

Abstract
The noncanonical NF-κB pathway forms a major arm of NF-κB signaling that mediates important biological functions, including lymphoid organogenesis, B lymphocyte function, and cell growth and survival1-3. Activation of the noncanonical NF-κB pathway involves degradation of an inhibitory protein, TNF receptor associated factor 3 (TRAF3), but how this signaling event is controlled is still unknown1,2. Here we have identified the deubiquitinase Otud7b as a pivotal regulator of the noncanonical NF-κB pathway. Otud7b deficiency in mice has no appreciable effect on canonical NF-κB activation but causes hyper-activation of noncanonical NF-κB. In response to noncanonical NF-κB stimuli, Otud7b binds and deubiquitinates TRAF3, thereby inhibiting TRAF3 proteolysis and preventing aberrant noncanonical NF-κB activation. Consequently, the Otud7b deficiency results in B-cell hyperresponsiveness to antigens, lymphoid follicular hyperplasia in the intestinal mucosa, and elevated host-defense ability against an intestinal bacterial pathogen, Citrobacter rodentium. These findings establish Otud7b as a crucial regulator of signal-induced noncanonical NF-κB activation and suggest a mechanism of immune regulation that involves Otud7b-mediated deubiquitination and stabilization of TRAF3.
Keywords: NF-κB, noncanonical NF-κB, Otud7b, TRAF3, ubiquitination, lymphoid follicular hyperplasia
Unlike the canonical NF-κB pathway, which depends on degradation of IκBα, the noncanonical NF-κB pathway depends on the inducible processing of p100, a process that not only generates p52 but also leads to nuclear translocation of the p52/RelB dimer2. The processing of p100 is triggered through its phosphorylation by the NF-κB-inducing kinase (NIK) together with a downstream kinase, IκB kinase alpha (IKKα)4,5. The steady-state noncanonical NF-κB activity is tightly controlled by TRAF3, which constantly targeting NIK to the TRAF2-cIAPs E3 complex for degradation6-8. Activation of noncanonical NF-κB involves signal-induced TRAF3 proteolysis and NIK accumulation, but how this event is regulated is unknown6. The deubiquitinases (DUBs) CYLD and A20 are vital regulators of the canonical NF-κB pathway, but they are not involved in the regulation of noncanonical NF-κB signaling9,10. Another DUB, Otud7b (also called Cezanne), was identified based on its sequence homology with A20 within their ovarian tumor (OTU) domain11. We studied the function of Otud7b using Otud7b knockout (KO) mice (Supplementary Fig. 1, 2). In contrast to the postnatal lethality of A20-deficient mice12, the Otud7b-KO mice did not show obvious defects in survival, although they had moderately reduced bodyweight (Supplementary Fig. 3). The development of B and T cells appeared to be also normal in the Otud7b-KO mice (Supplementary Fig. 4). These phenotypes suggest fundamental differences between Otud7b and A20 in signaling functions.
The Otud7b deficiency had no appreciable effect on canonical NF-κB activation in mouse embryonic fibroblasts (MEFs), bone marrow-derived macrophages (BMDM), or B cells (Supplementary Fig. 5). Although Otud7b has a related molecule, Otud7a, Otud7a was detected mainly in the central nervous system (Supplementary Fig. 6), arguing against the possibility of functional redundancies. We next examined the role of Otud7b in regulating NF-κB activation by three well-characterized noncanonical NF-κB-stimulating receptors: lymphotoxin-beta receptor (LTβR), CD40, and BAFF receptor (BAFFR)1,2. The Otud7b deficiency greatly enhanced the NF-κB activation by agonistic anti-LTβR and anti-CD40 antibodies or BAFF (Fig. 1a-c), a result that was not due to differential expression of the receptors (Supplementary Fig. 7). The heightened NF-κB DNA-binding activity in Otud7b-KO cells was associated with a marked increase in nuclear p52 and RelB and concomitant loss of cytoplasmic p100 (Fig. 1d-f). On the other hand, the Otud7b deficiency did not promote the activation of canonical IKK or MAP kinases (Supplementary Fig. 8), suggesting a specific role for Otud7b in noncanonical NF-κB regulation. We also found that Otud7b expression was induced by the noncanonical NF-κB signals (Supplementary Fig. 9), indicating a negative-feedback function.
Figure 1. Otud7b negatively regulates the noncanonical NF-κB pathway.
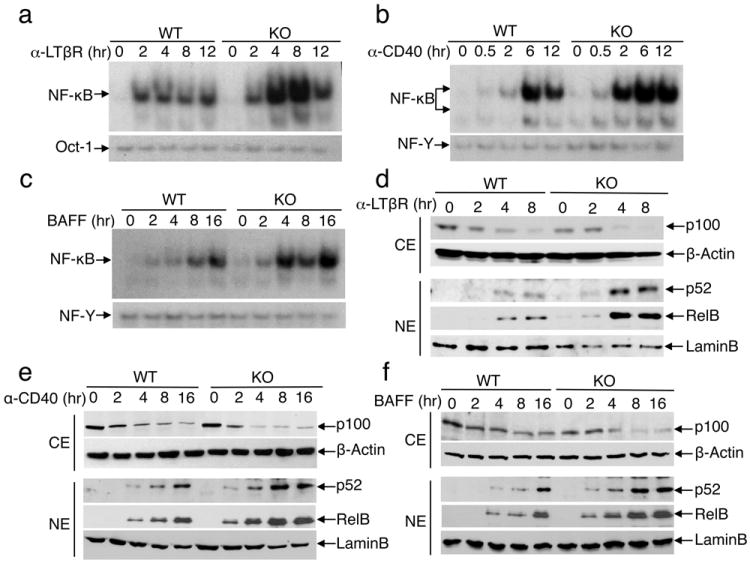
a-c, EMSA of NF-κB and control DNA-binding factors (Oct-1, NF-Y) using nuclear extracts isolated from WT and Otud7b-KO primary MEFs (a) or B cells (b, c) stimulated with the indicated inducers. d-f, Immunoblot (IB) assays using cytoplasmic (CE) and nuclear (NE) extracts of WT and Otud7b-KO primary MEFs (d) or B cells (e, f) that were stimulated as indicated. Data are representative of three independent experiments.
Compared with WT cells, the Otud7b-KO cells displayed a markedly higher level of TRAF3 degradation and NIK accumulation in response to the noncanonical NF-κB inducers (Fig. 2a,b and Supplementary Fig. 10). This effect was specific for the noncanonical NF-kB pathway, since the Otud7b deficiency did not alter the fate or function of TRAF3 in the type I interferon (IFN-I) pathway (Supplementary Fig. 11, 12)13,14. The loss of Otud7b promoted anti-CD40-stimulated TRAF3 ubiquitination (Fig. 2c). Although the recombinant OTU domain of Otud7b preferentially hydrolyzes K11-linked ubiquitin chains in vitro15, we did not detect appreciable levels of K11 ubiquitination of TRAF3 (Supplementary Fig. 13), which might be due to the quality of the anti-K11 ubiquitin antibody or the dominant K48 ubiquitination of TRAF37. On the other hand, the K48 ubiquitination of TRAF3 was markedly enhanced in the Otud7b-deficient cells (Fig. 2c). Since Otud7b also displays K48-specific DUB function in transfected cells16, it is conceivable that the specificity of Otud7b may be regulated by cofactors in vivo, as implicated for A2010. The Otud7b-mediated TRAF3 deubiquitination required its DUB activity since a catalytically inactive Otud7b mutant, C194S/H358R (CH), failed to deubiquitinate TRAF3 (Fig. 2d,e). Consistently, the WT Otud7b, but not Otud7b CH, suppressed the activation of noncanonical NF-κB signaling (Fig. 2f).
Figure 2. Otud7b negatively regulates TRAF3 degradation by affecting TRAF3 ubiquitination.
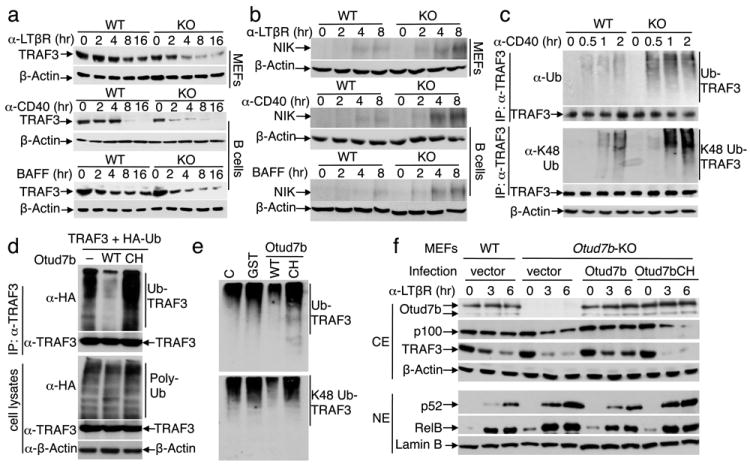
a, b, IB analyses of TRAF3 and NIK in WT and Otud7b-KO MEFs or B cells, stimulated as indicated. c, Endogenous TRAF3 was isolated by IP, under denaturing conditions, from the WT or Otud7b-KO B cells treated with anti-CD40 plus MG132. Total and K48-ubiquitination of TRAF3 were detected by IB using anti-ubiquitin and anti-K48-ubiquitin antibodies, respectively. d, HEK293 cells were transfected with the indicated expression vectors and subjected to TRAF3 ubiquitination (upper) and protein expression (lower) assays. e, TRAF3 was transfected with HA-ubiquitin or K48 ubiquitin in HEK293 cells. Purified TRAF3-ubiquitin conjugates were incubated with buffer control (C), GST, GST-Otud7b WT, or GST-Otud7bCH recombinant proteins followed by anti-HA IB. f, IB analysis of subcellular extracts from WT or Otud7b-KO MEFs, stably infected with vector, Otud7b, or Otud7bCH and stimulated as indicated. Data are representative of at least three independent experiments.
In response to CD40 or LTβR stimulation, Otud7b was rapidly recruited to TRAF3 (Fig. 3a), and this molecular interaction was coupled with the receptor recruitment of Otud7b along with TRAF3, TRAF2, and c-IAP2 (Fig. 3b,c). This finding was intriguing, since TRAF3 ubiquitination is mediated by c-IAP following their recruitment into the stimulating receptors2,7. While the Otud7b deficiency did not affect the recruitment of the TRAFs and c-IAP (Fig. 3c), TRAF3 knockdown blocked the receptor recruitment of Otud7b (Supplementary Fig. 14). The ubiquitin-association (UBA) domain, but not the zinc finger (ZF) domain, of Otud7b was required for its association with TRAF3 and receptor recruitment (Fig. 3d). The UBA domain of Otud7b was also indispensable for negatively regulating noncanonical NF-κB signaling (Supplementary Fig. 15). These findings indicate that Otud7b engages TRAF3 in response to cellular stimuli and, thereby, is recruited to the receptor complex, where it may mediate TRAF3 deubiquitination.
Figure 3. Otud7b inducibly interacts with TRAF3 and is recruited to the receptor complexes.
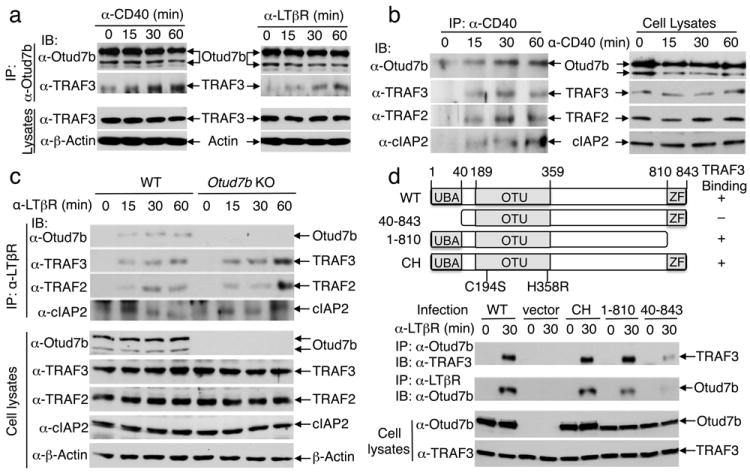
a, M12 B cells (left) and WT MEFs (right) were stimulated and subjected to Otud7b/TRAF3 co-IP (upper) and direct IB (lower) assays. b, M12-hCD40 cells were stimulated with anti-human CD40. CD40 was isolated by IP, and its associated proteins identified by IB (left panel). Cell lysates were subjected to direct IB (right panel). c, WT and Otud7b-KO MEFs were stimulated and subjected to identification of LTβR-associated proteins (upper) and analysis of protein expression (lower). d, Otud7b-KO MEFs, stably infected with Otud7b WT or the indicated Otud7b mutants, were stimulated with anti-LTβR and subjected to co-IP assays to detect Otud7b-TRAF3 interaction (panel 1) and Otud7b recruitment to LTβR (panel 2) or direct IB assays (bottom two panels). Data are representative of two to three independent experiments.
Our finding that Otud7b controls signal-induced, but not steady-state, activation of noncanonical NF-κB suggests specific roles for this DUB in the regulation of immune functions. The Otud7b-KO mice did not show obvious structural abnormalities of secondary lymphoid organs or the thymus, although the medullary thymic epithelial cells of the Otud7b-KO mice had elevated expression of the autoimmune regulator (AIRE) and several other related genes, indicating a higher level of maturation (data not shown). On the other hand, the Otud7b-KO mice displayed a remarkable phenotype in the mucosal tertiary lymphoid structures, characterized by a marked increase in the size and number of B cell-containing colonic patches (CLPs) (Fig. 4a and Supplementary Fig. 16). Consistently, the Otud7b-KO mice had a significant increase in fecal IgA concentration compared to the WT mice (Fig. 4b). Intestine is an organ that maintains dynamic host-microbiota homeostasis17, and unlike the secondary lymphoid organs, whose development is programmed in the fetus, the formation and maintenance of intestinal tertiary lymphoid tissues occur in adults and involve microbial triggers and LTβR signaling18-21. Indeed, injection of Otud7b-KO mice with a LTβR fusion protein (LTβR-Ig), known to block the interaction of LTβR with its ligands18, suppressed the lymphoid hyperplasia (Fig. 4c and Supplementary Fig. 17). Consistent with the involvement of chemokines in LTβR-mediated induction of lymphoid follicles, the intestine of Otud7b-KO mice had elevated expression of two major chemokines, CXCL12 and CXCL13, and the cell adhesion molecule MAdCAM1, which was suppressed upon LTβR-Ig injection (Fig. 4d). The LTβR-mediated induction of these genes was also enhanced in the Otud7b-KO MEFs (Fig. 4e).
Figure 4. Otud7b negatively regulates intestinal lymphoid homeostasis, anti-bacterial immunity, and B-cell responses.
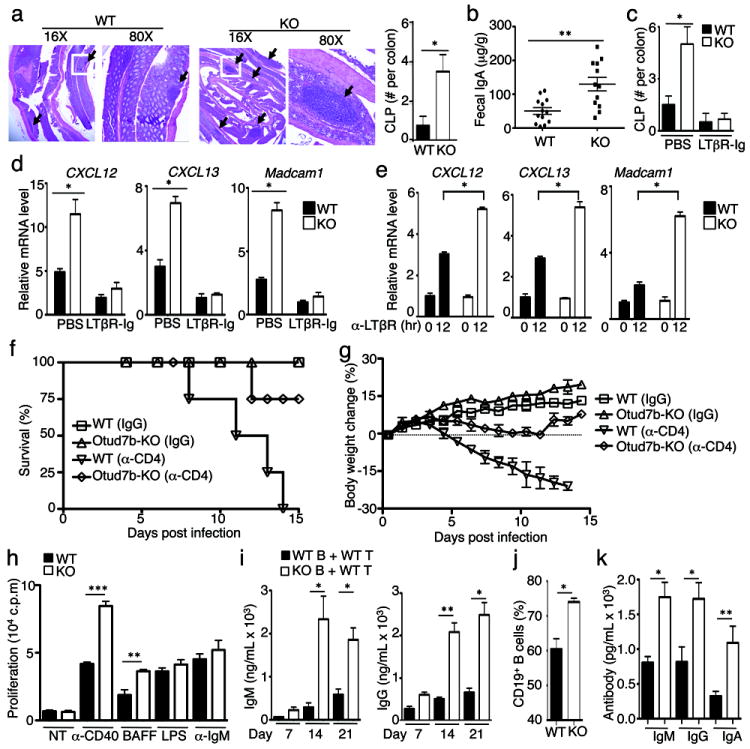
a, H&E picture (left) and summary graph (right) of WT and Otud7b-KO colon (n=4). Black arrows point to CLPs, and the squared area is enlarged in the 80X pictures. b, ELISA of IgA in the feces of WT and Otud7b-KO mice (μg IgA per gram of feces, n=13). c, Summary of CLP numbers in WT and Otud7b-KO mice (n=4) treated with PBS or LTβR-Ig. d, e, QPCR assays using RNAs prepared from colonic tissues (d) and anti-LTβR-stimulated MEFs (e). f, g, Survival (f) and body weight (g) of C. rodentium-infected WT or Otud7b-KO mice that were also injected with a control IgG or an anti-CD4 antibody on day 0, 5, and 10 (n=4). h, Proliferation assays of splenic B cells cultured without (NT) or with the indicated inducers in vitro. In vitro proliferation assays of splenic B cells. i, ELISA of serum NP-specific antibodies in NP-KLH-immunized Rag1-KO mice adoptively transferred with WT or Otud7b-KO B cells plus WT T cells. j, k, Mice were injected with BAFF (30mg/kg) i.v. every other day for 2 weeks and sacrificed on day 15 for flow cytometric analysis of B-cell frequency in splenocytes (j) and ELISA of the serum antibodies (k) (n=4). Data are representative of two-three independent experiments. Bar graphs are presented as mean±S.D. values. *p<0.05; **p<0.01; ***p<0.001.
The results described above prompted us to examine whether Otud7b negatively regulates mucosal immunity against infections. Indeed, the Otud7b-KO mice were considerably more resistant to a well-defined intestinal bacterial pathogen, C. rodentium22, as shown by the significantly reduced bacterial load in organs and feces and the ameliorated crypt hyperplasia in the colon (Supplementary Fig. 18). In order to compare the C. rodentium-induced lethality in the WT and Otud7b-KO mice, we took advantage of the recent finding that depletion of CD4 T cells in WT mice renders the mice sensitive to C. rodentium-induced lethality (Fu YX et al, unpublished data). Under these conditions, all of the WT mice died of C. rodentium infection by day 14, whereas 75% of the KO mice survived the infection (Fig. 4f). Consistently, the Otud7b-KO mice also experienced much milder bodyweight loss and had a significantly reduced fecal bacterial load compared to the WT mice (Fig. 4g and Supplementary Fig. 19). Collectively, these findings suggest the intriguing possibility that inhibition of Otud7b may enhance mucosal immunity against C. rodentium infection.
Deregulated noncanonical NF-κB activation in B cells, as seen in the B cell-conditional TRAF3-KO mice, is associated with B-cell hyperplasia and aberrant antibody production23. Consistently, the Otud7b-deficient B cells displayed elevated proliferative and survival ability when stimulated in vitro (Fig. 4h and Supplementary Fig. 20) and were hyperresponsive to antigen-stimulated antibody production when adoptively transferred into Rag1-KO mice along with WT T cells (Fig. 4i). We found that the Otud7b-KO mice did not show abnormal B-cell homeostasis under non-transferred conditions (Supplementary Fig. 4). This was likely due to attenuated activation of the Otud7b-deficient T cells (data not shown), a phenotype that has also been observed with the TRAF3-deficient T cells24,25. Interestingly, upon injection with low doses of recombinant BAFF, the Otud7b-KO mice displayed overt B-cell hyperplasia (Fig. 4j), coupled with accumulation of various antibody isotypes (Fig. 4k), thus further suggesting a negative role for Otud7b in regulating B-cell responses.
In conclusion, our findings establish Otud7b as a pivotal negative regulator of the noncanonical NF-κB pathway. Although another DUB, DUBA, has been implicated in TRAF3 deubiquitination, DUBA regulates the K63 ubiquitination and non-degradative function of TRAF3 in the PPR-induced IFN-I signaling pathway26. To date, Otud7b is the only DUB known to regulate TRAF3 degradation and noncanonical NF-κB signaling. Otud7b is unique in that it specifically regulates the signal-induced noncanonical NF-κB activation. These unique functions of Otud7b make it an attractive candidate to be exploited as a therapeutic target to boost mucosal immunity and treat diseases associated with the noncanonical NF-κB pathway.
METHODS
Mice
Otud7b-KO mice (in C57BL/6×129/sv genetic background) were generated by a conventional gene-targeting approach (Deltagen, Inc) (Supplementary Fig. 1) and backcrossed for 4 generations to the C57BL/6 background. Otud7b+/− heterozygous mice were bred to generate age-matched WT (+/+) and KO (−/−) experimental mice. Mice were maintained in specific pathogen-free facility, and all animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
Plasmids, antibodies, and reagents
HA-tagged Otud7b (pHR6-Cezanne) was provided by Dr. Paul Evans28, and Otud7b C194S/H358R (Otud7bCH), harboring point mutations in two conserved residues of the catalytic domain, was created site-directed mutagenesis (Agilent Technologies). Otud7b WT, Otud7bCH and Otud7b 1-810 and Otud7b 40-843 were cloned into pCLXSN(GFP) vector29 for MEF infections. Otud7b and Otud7bCH were cloned into pGEX-4T-1 bacterial expression vector for producing glutathione-S-transferase (GST) fusion proteins in E.coli. The HA-tagged TRAF3 and ubiquitin vectors have been described previously6, and the HA-ubiquitin K48 (where all of the lysines, except for lysine 48, were substituted with alanines) was provided by Dr. Zhijian Chen.
Murine LTβR antibody (ACH6) was provided by Dr. Jeffrey Browning (Biogen). Anti-c-IAP2 (H-85) and anti-ubiquitin (P4D1) were from Santa Cruz Biotech, anti-K48 ubiquitin and anti-K11 ubiquitin were from Millipore, and anti-Otud7b was from Proteintech. Other antibodies for cell stimulation, IB, and flow cytometry were as described6,27. Recombinant LTβR-Ig was described previously30. 5’ppp-dsRNA was from Invivogen, and other agents were as described27.
Cell culture and stimulation
HEK-293 cell culture and transfection, splenic B-cell purification, and the generation of BMDM and MEFs were as described27. Thymic stromal cells were isolated as reported 31. For B-cell proliferation assays, the cells were stimulated with anti-murine CD40 (500 ng/ml), BAFF (200 ng/ml), LPS (100 ng/ml) or anti-mouse IgM (10 μg/ml) for 40 hours and then pulsed for 8 hours with 3H-thymidine. Primary MEFs were immortalized by infection with a retroviral vector encoding the adenoviral E1A proto-protein (pCL-E1A). For Otud7b reconstitution, the immortalized MEFs were infected with pCLXSN(GFP) encoding Otud7b or its mutants and screened based on GFP expression.
For gene induction and signaling studies, BMDMs were stimulated with LPS (100 ng/ml), poly I:C (10 μg/ml), R848 (2.5 μg/ml), or CpG ODN1668 (1μg/ml), B cells were stimulated with anti-murine CD40 (500 ng/ml), BAFF (200-400 ng/ml), LPS (5 μg/ml), or anti-IgM (10 μg/ml), and MEFs were stimulated with anti-LTβR (500 ng/ml), LPS (1μg/ml), TNF-α (50 ng/ml), IL-1β (10 ng/ml), or lipofectamine-transfected poly I:C (2.5μg /ml) or dsRNA (1μg/ml) (for stimulation of RIG-I).
Receptor recruitment assays
The recruitment of Otud7b and other signaling molecules to CD40 was detected as previously described32. Briefly, M12 cells stably expressing human CD40 (M12-hCD40) (2×107) were stimulated with anti-hCD40 (in 1 ml of growth medium) and lysed in 600 μl of a lysis buffer. The CD40 complexes were precipitated by protein G-agarose and analyzed by IB. The recruitment of proteins to LTβR was detected in a similar way, except for the use of MEF cells stimulated with anti-LTβR antibody22.
Ubiquitination and deubiquitination assays
TRAF3 was isolated by IP under denaturing conditions27 to inactivate DUBs and disrupt protein complexes, and the ubiquitinated TRAF3 was detected by IB using pan-ubiquitin or chain-specific ubiquitin antibodies. For transfection models, TRAF3 was transfected into HEK293 cells along with HA-ubiquitin or HA-ubiquitin K48. Following TRAF3-denaturing IP, the TRAF3-ubiquitin conjugates were detected by IB using anti-HA. In deubiquitination assays, the isolated TRAF3-ubiquitin conjugates were incubated with purified GST-Otud7b or GST-Otud7bCH recombinant proteins in a deubiquitination buffer33 for 16 hours and then subjected to IB using anti-HA.
Flow Cytometry
Cell suspensions were subjected to flow cytometry analyses using a LSRII flow cytometer (BD Biosciences), as described previously34. The data were analyzed using FlowJo software.
Real-time quantitative reverse PCR (QPCR)
RNA preparation and QPCR assays were as described27 using gene-specific primers listed in Supplementary Table 1.
B-cell adoptive transfer, immunization, and ELISA
B220+ B cells and CD90.2+ T cells were isolated from the splenocytes of WT or Otud7b-KO mice using magnetic beads (Miltenyi Biotec). The isolated cells were >95% pure, as determined by flow cytometry. WT T cells (5 × 106) were mixed with either WT or Otud7b-KO B cells (5 × 106) and then injected via a tail vein into Rag1-KO mice. After 16 h, the recipient mice were subjected to immunization with NP-KLH and the sera were collected on day 7, day 14 and day 21. ELISA was used to detect the different isotopes of antibodies (Southern Biotech). For measuring fecal IgA concentration, feces were taken from age-matched WT and Otud7b-KO mice, weighted, and homogenized in PBS. IgA level was detected by ELISA.
LTβR-Ig treatment
To block the LTβR signaling, WT or Otud7b-KO mice were injected i.p. with 100 μL of PBS or LTβR-Ig (1 μg/μL) on day 0 and day5 and sacrificed on day10. Colons were collected for histological examination.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from indicated cells and subjected to EMSA using the following 32P-radiolabeled oligonucleotide probes:
NF-κB: CAACGGCAGGGGAATTCCCCTCTCCTT
NF-Y: AAGAGATTAACCAATCACGTACGGTCT
Oct-1: TGTCGAATGCAAATCACTAGAA
C. rodentium infection
C. rodentium strain DBS100 (ATCC 51459) was cultured by shaking at 37°C overnight in LB broth. The concentration of bacteria was assessed by measuring OD at 600 nm and confirmed by plating serially diluted bacterial cultures for colony forming unit (CFU) determination. Mice were orally injected with 2-4.5 × 109 C. rodentium in a total volume of 200 μl per mouse. For CD4 T-cell depletion, the mice were also injected i.p. with either a control rat IgG or a rat anti-mouse CD4 antibody (GK1.5, 50 μg/mice) on day 0, 5, and 10. Mice were allowed to access to food and water after the inoculation and sacrificed at the indicated times after infection. Colons were isolated and fixed for H&E staining to evaluate tissue pathology. Spleen, liver, and feces were collected, weighed, and homogenized. The homogenates were serially diluted and plated on MacConkey agar plates, and the plates were incubated at 37°C for 24 hours followed by counting the C. rodentium colonies (pink colonies).
CLP staining
Whole-mount immunofluorescence staining of colonic lymphoid follicles was performed essentially as described35. Colons were collected, flushed and opened longitudinally along the mesenteric border. The tissues were incubated with shaking at room temperature for 60 min in Hank’s balanced-salt solution containing 5 mM EDTA, fixed in 4% paraformaldehyde, and rinsed. After blocking nonspecific epitopes with a blocking buffer35 containing 5% BSA, colons were stained with Alexa Fluor 594-conjugated anti-B220 antibody. Pictures were taken by Leica SP5 RS confocal microscope and analyzed by SlideBook 5.0 software. The B220+ lymphoid follicles larger than 10,000 μm2 were considered as CLPs.
Statistical Analysis
Two-tailed unpaired t tests were performed using Prism software. P values less than 0.05 was considered significant, and the level of significance was indicated as *P< 0.05, **P<0.01), and ***P<0.001.
Supplementary Material
Acknowledgments
We thank Drs. Zhijian Chen and Paul Evans for expression vectors and Dr. Jeffrey Browning and Biogen for the anti-LTβR antibody. We also thank the personnel from the flow cytometry, DNA analysis, animal facility, and histology core facilities at The MD Anderson Cancer Center for technical assistance. This study was supported by grants from the National Institutes of Health (AI057555, AI064639, and GM84459 to S.C.S.; CA137059 to T.Z.; T32CA009598 to G.C.B.) and the Sister Institution Network Fund of MD Anderson Cancer Center.
Footnotes
AUTHOR CONTRIBUTIONS
H.H. designed the study, performed experiments, analyzed data, and wrote part of the manuscript; G.C.B., J.-H.C., N.P.-O., J.J., A.Z., Y. X., X.C. and M.C. contributed to the performance of the experiments, Y.-X.F. contributed critical reagents; C.Z. and T.Z. were involved in the supervision of N.P.-O. and A.Z., respectively, and the discussion of results; and S.-C.S. designed the research and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–1179. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Razani B, Reichardt AD, Cheng G. Non-canonical NF-kappaB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011;244:44–54. doi: 10.1111/j.1600-065X.2011.01059.x. [DOI] [PubMed] [Google Scholar]
- 4.Senftleben U, et al. Activation of IKKa of a second, evolutionary conserved, NF-kB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 5.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 6.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 7.Vallabhapurapu S, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarnegar BJ, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. 2009;284:8217–8221. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunol Rev. 2012;246:107–124. doi: 10.1111/j.1600-065X.2012.01100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans PC, et al. Isolation and characterization of two novel A20-like proteins. Biochem J. 2001;357:617–623. doi: 10.1042/0264-6021:3570617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Häcker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 14.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 15.Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17:939–947. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enesa K, et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. 2008;283:7036–7045. doi: 10.1074/jbc.M708690200. [DOI] [PubMed] [Google Scholar]
- 17.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 18.Dohi T, et al. Elimination of colonic patches with lymphotoxin beta receptor-Ig prevents Th2 cell-type colitis. J Immunol. 2001;167:2781–2790. doi: 10.4049/jimmunol.167.5.2781. [DOI] [PubMed] [Google Scholar]
- 19.Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol. 2003;3:642–655. doi: 10.1038/nri1151. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 21.Bouskra D, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. 2010;32:403–413. doi: 10.1016/j.immuni.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–267. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 25.Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 2011;186:143–155. doi: 10.4049/jimmunol.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kayagaki N, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 27.Chang M, Jin W, Sun SC. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol. 2009;10:1089–1095. doi: 10.1038/ni.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans PC, et al. A novel type of deubiquitinating enzyme. J Biol Chem. 2003;278:23180–23186. doi: 10.1074/jbc.M301863200. [DOI] [PubMed] [Google Scholar]
- 29.Reiley W, Zhang M, Wu X, Graner E, Sun S-C. Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol Cell Biol. 2005;25:3886–3895. doi: 10.1128/MCB.25.10.3886-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders RA, Subudhi SK, Wang J, Pfeffer K, Fu YX. Contribution of the lymphotoxin beta receptor to liver regeneration. J Immunol. 2005;175:1295–1300. doi: 10.4049/jimmunol.175.2.1295. [DOI] [PubMed] [Google Scholar]
- 31.Lomada D, Liu B, Coghlan L, Hu Y, Richie ER. Thymus medulla formation and central tolerance are restored in IKKalpha-/- mice that express an IKKalpha transgene in keratin 5+ thymic epithelial cells. J Immunol. 2007;178:829–837. doi: 10.4049/jimmunol.178.2.829. [DOI] [PubMed] [Google Scholar]
- 32.Morrison MD, Reiley W, Zhang M, S SC. An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappaB signaling pathway. J Biol Chem. 2005;280:10018–10024. doi: 10.1074/jbc.M413634200. [DOI] [PubMed] [Google Scholar]
- 33.Hassink GC, et al. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009;10:755–761. doi: 10.1038/embor.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reiley WW, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ota N, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol. 2011;12:941–948. doi: 10.1038/ni.2089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.