divergence may prevent the identification of sites under positive selection by M8+BEB. In addition, the M8+BEB method may fail to recognize sites where selection is episodic. The MEME algorithm pools information over branches in the phylogenetic tree to gain power to detect episodic selection at a site, reducing the stringency of the analysis. Sites indicated as evolving under positive selection by MEME and M8+BEB were visually inspected, and only the ones that do not appear at low-quality alignment regions were reported.
3-D modeling
When possible, protein 3-D structure models were built based on fold recognition using the Protein Homology/analogY Recognition Engine v.2.0 (Phyre247) and the 3-D representation of molecular structures was obtained using Geneious v5.4.48 Amino acids predicted as evolving under positive selection where then mapped into the protein structure.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was supported by funds from the Ministerio de Ciencia e Innovación (MICINN) of Spain (grants AGL2008–03177 and AGL2011–29446) and the Junta de Castilla y León, Spain (grant SA-134A08). G.E.R was supported by a Formación de Personal Investigador graduate fellowship (BES–2009–013920), W.A.V was supported by a fellowship from the Juan de la Cierva Program (JCI–2009–05364) and M.R.T by the Ramón y Cajal Program (RYC-2006–001381).
Footnotes
Previously published online: www.landesbioscience.com/journals/psb/article/22362
References
- 1.Aguileta G, Refrégier G, Yockteng R, Fournier E, Giraud T. Rapidly evolving genes in pathogens: methods for detecting positive selection and examples among fungi, bacteria, viruses and protists. Infect Genet Evol. 2009;9:656–70. doi: 10.1016/j.meegid.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Bishop JG, Dean AM, Mitchell-Olds T. Rapid evolution in plant chitinases: molecular targets of selection in plant-pathogen coevolution. Proc Natl Acad Sci U S A. 2000;97:5322–7. doi: 10.1073/pnas.97.10.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth C, Liberles DA. A systematic search for positive selection in higher plants (Embryophytes) BMC Plant Biol. 2006;6:12. doi: 10.1186/1471-2229-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamora A, Sun Q, Hamblin MT, Aquadro CF, Kresovich S. Positively selected disease response orthologous gene sets in the cereals identified using Sorghum bicolor L. Moench expression profiles and comparative genomics. Mol Biol Evol. 2009;26:2015–30. doi: 10.1093/molbev/msp114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 6.Vargas WA, Martín JM, Rech GE, Rivera LP, Benito EP, Díaz-Mínguez JM, et al. Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotricum graminicola in maize. Plant Physiol. 2012;158:1342–58. doi: 10.1104/pp.111.190397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–86. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 9.Doehlemann G, Wahl R, Horst RJ, Voll LM, Usadel B, Poree F, et al. Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J. 2008;56:181–95. doi: 10.1111/j.1365-313X.2008.03590.x. [DOI] [PubMed] [Google Scholar]
- 10.Horst RJ, Doehlemann G, Wahl R, Hofmann J, Schmiedl A, Kahmann R, et al. Ustilago maydis infection strongly alters organic nitrogen allocation in maize and stimulates productivity of systemic source leaves. Plant Physiol. 2010;152:293–308. doi: 10.1104/pp.109.147702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 13.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–92. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- 14.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheideler M, Schlaich NL, Fellenberg K, Beissbarth T, Hauser NC, Vingron M, et al. Monitoring the switch from housekeeping to pathogen defense metabolism in Arabidopsis thaliana using cDNA arrays. J Biol Chem. 2002;277:10555–61. doi: 10.1074/jbc.M104863200. [DOI] [PubMed] [Google Scholar]
- 16.Britton K, Langridge S, Baker PJ, Weeradechapon K, Sedelnikova SE, De Lucas JR, et al. The crystal structure and active site location of isocitrate lyase from the fungus Aspergillus nidulans. Structure. 2000;8:349–62. doi: 10.1016/S0969-2126(00)00117-9. [DOI] [PubMed] [Google Scholar]
- 17.Dunn MF, Ramírez-Trujillo JA, Hernández-Lucas I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology. 2009;155:3166–75. doi: 10.1099/mic.0.030858-0. [DOI] [PubMed] [Google Scholar]
- 18.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–19. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 19.Tahlan K, Park HU, Wong A, Beatty PH, Jensen SE. Two sets of paralogous genes encode the enzymes involved in the early stages of clavulanic acid and clavam metabolite biosynthesis in Streptomyces clavuligerus. Antimicrob Agents Chemother. 2004;48:930–9. doi: 10.1128/AAC.48.3.930-939.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paradkar AS, Jensen SE, Mosher RH. Biotechnology of antibiotics, 2nd ed., revised and expanded. New York N.Y.: Informa Healthcare, 1997:241–277. [Google Scholar]
- 21.Bennett RN, Wallsgrove RM, Bennett RN, Wallsgrove RM. Secondary metabolites in plant defence mechanisms. New Phytol. 1994;127:617–33. doi: 10.1111/j.1469-8137.1994.tb02968.x. [DOI] [PubMed] [Google Scholar]
- 22.Tzin V, Galili G. New insights into the shikimate and aromatic amino acids biosynthesis pathways in plants. Mol Plant. 2010;3:956–72. doi: 10.1093/mp/ssq048. [DOI] [PubMed] [Google Scholar]
- 23.Simmons CR, Fridlender M, Navarro PA, Yalpani N. A maize defense-inducible gene is a major facilitator superfamily member related to bacterial multidrug resistance efflux antiporters. Plant Mol Biol. 2003;52:433–46. doi: 10.1023/A:1023982704901. [DOI] [PubMed] [Google Scholar]
- 24.Peng H, Han S, Luo M, Gao J, Liu X, Zhao M. Roles of multidrug transporters of MFS in plant stress responses. IJBBB. 2011;1:109–13. [Google Scholar]
- 25.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 26.Egner R, Bauer BE, Kuchler K. The transmembrane domain 10 of the yeast Pdr5p ABC antifungal efflux pump determines both substrate specificity and inhibitor susceptibility. Mol Microbiol. 2000;35:1255–63. doi: 10.1046/j.1365-2958.2000.01798.x. [DOI] [PubMed] [Google Scholar]
- 27.Loo TW, Clarke DM. Mutations to amino acids located in predicted transmembrane segment 6 (TM6) modulate the activity and substrate specificity of human P-glycoprotein. Biochemistry. 1994;33:14049–57. doi: 10.1021/bi00251a013. [DOI] [PubMed] [Google Scholar]
- 28.Conte SS, Lloyd AM. Exploring multiple drug and herbicide resistance in plants--spotlight on transporter proteins. Plant Sci. 2011;180:196–203. doi: 10.1016/j.plantsci.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Xia B, Jiang Y, Wu Q, Wang C, He L, et al. A new pathogenesis-related protein, LrPR4, from Lycoris radiata, and its antifungal activity against Magnaporthe grisea. Mol Biol Rep. 2010;37:995–1001. doi: 10.1007/s11033-009-9783-0. [DOI] [PubMed] [Google Scholar]
- 30.Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, et al. Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science. 1993;262:1432–6. doi: 10.1126/science.7902614. [DOI] [PubMed] [Google Scholar]
- 31.Romeis T. Protein kinases in the plant defence response. Curr Opin Plant Biol. 2001;4:407–14. doi: 10.1016/S1369-5266(00)00193-X. [DOI] [PubMed] [Google Scholar]
- 32.Song W-Y, Wang G-L, Chen L-L, Kim H-S, Pi L-Y, Holsten T, et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995;270:1804–6. doi: 10.1126/science.270.5243.1804. [DOI] [PubMed] [Google Scholar]
- 33.Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 2003;13:463–71. doi: 10.1016/S0962-8924(03)00167-3. [DOI] [PubMed] [Google Scholar]
- 34.Kajava AV, Kobe B. Assessment of the ability to model proteins with leucine-rich repeats in light of the latest structural information. Protein Sci. 2002;11:1082–90. doi: 10.1110/ps.4010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mondragón-Palomino M, Meyers BC, Michelmore RW, Gaut BS. Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res. 2002;12:1305–15. doi: 10.1101/gr.159402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prusky D, McEvoy JL, Leverentz B, Conway WS. Local modulation of host pH by Colletotrichum species as a mechanism to increase virulence. Mol Plant Microbe Interact. 2001;14:1105–13. doi: 10.1094/MPMI.2001.14.9.1105. [DOI] [PubMed] [Google Scholar]
- 37.Miyara I, Shnaiderman C, Meng X, Vargas WA, Díaz-Minguez JM, Sherman A, et al. Role of nitrogen-metabolism genes expressed during pathogenicity of the alkalinizing Colletotrichum gloeosporioides and their differential expression in acidifying pathogens. Mol Plant Microbe Interact. 2012;25:1251–63. doi: 10.1094/MPMI-01-12-0017-R. [DOI] [PubMed] [Google Scholar]
- 38.Anantharaman V, Aravind L. The GOLD domain, a novel protein module involved in Golgi function and secretion. Genome Biol 2002; 3:research0023.1–research0023.7. [DOI] [PMC free article] [PubMed]
- 39.Batalia MA, Monzingo AF, Ernst S, Roberts W, Robertus JD. The crystal structure of the antifungal protein zeamatin, a member of the thaumatin-like, PR-5 protein family. Nat Struct Biol. 1996;3:19–23. doi: 10.1038/nsb0196-19. [DOI] [PubMed] [Google Scholar]
- 40.Roberts WK, Selitrennikoff CP. Zeamatin, an antifungal protein from maize with membrane-permeabilizing activity. J Gen Microbiol. 1990;136:1771–8. doi: 10.1099/00221287-136-9-1771. [DOI] [Google Scholar]
- 41.Anžlovar S, Dermastia M. The comparative analysis of osmotins and osmotin‐like PR‐5 proteins. Plant Biol. 2003;5:116–24. doi: 10.1055/s-2003-40723. [DOI] [Google Scholar]
- 42.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 44.Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 45.Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–7. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19:950–8. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- 47.Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 48.Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, et al.. Geneious v5.4. 2011.