

Abstract
Ku70 was originally described as an auto-antigen, but it also functions as DNA repair protein in the nucleus and as an anti-apoptotic protein by binding to Bax in the cytoplasm, blocking Bax-mediated cell death. In neuroblastoma (NB) cells, Ku70’s binding with Bax is regulated by Ku70 acetylation such that increasing Ku70 acetylation results in Bax release, triggering cell death. While regulating cytoplasmic Ku70 acetylation is important for cell survival, the role of nuclear Ku70 acetylation in DNA repair is unclear. Here we demonstrated that Ku70 acetylation in the nucleus is regulated by the CREB-binding protein (CBP), and that Ku70 acetylation plays an important role in DNA repair in NB cells. We treated NB cells with ionization radiation and measured DNA repair activity as well as Ku70 acetylation status. Cytoplasmic and nuclear Ku70 were acetylated after ionization radiation in NB cells. Interestingly, cytoplasmic Ku70 was redistributed to the nucleus following irradiation. Depleting CBP in NB cells results in reducing Ku70 acetylation and enhancing DNA repair activity in NB cells suggesting nuclear Ku70 acetylation may have an inhibitory role in DNA repair. These results provide support for the hypothesis that enhancing Ku70 acetylation, through deacetylase inhibition, may potentiate the effect of ionization radiation in NB cells.
Keywords: acetyltransferase, histone deacetylase, Ku70, Bax, CBP, cell death
INTRODUCTION
Ku70 was first characterized as an autoantigen and subsequently it was also identified to be a nuclear DNA binding component of the non-homologous end joining (NHEJ) DNA repair complex [1]. When dimerized with Ku80, Ku70 binds to the broken end of double strand DNA breaks [2]. However, following studies have also shown that Ku70 is also present in the cytoplasm [3]. To date, one described function of cytoplasmic Ku70 is to bind Bax, an apoptotic protein, thereby blocking Bax-mediated cell death. The binding between Ku70 and Bax is regulated by Ku70 acetylation [4]. We have previously shown that inhibiting deacetylase activity in neuroblastoma (NB) cells increases Ku70 acetylation, resulting in Bax release that triggers Bax-dependent cell death [5]. Our studies further indicated that cytoplasmic Ku70 plays an important role in NB cell survival as Ku70 knock down or increased Ku70 acetylation by inhibiting HDAC activity induces NB cell death mediated by Bax [6].
In the nucleus, Ku70 [7] when dimerized with Ku80, binds and bridges two proximal broken DNA ends and facilitates the repair machinery through a cascade of reactions that involve DNA-dependent protein kinase and DNA ligase IV [8, 9]. Ku70 plays a critical role in this DNA repair activity as even partial knock down of Ku70 has been shown to enhance the radiosensitivity of human MCF10A cells [10]. Moreover, murine embryonic stem cells (ES) deficient in Ku70 are sensitive to radiation and have V(D)J recombination defects and deficiencies in DNA binding [11]. In cells with targeted deletion of Ku70, the Ku80 partner is unstable as is the Ku70 partner in Ku80 deficient cells [11, 12]. The two DNA binding domains of Ku70 present in the NH2 and COOH termini are required for high affinity DNA binding. In addition, the COOH terminal of Ku70 also binds to Bax and prevents apoptotic translocation of Bax to the mitochondria [13]. Thus, Ku70 mediates cytoprotective functions through two distinct mechanisms: DNA repair in the nucleus and blocking Bax-mediated cell death in the cytoplasm.
While we and others have shown that acetylation regulates the binding between cytoplasmic Ku70 and Bax [14], the effect of Ku70 acetylation in the nucleus remains unclear. We have previously shown that in NB cells, acetylation of Ku70 by CBP at lysines 539 and 542 resulted in Bax release from Ku70, followed by Bax translocation to mitochondria [5]. Computer docking analysis indicated the presence of multiple lysine residues that form a positively charged lining for interaction with broken DNA ends at the DNA binding cradle of Ku70 [4, 15]. Additional studies carried out in prostate cancer cells using site directed mutagenesis to replace the lysine residues at K282, K338, K539 or K542 with glutamine showed that in addition to the above mentioned lysine residues, namely K539 and K542, two other lysine residues, K282 and K338, also take part in binding broken-end DSB DNA [16]. The fact that the K539 and K542 acetylation by CBP are responsible for Bax-dependent cell death in NB cells and the same lysine residues are involved in binding to broken-end DSB DNA prompted us to investigate the role of Ku70 acetylation by CBP in response to IR-induced DNA damage.
Our results demonstrate that IR does not affect Ku70 expression in NB cells but IR induces Ku70 redistribution from the cytoplasm to the nucleus. When NB cells are subjected to IR, the more aggressive neuroblastic (N-type) NB cells undergo cell death while the less aggressive stromal (S-type) NB cells demonstrate IR resistance. Moreover, the DNA repair activity, as measured by phosphorylated H2AX (γH2AX) expression and by the Comet assay, is more efficient in the S-type cells compared to the N-type cells. The possibility that increased acetylation of Ku70 might block Ku70 DNA binding activity necessary for NHEJ repair, along with previous work showing N-type cells express higher levels of CBP, led us to hypothesize that Ku70 acetylation by CBP may play a critical role in IR-induced killing of N-type NB cells. This model is supported by evidence that knocking down CBP in N-type cells led to a reduction of Ku70 acetylation, increased DNA repair activity, and NB cell survival following IR. These results suggest a critical role for CBP in Ku70 acetylation following IR-induced DNA damage in NB cells and provide further support for the development of NB therapeutic strategies that target Ku70 acetylation.
MATERIALS AND METHODS
Cell Culture and irradiation
Human NB cell lines SH-SY5Y, IMR32, SH-EP1, and the fibroblast cell line IMR90 were cultured in modified Eagle’s medium (MEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin, and the cells were maintained at 37°C in a humidified 5% CO2 incubator. The cells were irradiated over the clinically relevant dosage ranging from 0 to 20 Gy using Philips 250 orthovoltage unit at the Irradiation Core of the University of Michigan Cancer Center.
Cell Viability Assay
The viability of the human NB cell lines SH-SY5Y, SH-EP1, IMR32, and the fibroblast cell line IMR 90 was determined 24 and 48 hours after exposure to IR by MTT assay or sulforhodamine assay as previously described [5]. The viability of the CBP siRNA, control siRNA and mock transfected SH-SY5Y cells were similarly measured by MTT after IR exposure. All experiments were carried out in triplicate and the average values and standard deviations were calculated.
siRNA mediated silencing of CBP
SH-SY5Y cells were plated at a density of 2X106 cells per 10 cm plate 24 hours before transfection. The following day the cells were transfected either with smart pool CBP siRNA, or the scrambled non-targeting siRNA (Dharmacon Inc.) using nucleofactor kit V (Lonza) as per the manufacturers instruction. Mock transfection as well as the non-targeting scrambled siRNA transfection served as controls. The knock down of CBP was determined 72 hours post-transfection by immunoblotting using anti-CBP antibodies.
Immunoblot Analysis
For immunoblot analyses, whole cell extracts were prepared from human NB cell lines following IR treatment. The proteins were separated by SDS-PAGE, transferred to a PVDF membrane, and immunoblotted for γH2AX, CBP, Bax, or control GAPDH antibodies. The presence of proteins were visualized using Enhanced Chemiluminescence plus.
Fractionation and Immunoprecipitation
The SH-SY5Y cells and the CBP-knockdown SH-SY5Y cells were fractionated into cytosolic and nuclear fractions using low salt and high salt buffer as previously described [17]. The fractions were immunoprecipitated using an anti-acetyl-lysine antibody (Santa Cruz) or K103 acetylated lysine antibody (Cell Signaling) in CHAPS buffer and complexes were immunoblotted with Ku70 antibody [3].
Clonogenic Assay
To determine the survivability of NB cells after various treatments, we used a clonogenic assay [18]. Cells were trypsinized and seeded in triplicate into 60mm dishes (400 cells for un-irradiated control, and 40000 for irradiated cells) and allowed to grow undisturbed for one week. Colonies were stained with crystal violet, and counted manually using a Lieca microscope. The percentage of surviving fraction at different doses is calculated as previous described [18].
Immunofluorescent microscopy and quantification of γH2AX foci
NB cells and controls were seeded in chamber slides. At Different time points following irradiation at 2Gy, cells were fixed with 4% paraformaldehyde for 15 min, washed with PBS, and permeabilized in 0.2% Triton-X100. After blocking with 5% normal goat serum (NGS) for 1hr samples were incubated with anti–phospho-histone γH2AX (Ser139, clone JBW301) mouse monoclonal antibody (Millipore) at a 1:500dilution in 2.5% NGS-PBS overnight at 4°C, followed by incubation with the secondary antibody Alexa Fluor® 568 goat anti-mouse-IgG (1:1000) for 1hr. Cells were then washed with PBS and mounted using mounting solution with DAPI (Invitrogen). Images of γH2AX foci and nuclei were taken using Leica DMR fluorescent microscope at 40X. Quantitative analysis of foci was carried out using Image-J as previously described [19]. To test for variation between experiments, at least 100 cells from three different experiments were scored for each data point. The mean number of foci per cell and the standard deviation from three independent measures were calculated.
The Comet Assay
The neutral comet assay was performed using a Trevigen Comet Assay Kit (4250-050-K, Trevigen, Gaithersburg, MD) according to the manufacture’s protocol. SH-SY5Y, SH-EP1, CBP-knocked down SH-SY5Y cells, or control siRNA treated SH-SY5Y cells were exposed to radiation dose of 10Gy and subjected to comet assay at the indicated time points of 0, 1, or 3 hrs. The comet images were captured using fluorescent microscopy after staining with SYBR green. Average comet tail moment (percentage of DNA in tail length) was scored for three fields (more than 50 comets in each field) using the Comet Score software (TriTek, Sumerduck, VA, USA). The results are expressed as MEAN±S.D.
RESULTS
Ku70 is acetylated in the cytoplasm and in the nucleus after irradiation
We have shown previously that acetylation of cytoplasmic Ku70 triggers Bax release and NB cell apoptosis [5]. However, the role of Ku70 acetylation in NB DNA nuclear repair responses is unknown. In this study we have determined that IR induces Ku70 acetylation in SH-SY5Y cells, N-type NB cells (Fig. 1A, left), but not in SH-EP1 cells, S-type NB cells, (Fig. 1A, right). Also in SH-SY5Y N-type cells, both cytoplasmic and nuclear Ku70 are acetylated 24 hours after 10Gy IR (Fig. 1B). Interestingly, when we determined the level of Ku70 in the cytoplasm and in the nucleus at 0, 2, 4, and 6 hours after IR, we found the level of Ku70 is similar in the cytoplasm and in the nucleus of untreated cells (0 hour) (Fig. 1C). Two hours after IR, however, there is a visible reduction in the cytoplasmic Ku70 level, and simultaneously, nuclear Ku70 level increases. These changes of Ku70 level between the cytoplasm and the nucleus are evident for 6 hours after treatment suggesting DNA-damage induced by IR causes Ku70 to translocate from the cytoplasm to the nucleus.
Figure 1.

Ionization radiation induces Ku70 acetylation and translocation from cytoplasm to nucleus. (A) N-type SH-SY5Y and S-type SH-EP1 cells received 10Gy radiation, and 24 hours later, lysates were immunoprecipitated using an anti-acetyl-lysine antibody (Cell Signaling) and probed for Ku70. (B) SH-SY5Y cells were irradiated at 10Gy and 24 hours later, lysates were fractionated into cytoplasmic and nuclear fractions. Both fractions were immunoprecipitated using an anti-acetyl-lysine antibody (Santa Cruz) and probed for Ku70. (C) SH-SY5Y cells were irradiated at 10Gy. The irradiated cells were harvested 2, 4, or 6 hours later. Lysates were fractionated into cytoplasmic and nuclear fractions and then immunoblotted for Ku70, β-tubulin as a cytoplasmic marker, and CREB as a nuclear maker.
Kinetics of DNA repair in NB cells in response to irradiation
To investigate the DNA damage response of NB cells to IR, we utilized phosphorylation of histone H2AX (γH2AX) as a marker of DNA damage [20]. Interestingly, when we compared the kinetics of γH2AX expression using immunoblot analyses in N-type verses S-type NB cells, we found that in SH-SY5Y N-type cells the rate of disappearance of γH2AX is prolonged up to 8 hours after 10Gy IR treatment (Fig. 2A, left). In contrast, in SH-EP1 S-type cells, γH2AX disappears within 2 hours of treatment (Fig. 2A, right). These results suggest that repair of DNA damage is much faster in SH-EP1 cells compared to that of SH-SY5Y cells. Similar results were obtained when we used a lower dose of IR, 2Gy, and counted the total foci of γH2AX of over 100 cells for each treatment. The immunocytochemistry staining at 0, 3, and 7 hours after 2 Gy IR treatment is shown in Fig. 2B. The compilation of the total γH2AX foci counts at various times after IR treatment is shown in Fig. 2C for SH-SY5Y cells and SH-EP1 cells. The results are consistent with the immunoblot data (shown in Figure 2A) when a higher dose (10Gy) of IR is used in that S-type NB cells have a faster kinetics of repairing DNA than that of the N-type NB cells. Similar results were also obtained when a direct measurement of DNA breaks using a Comet Assay, which measures the DNA breaks at the time when the cells are lysed [21]. Results shown in figs. 2D and 2E demonstrate that SH-EP1 has a faster kinetics of DNA repair (measuring at 1 and 3 hrs following IR) than that of SH-SY5Y cells after 10Gy treatment.
Figure 2.
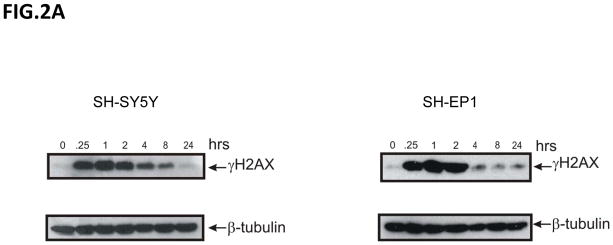



Kinetics of DNA repair in neuroblastoma cells in response to irradiation. (A) SH-SY5Y and SH-EP1 cells were irradiated at 10Gy. At various times as indicted after IR, lysates were collected and immunoblotted for γH2AX and β-tubulin. (B) SH-SY5Y and SH-EP1 cells were irradiated at 2 Gy. At various times as indicated after IR, immunofluorescence for γH2AX was conducted. Only 0, 3, 7 hours are shown. (C) Compilation of the number of foci of γH2AX per cell after IR as shown in (B) at times indicated. The results are expressed as Mean±SD (N=3, 100 foci per sample). (D and E) SH-SY5Y cells and SH-EP1 cells were irradiated at 10Gy. At 0, 1, and 3 hours later, the cells were subjected to the Comet assay as described. The fluorescent microscopy staining with SYBR green was shown in (D). The average comet tail moment (percentage of DNA in tail length) was scored in three different fields (at least 50 comets per field) shown in (E). The results are expressed as Mean±SD (n=3).
When we determined cell viability using MTT assay following IR, we found that in N-type NB cells (SH-SY5Y and IMR32 cells) viability decreases proportional to the IR dose (5, 10, or 20 Gy) used. In contrast, in SH-EP1 cells, similar to the IMR90, a fibroblast type cell line, the viability dropped minimally following (down to 85% of control) various IR treatments (Fig. 3A). Similar results were obtained when a clonogenic assay was used to assess the survivability of SH-SY5Y cells and SH-EP1 cells following various doses of IR (Fig. 3B).
Figure 3.

Irradiation reduces neuroblastoma cell viability. (A) SH-SY5Y, IMR32, SH-EP1, and IMR90 cells were irradiated at 5, 10, or 20 Gy. Cell viability was determined by MTT assay after 24 or 48 hours. Results are expressed as the percentage of viable cells compared with un-irradiated control cells (Mean±SD, N=3). (B) SY-SH5Y cells and SH-EP1 cells were irradiated at various doses of IR as indicated. Immediately after irradiation, cells were plated. One week after plating, the colonies were fixed with glutaraldehyde (6.0% v/v), stained with crystal violet (0.5%w/v), and counted using a microscope. The results are expressed as the percentage of the number of colonies (Mean±SD, N=3) compared to the number of colonies of un-irradiated. (C) SH-SY5Y cells (left panel) and SH-EP1 cells (right panel) were irradiated at 10Gy. Twenty-four hours later, cell lysates were immunoprecipitated with anti-Bax antibodies, and then immunoblotted for Ku70 or Bax.
The reduction of cell viability after IR seen in SH-SY5Y cells but not in SH-EP1 cells is consistent with our previous findings showing that cytoplasmic Ku70 is acetylated in SH-SY5Y cells but not in SH-EP1 cells (Fig. 1). When we immunoprecipiated Bax after 10Gy IR in these two cell types, we found much less Ku70 is associated with Bax in the SH-SY5Y cells compared to that of SH-EP1 cells. These results indicate that Bax is released from Ku70 after IR in SH-SY5Y cells (Fig. 3C, left panel) but not in SH-EP1 cells (Fig. 3C, right panel).
Interestingly, when we further studied SH-EP1 cells that survived IR treatment, we found that they demonstrated resistance to the effects of cisplatin, a chemotherapeutic agent known to induce cell death in NB cells (Fig. 4A) [22]. Furthermore, the SH-EP1 cells had faster growth rates in low serum conditions (0.1 or 0.5%) when compared to un-irradiated cells (Fig. 4B).
Figure 4.
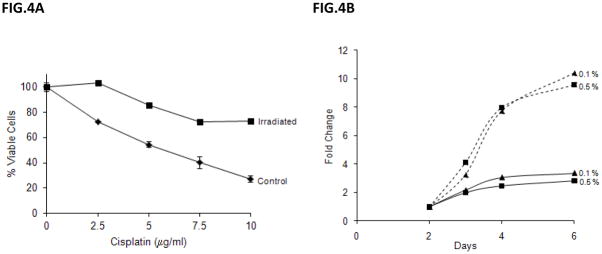
Irradiated SH-EP1 cells are more resistant to cisplatin and grow better in low serum media. (A) SH-EP1 cells are irradiated at 10Gy. Twenty-four hours later, the un-irradiated or irradiated cells were treated with various doses of cisplatin as indicated. Cell viability was determined using sulforhodamine assay 24 hours after drug treatment. Results are expressed as the percentage of viable cells compared with un-irradiated control cells (Mean±SD, n=3). (B) SH-EP1 cells were irradiated at 10Gy, and 24 hours later the cells were plated at serum concentrations of 0.1% and 0.5%. Cell viability was determined at day 2, 3, 4, or 6 using sulforhodamine assay. Results are expressed as the fold of viable cells compared with the day 2 value (equals to 1 fold) (Mean±SD, n=3). The solid lines are control cells and the broken lines are irradiated cells.
CBP regulates Ku70 acetylation in response to irradiation in NB cells
We have shown previously that CBP regulates Ku70 acetylation in NB cells. Over expression of CBP or CBP knock down will alter the response of histone deacetylase inhibitor treatment that induces cell death in NB cells [6]. In this current study, we tested whether CBP regulates Ku70 acetylation in response to DNA damage induced by IR. Since we did not observe Ku70 acetylation in S-type cells (Fig. 1A) after IR, we focused our studies on SH-SY5Y cells.
To test whether CBP regulates Ku70 acetylation following IR treatment in NB cells, we knocked down CBP using CBP specific siRNA and tested (Fig. 5A) whether Ku70 is acetylated in response to IR (10 Gy) (Fig. 5A). Twenty-four hours after IR treatment, scrambled siRNA or CBP siRNA transfected cells with and without exposure to IR were fractionated to separate the cytoplasmic and nuclear fractions. Samples were then immunoprecipitated using an anti-acetyl-lysine antibody (Santa Cruz) and then immunoblotted for Ku70. As shown in Fig. 5B, CBP knock down reduces the level of Ku70 acetylation in both the cytosolic and nuclear fractions following IR.
Fig. 5.
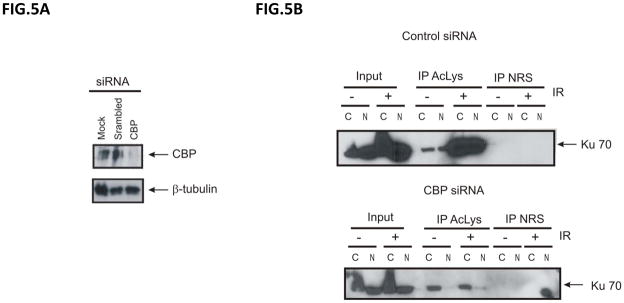
CBP regulates Ku70 acetylation in response to irradiation in NB cells. SH-SY5Y cells were transfected with either control scrambled siRNA or CBP siRNA. Forty-eight hours after transfection the cells were either collected to determine the depletion of CBP (A) or (B) irradiated at 10Gy. Twenty-four hours following IR, lysates were fractionated into cytoplasmic and nuclear fractions. Each fraction was immunoprecipitated using an anti-acetyl-lysine antibody, and immunoblotted for Ku70.
To determine if there was a change in IR-induced DNA damage repair activity following CBP knockdown, we treated scrambled siRNA control or CBP siRNA transfected SH-SY5Y cells with IR (2Gy), and the kinetics of γH2AX was determined by immunocytochemistry using γH2AX specific antibodies (Fig. 6A). The results shown in Fig. 6B are the number of γH2AX foci per nucleus (100 nucleus). These results show that the CBP-knockdown cells show a faster kinetics of the disappearance of γH2AX compared to the scrambled siRNA control. These results suggest that CBP may be regulating the DNA damage response, possibly by regulating Ku70 acetylation, induced by IR. Similar results were obtained when the Comet Assay was used to determine the total DNA breaks at the time of cell lysis (Figs. 6C and 6D). A clonogenic assay was also carried out simultaneously to identify the survival ability after IR-induced DNA damage when CBP is depleted. Results in figure 7A show CBP-knock down SH-SY5Y cells demonstrating significantly higher survival when exposed to various doses of IR compared to control transfected cells. Interestingly, transfection of a Ku70 mutant which has K539 and K542 converted to arginine (K539/542R) has no effect on IR induced reduction in cell viability in SH-SY5Y cells (fig. 7B), suggesting that acetylation of K539and K542 may not be responsible in regulating SH-SY5Y cell DNA repair following IR treatment.
Figure 6.



CBP depletion shortens the DNA repair time in neuroblastoma cells. (A) SH-SY5Y cells transfected with CBP siRNA or control siRNA were irradiated at 2Gy. At different times following IR, immunofluorescence for γH2AX was conducted as described in the Method section. Zero, 3, 5 and 7 hour after IR are shown in (A). (B) Compilation of the number of foci ofγH2AX per cells after IR described in (A) at times indicated. The results are expressed as Mean±SD (N=3, 100 foci per sample). (C) and (D) SH-SY5Y cells were transfected with CBP siRNA or control siRNA. Two days after transfection, the cells were irradiated at 10Gy. At 0, 1, and 3 hours after IR the cells were subjected to the Comet assay as described. The fluorescent microscopy staining with SYBR green was shown in (C). The average comet tail moment (percentage of DNA in tail length) was scored in three different fields (at least 50 comets per field) shown in (D). The results are expressed as Mean±SD (n=3).
Figure 7.

CBP depletion increases the survival of neuroblastoma cells after irradiation. (A) SH-SY5Y cells were transfected with control siRNA or CBP specific siRNA. Two days after transfection, the cells were irradiated at various doses of irradiation. Immediately after irradiation, cells were plated. One week after plating, the colonies were fixed with glutaraldehyde (6.0% v/v), stained with crystal violet (0.5%w/v), and counted using a microscope. The results are expressed as the percentage of the number of colonies (Mean±SD, N=3) compared to the number of colonies of IR-treated without siRNA transfection. (B) SH-SY5Y cells were transfected with a Ku70 K539/K542R expression vector. Two days after transfection, the cells were treated with various doses of irradiation as indicated. One day after irradiation, the cell viability was determined by MTT assay. Results are expressed as the percentage of viable cells compared with un-irradiated control cells (Mean±SD, N=3).
DISCUSSION
Of the various types of DNA damage, DNA double strand breaks (DSB) induced by ionization radiation is most toxic to dividing cells [23]. Efficient repair of DNA DSB is critical for maintaining genomic stability and cell viability [24]. In mammalian cells, DNA repair activity in response to IR-induced DSBs occurs via two routes: homologous recombination (HR) and NHEJ [25]. HR is an accurate form of DNA repair that is restricted to DNA repair occurring at S-phase following DNA replication. In contrast, NHEJ operates throughout the cell cycle and is considered the major pathway for the IR induced DSB DNA repair [26–28].
The Ku70-Ku80 heterodimer plays a key role in the NHEJ DNA repair pathway. The initial step in the NHEJ pathway is detection of the DSB by Ku heterodimer, followed by recruitment of the catalytic subunit of DNA-dependent protein kinase (DNA-PKc) to form the active DNA-protein kinase dependent holoenzyme [29]. Deficiency of either NHEJ proteins in cells or mutations in Ku70, Ku80, or DNA-PKc leads to IR sensitization [23]. Previous studies have shown that Ku70 acetylation blocks Ku70 binding to broken-end DSB DNA [16]. In this study, we have shown that Ku70 is acetylated upon IR not only in the cytosol but also in the nucleus of N-type, but not in S-type, NB cells. We have also shown that in N-type, but not in S-type, NB cells, Ku70 acetylation in the cytosol releases Bax from Ku70 resulting in Bax translocation to the mitochondria causing cell death [5]. However, expression of Ku70 K539/542R mutant does not rescue SH-SH5Y cell death induced by IR suggesting that Bax may not regulate cell death triggered by IR in this cell type.
Our results have also shown that N-type cells demonstrate prolonged repair activity and more apoptotic cell death when compared to the S-type NB cells following IR, suggesting that regulation of Ku70 acetylation may play a role in NB cell survival. We have shown that the irradiated SH-EP1 cells demonstrate resistance to the chemotherapy drug cisplatin, and that they grow better in lower serum conditions. These results suggest that IR induces cell death of least survivable SH-EP1 cells, and the remaining SH-EP1 cells, having a faster DNA repair rate and low Ku70 acetylation following IR, they adopt a better survival mechanism that is resistant to DNA damage compounds like cisplatin. However, whether this survival mechanism in SH-EP1 cells is related to Ku70 acetylation affecting its DNA binding for repair remains to be tested.
Our results show that CBP acetylates Ku70 in the cytosol and in the nucleus of NB cells; depleting CBP lowers Ku70 acetylation in both compartments. Consistent with our model, the repair activity of irradiated, CBP-depleted SH-SY5Y cells is more robust than control cells, and CBP-depleted cells have higher survivability, judging by the increase of colonies of CBP-depleted cells upon IR in the clonogenic assay. These results suggest that CBP and deacetylases regulate Ku70 functions in NB cells. However, this mechanism may be cell type specific. Ogiwara et al. have demonstrated that, in human lung cancer H1299 cells, depletion of CBP and p300 results in repressing DNA repair activity, which is the opposite of what we report here [30]. Together, their results as well as ours suggest that the role of CBP in DNA repair activity may be cell context specific. What regulates this specificity remains to be determined.
Taken together our data for the first time in NB cells identify Ku70 acetylation in response to IR- induced DNA damage to be responsible cell death exposed to IR. Our previous study showing low level CBP expression in S-type cells compared to that in N-type cells indicates that the resistance of S-type NB cells to radiation and faster DNA repair activity may be due to low level CBP expression [6]. These results also provide novel insight into the differences between N-type and S-type NB cells in response to IR-induced DNA damage. These findings provide a rationale for testing modulators of Ku70 acetylation in clinical trials along with radiotherapy or DNA damaging agents for the treatment of NB.
Acknowledgments
This work was supported in part by the National Institutes of Health grants DK067102 (R.P.S.K.), the Janette Ferrantino Hematology Research Fund (V.P.C.), the Ravitz Foundation (V.P.C.), Hope Street Kids Foundation (C.S.), and the A. Alfred Taubman Medical Research Institute (V.P.C., R.P.S.K., A.W.O.). This work used the Sequencing Core of the Michigan Diabetes Research and Training Center, which was funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant NIH5P60 DK20572.
References
- 1.Featherstone C, Jackson SP. Ku, a DNA repair protein with multiple cellular functions? Mutat Res. 1999;434(1):3–15. doi: 10.1016/s0921-8777(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 2.Hammel M, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285(2):1414–23. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawada M, et al. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5(4):320–9. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- 4.Cohen HY, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13(5):627–38. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 5.Subramanian C, et al. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102(13):4842–7. doi: 10.1073/pnas.0408351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramanian C, et al. CREB-binding protein is a mediator of neuroblastoma cell death induced by the histone deacetylase inhibitor trichostatin A. Neoplasia. 2007;9(6):495–503. doi: 10.1593/neo.07262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weterings E, van Gent DC. The mechanism of non-homologous end-joining: a synopsis of synapsis. DNA Repair (Amst) 2004;3(11):1425–35. doi: 10.1016/j.dnarep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Dong X, Reeves WH. A model for Ku heterodimer assembly and interaction with DNA. Implications for the function of Ku antigen. J Biol Chem. 1998;273(47):31068–74. doi: 10.1074/jbc.273.47.31068. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Lieber MR. Protein-protein and protein-DNA interaction regions within the DNA end-binding protein Ku70-Ku86. Mol Cell Biol. 1996;16(9):5186–93. doi: 10.1128/mcb.16.9.5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandersickel V, et al. The radiosensitizing effect of Ku70/80 knockdown in MCF10A cells irradiated with X-rays and p(66)+Be(40) neutrons. Radiat Oncol. 2010;5:30. doi: 10.1186/1748-717X-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y, et al. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci U S A. 1997;94(15):8076–81. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nussenzweig A, et al. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382(6591):551–5. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 13.Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol. 2003;5(4):352–7. doi: 10.1038/ncb955. [DOI] [PubMed] [Google Scholar]
- 14.Nothwehr SF, Martinou JC. A retention factor keeps death at bay. Nat Cell Biol. 2003;5(4):281–3. doi: 10.1038/ncb0403-281. [DOI] [PubMed] [Google Scholar]
- 15.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412(6847):607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 16.Chen CS, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–27. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian C, et al. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia. 2011;13(8):726–34. doi: 10.1593/neo.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franken NA, et al. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 19.Cai Z, Vallis KA, Reilly RM. Computational analysis of the number, area and density of gamma-H2AX foci in breast cancer cells exposed to (111)In-DTPA-hEGF or gamma-rays using Image-J software. Int J Radiat Biol. 2009;85(3):262–71. doi: 10.1080/09553000902748757. [DOI] [PubMed] [Google Scholar]
- 20.Taneja N, et al. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279(3):2273–80. doi: 10.1074/jbc.M310030200. [DOI] [PubMed] [Google Scholar]
- 21.Singh NP, et al. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–91. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 22.Bian X, et al. Chemotherapy-induced apoptosis of S-type neuroblastoma cells requires caspase-9 and is augmented by CD95/Fas stimulation. J Biol Chem. 2004;279(6):4663–9. doi: 10.1074/jbc.M306905200. [DOI] [PubMed] [Google Scholar]
- 23.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–50. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27(4):589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cahill D, Connor B, Carney JP. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci. 2006;11:1958–76. doi: 10.2741/1938. [DOI] [PubMed] [Google Scholar]
- 26.Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7(11):861–9. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- 27.Helleday T, et al. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6(7):923–35. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 28.O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7(1):45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 29.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18(1):114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 30.Ogiwara H, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30(18):2135–46. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]