

Abstract
Successful implantation and maintenance of pregnancy require the transformation of uterine endometrial stromal cells into distinct decidualized cells. Although estrogen and progesterone (P4) receptors are known to be essential for decidualization, the roles of steroid receptor coregulators in this process remain largely unknown. In this study, we have established a key role for the coregulator, repressor of estrogen receptor activity (REA), in the decidualization of human endometrial stromal cells (hESCs) in vitro and of the mouse uterus in vivo. Our studies revealed that the level of REA normally decreases to half as hESC decidualization proceeds and that uterine reduction of REA in transgenic heterozygous knockout mice or small interfering RNA knockdown of REA in hESC temporally accelerated and strongly enhanced the differentiation process, as indicated by changes in cell morphology and increased expression of biomarkers of decidualization, including P4 receptor. Findings in hESC cultured in vitro with estradiol, P4, and 8-bromo-cAMP over a 10-day period mirrored observations of enhanced decidualization response in transgenic mice with heterozygous deletion of REA. Importantly, gene expression and immunohistochemical analyses revealed changes in multiple components of the Janus kinase/signal transducer and activator of transcription pathway, including marked up-regulation of signal transducer and activator of transcription 3 and IL-11, master regulators of decidualization, and the down-regulation of several suppressor of cytokine signaling family members, upon reduction of REA. The findings highlight that REA physiologically restrains endometrial stromal cell decidualization, controlling the timing and magnitude of decidualization to enable proper coordination of uterine differentiation with concurrent embryo development that is essential for implantation and optimal fertility.
The steroid hormones estradiol (E2) and progesterone (P4), acting through their receptor proteins, coordinate early uterine events during pregnancy (1). Before implantation, these hormone receptors direct changes in the uterine endometrium to establish its receptivity for blastocyst attachment and implantation. The endometrial stromal cells undergo a series of biochemical and morphological transformations, a process known as decidualization, to generate a differentiated maternal tissue that can support embryo implantation and maintenance of pregnancy (2, 3). Although studies have shown that estrogen and P4 receptors (PGRs) are essential for decidualization (4–8), the roles of steroid receptor coregulators, and especially steroid receptor corepressors, in this process remain less well understood.
The controlling role of steroid hormones and their nuclear receptors (4, 8) and the requirement of estrogen and P4-signaling pathways for successful pregnancy have been proven in studies using genetic mutant mouse models lacking estrogen receptor α (ESR1) (5) and PGR (7). In addition, the P4-PGR signaling pathway is essential for the differentiation of human endometrial stromal cells (hESCs), and cross-talk between PGR and cAMP-dependent pathways during decidualization is emerging from recent studies (11).
It is well recognized that the cellular activity of these steroid hormone receptors is not only dependent on their ligands but also on their interactions with coregulators (12). Although a few of these coregulators have been shown to be required for steroid hormone actions in the uterus, such as Kruppel-like factor 9 (13) and the steroid receptor coactivators (14–16), the mechanisms and events defining how interactions between coregulators and steroid hormone receptors impact uterine function still remain largely unknown.
Because the pregnant uterus progresses through physiological stages in which steroid receptor functions are alternately switched on or switched off in various tissue compartments in the uterus, the modulatory roles of these coregulator proteins on receptor function are extremely significant. Indeed, recent gene knockout studies have revealed an essential modulatory role of the coactivator steroid receptor coactivator-2 in uterine decidualization (14). However, little information is available regarding the role of corepressors in modulating steroid receptor activity in the uterus, although knockout studies suggest that these repressive proteins might be playing equally important or even more central roles in uterine function than coactivators (17, 18). We recently characterized a unique corepressor, repressor of estrogen receptor activity (REA), that regulates estrogen receptor activity, and we generated a conditional tissue-specific REA knockout mouse model and established that REA controls critical uterine functions during early gestation. Conditional homozygous mutant mice, in which REA was ablated in PgR-positive uterine cells, were viable but were found to be infertile due to severely compromised uterine development and failure of implantation and decidualization (17). By contrast, uteri of mice with heterozygous knockout of only one REA allele, which contained half the wild-type level of REA, showed a hyperproliferative response to E2 treatment and subfertility (19), indicating that the normal gene dosage of REA is required for optimal uterine function and fertility.
In our current study, to understand how REA regulates fertility and might impact uterine decidualization, we have examined the role of REA during decidualization using both a human in vitro endometrial stromal cell differentiation system and a mouse artificial decidualization model in transgenically modified REA mice in vivo. The findings highlight that REA is required to provide precise control of the timing and magnitude of uterine decidualization, aspects that are known to be central in coordinating uterine receptivity with embryo development for the successful establishment and maintenance of pregnancy.
Materials and Methods
Primary hESC cultures, in vitro differentiation, and small interfering RNA (siRNA) studies
Our studies involving human endometrial biopsies and endometrial cell cultures were approved by the Institutional Review Boards of the University of Illinois, Emory University, and Wake Forest University. All protocols adhere to the regulations set forth for the protection of human subjects participating in clinical research, including the establishment of a data and safety monitoring plan. Endometrial samples were obtained by Pipelle biopsy from fertile volunteers providing written informed consent.
hESCs were isolated from biopsies taken from the early proliferative stage endometrium of regularly cycling women on no hormonal medications, as described before (20). Cells were cultured in DMEM/F-12 medium (Invitrogen, Carlsbad, California) containing 5% charcoal-stripped fetal bovine serum. To induce in vitro decidualization, hESCs were treated with a hormone cocktail containing 10nM E2, 1μM P4, and 0.5mM 8-bromo-cAMP (8-Br-cAMP) (Sigma Chemical Co, St. Louis, Missouri) for up to 10 days, and media were changed every 48 hours. For siRNA experiments, hESCs were transfected with REA siRNA (si-REA) or GL3 luciferase control siRNA (si-GL3) (Dharmacon, Lafayette, Colorado) following the Silent-Fect kit protocol (Bio-Rad Laboratories, Hercules, California). After 48 hours of transfection, hESCs were exposed to the hormone cocktail for differentiation. The scheme for this in vitro cell culture decidualization protocol is presented in figure 2 below.
Animals and artificial decidualization model
All animals were maintained in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and all procedures were approved by the University of Illinois Institutional Animal Care and Use Committee. REA heterozygous (REA+/−) animals were maintained, genotyped, and received hormone treatments to induce decidualization as described previously (19, 21). Briefly, ovariectomized REA heterozygous or wild-type C57BL/6 mice (8 wk old) were injected sc with 100 ng of E2 for 3 days. After 2 days of rest, mice received 1 μg of P4 and 10 ng of E2 for 3 days. At 6 hours after the third P4 plus E2 injection, 1 uterine horn of each mouse was mechanically stimulated by an intraluminal injection of 20 μL of sesame oil. REA+/− or wild-type mice (n = 5 per group) were killed 72 hours after stimulation.
Isolation of mouse uterine stromal cells
Uterine stromal cells were isolated as described, with slight modification (22). Briefly, at 72 hours after intrauterine oil injection, the uterine horns were dissected, slit longitudinally, and then cut into small (5–6 mm) pieces. The tissues were digested in Hanks' balanced salt solution (HBSS) containing 6-g/L dispase (Invitrogen), and 25-g/L pancreatin (Invitrogen) for 1 hour at room temperature and then 10 minutes at 37°C. The tissue-digestion mixture was gently washed in HBSS, and the supernatant containing the endometrial epithelial clumps was discarded. The partially digested tissues were further digested in HBSS containing 0.5-g/L collagenase I (Invitrogen) for 45 minutes at 37°C and then vortexed to disperse endometrial stromal cells. The resultant cell suspension was filtered through a 70-μm gauze filter (Millipore, Decatur, Illinois) and centrifuged at 1000 rpm for 5 minutes. Cells were then resuspended in DMEM-F12 with 1% fetal bovine serum and seeded in 6-well culture plates. After a 30-minute adherence to the dish, the medium was changed to remove any epithelial cells, and purified stromal cells were utilized for further analysis.
Western blot analysis
Immunoblotting was performed as previously described (18). Briefly, cell extracts were prepared from hESCs with lysis buffer (25mM Tris-HCl [pH 7.4], 150mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 1% sodium dodecyl sulfate) supplemented with EDTA-free protease inhibitors (Roche, Indianapolis, Indiana) and phosphatase inhibitors (Roche). BCA protein assay kit (Pierce, Rockford, Illinois) was used to determine protein concentrations. Proteins (30 μg) were loaded and transferred onto nitrocellulose membranes, after separation on SDS-PAGE gels and subjected to immunoblotting with antibodies to REA (Millipore Co, Billerica, Massachusetts) and β-actin (Sigma-Aldrich, St. Louis, Missouri).
Immunocytochemistry and histological analyses
For immunofluorescence, cultured hESCs were fixed in formalin solution (Sigma, St Louis, Missouri) and stained as described (22), using primary antibodies to REA (AB10198; Millipore Co), PGR (M3568; Dako Co, Glostrup, Denmark), signal transducer and activator of transcription (STAT)3 (no. 9139; Cell Signaling, Beverly, Massachusetts), phospho-STAT3 (Tyr705, no. 9145; Cell Signaling), and suppressor of cytokine signaling (SOCS)3 (ab16030; Abcam, Cambridge, Massachusetts). Paraffin-embedded mouse endometrial tissue sections were subjected to hematoxylin and eosin staining or immunohistochemistry as described previously (18). Incubations with these primary antibodies were performed overnight at 4°C. Fluorescence (Cy3 or Delight 488)-conjugated antimouse IgG or antirabbit IgG (The Jackson Laboratory, Bar Harbor, Maine) was used as secondary antibody. The Phalloidin staining reagent (Alexa Fluor 488 Phalloidin, catalog no. A12379; Invitrogen) selectively stains F-actin. 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml in PBS) was used for counter staining.
RNA isolation and real-time RT-PCR analysis
Total RNA was extracted from cultured hESC or isolated mouse uterine stromal cells using the TRIzol RNA purification kit (Invitrogen) according the manufacturer's instructions. Reverse transcription was performed using a cDNA synthesis kit (Invitrogen) as recommended. The expression of mRNAs was examined by real-time RT-PCR (q-PCR) analysis using SYBR-Green Supermix (Bio-Rad Laboratories) and gene-specific primers (Table 1). The gene expression level at any given time point or under a given condition was quantified as fold change (mean ± SD) relative to that at 0 hours or control condition after normalization to the internal control 36B4, a constitutive gene encoding a ribosomal protein.
Table 1.
Primer Sets Used for q-PCR of Genes Studied
| Gene symbol | Forward | Reverse |
|---|---|---|
| GP130 (human) | CGGACAGCTTGAACAGAATGT | ACCATCCCACTCACACCTCA |
| IGFBP-1 (human) | CTATGATGGCTCGAAGGCTC | TTCTTGTTGCAGTTTGGCAG |
| IL11 (human) | CGAGCGGACCTACTGTCCTA | GCCCAGTCAAGTGTCAGGTG |
| JAK1 (human) | CTTTGCCCTGTATGACGAGAAC | ACCTCATCCGGTAGTGGAGC |
| P57 (human) | GCGGCGATCAAGAAGCTGT | GCTTGGCGAAGAAATCGGAGA |
| PGR (human) | TGTCGAGCTCACAGCGTTTCT | AGTGCCCGGGACTGGATAA |
| PRL (human) | CTACATCCATAACCTCTCCTCA | GGGCTTGCTCCTTGTCTTC |
| PRLR2 (human) | TCTCCACCTACCCTGATTGAC | CGAACCTGGACAAGGTATTTCTG |
| REA (human) | GCACTGAGCAAGAACCCTGG | CTGGCGATCGTCTTGGAGAT |
| SOCS1 (human) | CACGCACTTCCGCACATTC | TAAGGGCGAAAAAGCAGTTCC |
| SOCS3 (human) | CCTGCGCCTCAAGACCTTC | GTCACTGCGCTCCAGTAGAA |
| SOCS5 (human) | GTGCCACAGAAATCCCTCAAA | TCTCTTCGTGCAAGTCTTGTTC |
| STAT3 (human) | CAGCAGCTTGACACACGGTA | AAACACCAAAGTGGCATGTGA |
| TXN (human) | GTGAAGCAGATCGAGAGCAAG | CGTGGCTGAGAAGTCAACTACTA |
| 36B4 (human) | GTGTTCGACAATGGCAGCAT | GACACCCTCCAGGAAGCGA |
| Bmp2 (mouse) | AAAGCGTCAAGCCAAACACA | ACCCCACATCACTGAAGTCCA |
| Cx43 (mouse) | CTATCGTGGATCAGCGACCTTC | CACGGGAACGAAATGAACACC |
| Esr1 (mouse) | TGAATGGCTAGGCTTCTCTTG | GCTCTCCCAGTTTCCACATC |
| Prl8a2 (mouse) | AGTCTGAACTCATCCTGCTTGG | TTGATGCAGCTTTCTCCCACAG |
| REA (mouse) | AGTGCTGCCGTCCATTGTTAA | TCTTCGGATCAACAGGGACAC |
| Stat3 (mouse) | CCTGAAGACCAAGTTCATCTGTGT | CACACAAGCCATCAAACTCTGGTC |
| 36b4 (mouse) | CATCACCACGAAAATCTCCA | TTGTCAAACACCTGCTGGAT |
Gene expression microarray analysis
Independent hESC lines were established from individual biopsies of proliferative stage endometria, each cell line being from a different woman. These cultures were transfected with control (GL3 luciferase) or REA-specific siRNA for 24 hours. siRNAs were then removed, and cells were then treated for 24 or 96 hours in the presence of the hormone cocktail containing 10nM E2, 1μM P4, and 0.5mM 8-Br-cAMP. Cells were harvested, and total RNA was prepared with TRIzol reagent (Invitrogen). RNA integrity was verified using an Agilent 2100 Bioanalyser (Agilent Technologies, Inc, Santa Clara, California) at the Biotechnology Center of the University of Illinois, Urbana.
RNA samples were processed for microarray hybridization using GeneChip Human Genome U133 A Plus 2.0 arrays (Affymetrix, Inc, Santa Clara, California), following established Affymetrix protocols as described (23). Quality control assessment, data processing, and statistical analysis were done in R, a software module for statistical computing and graphics, as described previously (24). Fold differences in gene expression and false discovery rates set at more than or equal to 1.8 and less than 0.05, respectively, were considered significant. The significantly regulated genes were further analyzed by gene ontology and pathways using PANTHER Classification Software. Gene expression microarray data are available from the Gene Expression Omnibus under accession number GSE42376.
Statistical analysis
RNA and protein samples were prepared from at least 3 separate primary cultures and were subjected to the same experimental treatments. The real-time RT-PCR results are expressed as mean ± SD of 3 separate experiments. Statistical significance was assessed by ANOVA, and a significance level of P < .05, indicated by an asterisk in the figures, was considered significant.
Results
REA expression decreases during differentiation of hESC
We first examined the expression of REA during the time course of hESC differentiation and found that REA expression decreased as differentiation of hESC proceeded. REA mRNA decreased to approximately half (relative to the undifferentiated state) by 2 days and remained at this level throughout the remainder of the 8 days examined (Figure 1A). This decrease was also observed for REA protein, monitored by Western blotting and immunofluorescence cytochemistry (Figure 1, B and C) over the 8-day time period.
Figure 1.

REA expression during hESC differentiation. Primary hESC cultures were exposed to a hormone cocktail containing 10nM E2, 1μM P4, and 0.5mM 8-Br-cAMP to initiate in vitro differentiation. REA decreased as hESC differentiation proceeded as shown by q-PCR (A), Western blot (B), and immunofluorescence cytochemistry (C) analyses. *P < .05.
REA knockdown both accelerates and enhances hESC differentiation
To examine whether REA levels might affect hESC differentiation, we reduced REA expression by siRNA knockdown, as schematized in Figure 2A. The knockdown of REA was confirmed at the mRNA level (Figure 2B) and also at the protein level by Western blotting (Figure 2C) and cell immunofluorescence microscopy (Figure 2D).
Figure 2.

Impact of REA knockdown on hESC differentiation. siRNA transfection was performed to assess the impact of REA on hESC differentiation. (A) Schematic of the time line of siRNA transfection and in vitro hESC differentiation. Knockdown of REA was assessed by q-PCR (B), Western blot (C), and immunofluorescence (D) analyses at the times indicated. *P < .05. Fold change in mRNA in B is shown relative to stromal cells transfected with si-GL3 and before hormone cocktail (d 0), which is set at 1.0.
Interestingly, morphological analysis by phase contrast microscopy and Phalloidin fluorescent staining of cytoskeletal F-actin revealed that hESCs with decreased REA started to display a round shape and cytoskeletal changes typical of decidual transformation as early as day 4 of differentiation, 3–4 days in advance of that seen in wild-type cells (Figure 3A). Moreover, evidence that knockdown of REA accelerated differentiation of the stromal cells was afforded by precocious and enhanced expression of 2 hESC differentiation markers, prolactin (PRL) and IGF binding protein 1 (IGFBP-1), in cells in which REA was reduced (Figure 3B). The increased expression level of these 2 differentiation markers was greatest at day 4, with a 6-fold increase in PRL and a 5-fold increase of IGFBP-1 expression over that of control si-GL3 transduced cells. These elevated expression levels then declined to those of control si-GL3 cells by day 10 for PRL and day 8 for IGFBP-1. Because PGR is well known to be an essential regulator of hESC differentiation (6, 25), we also investigated the impact of REA on PGR. Interestingly, expression of PGR was greatly increased as well as being temporally accelerated by REA knockdown. As shown in Figure 3B, the enhancement of PGR mRNA expression was 5-fold over that of control cells at day 2, and this peak expression level occurred 4 days earlier than in control cells. Immunofluorescence analyses for PGR also confirmed increased PGR protein in si-REA vs control si-GL3 hESCs, monitored at day 4 of differentiation (Figure 3C). Further, the expression of ESR1 target genes, such as PGR, cyclin-dependent kinase inhibitor 1C (P57), and thioredoxin (TXN) (Figure 3D) were increased with reduction in REA, which might contribute to acceleration and enhancement of hESC differentiation. Thus, REA knockdown appeared to accelerate and also enhance hESC differentiation, based on the analysis of cell morphology and expression of differentiation markers such as PRL, IGFBP-1, and PGR.
Figure 3.

REA reduction both accelerates and enhances hESC differentiation. Stromal cell differentiation was accelerated in time and enhanced in magnitude after the reduction of REA by siRNA knockdown, as indicated by (A) phase contrast microscopy (left) and Phalloidin/DAPI fluorescent staining (right), as well as (B) gene expression of hESC differentiation markers, PRL, IGFBP-1, and PGR, by q-PCR. *P < .05. C, Up-regulation of PGR in differentiating hESC after REA knockdown was further confirmed by immunofluorescence, monitored after 4 days of exposure to the decidualization hormone cocktail. D, Expression of the estrogen-regulated genes PGR, P57, and TXN in hESC treated with control si-GL3 or si-REA and then exposed to the hormone cocktail for 24 hours before RNA isolation and analysis by q-PCR. Scale bar, 100 μm. P < .05.
Reduction of REA enhances the decidualization response in the mouse uterus in vivo
We wished to determine whether the effects of REA that we observed on the differentiation of hESCs in vitro were also observed in the uteri of mice in vivo. Therefore, we examined the decidualization response in knockout mice heterozygous for REA deletion (REA+/−). These mice, as described previously (19), express half the wild-type control level of REA. After hormonal preparation, decidualization was induced by an artificial oil stimulus in 1 uterine horn with the contralateral horn remaining unperturbed as the control, as shown in Figure 4A. An enhanced decidualization response, monitored at 72 hours, was observed in REA+/− mice after the artificial decidualization stimulus, as indicated by uterine morphology (Figure 4B) and increased uterine horn weight in REA+/− vs REA+/+ mice (Figure 4C). Hematoxylin and eosin staining and immunohistochemical staining for REA (Figure 4D) also documented the enhanced decidualization response in the REA heterozygous mouse uterus and revealed that REA is normally expressed throughout the decidualized uterus and is present at a reduced level in the REA+/− decidualized uterus.
Figure 4.
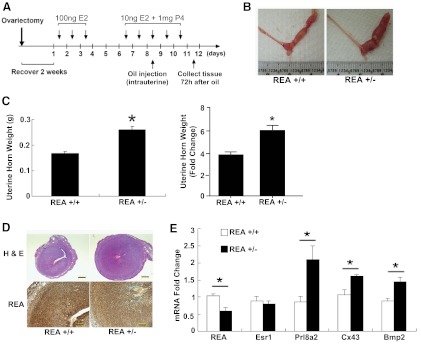
Enhanced decidualization response in REA+/− mice. The mouse model of artificial decidualization response was used to study the role of REA in stromal decidualization in vivo. A, Schematic of the decidualization protocol. Enhanced decidualization response in REA+/− mice was evident after the artificial decidualization (oil) stimulus for 72 hours, indicated by uterine morphology (B), uterine horn wet weight, shown as gross weight (left) and uterine horn weight fold change (right) relative to the noninjected horn (C), uterine hematoxylin and eosin histology and REA immunohistochemistry (D), as well as gene expression profiling of decidual Prl8a2, Cx43, and Bmp2 in isolated uterine stromal cells (E). The approximately 50% reduction of REA mRNA and protein in the heterozygous REA+/− uterus was confirmed by immunohistochemistry (D) and q-PCR (E). Scale bar in hematoxylin and eosin stain panels in D, 500 μm; in REA panels in D, 200 μm. *P < .05.
In endometrial stromal cells isolated from REA+/− uteri, where REA mRNA was 50% reduced (Figure 4E), expression levels of the decidualization markers decidual/trophoblast PRL-related protein (Prl8a2), connexin 43 (Cx43), and bone morphogenetic protein 2 (Bmp2) transcripts were significantly increased. As expected, the expression of the estrogen receptor, Esr-1, did not differ in stromal cells from REA wild-type (+/+) or REA heterozygous (+/−) uteri. These data further support the observations in hESC in vitro in Figure 2, B–D, by documenting that the decidualization response was likewise enhanced by reduced REA at the in vivo level.
STAT3 expression and activity are associated with changes in REA level during hESC decidualization in vitro
To investigate the underlying molecular pathways through which REA impacts hESC differentiation, global gene expression profiling was performed in differentiating hESC by microarray analysis. The expression level of 237 genes was increased by REA knockdown, including 109 genes at 24 hours and 165 genes at 96 hours after exposure to the cocktail of E2, P4, and 8-Br-cAMP. On the other hand, there were 345 genes down-regulated by REA knockdown, with 270 genes at 24 hours and 182 genes at 96 hours. Among these genes, the most striking were changes in the expression of multiple components of the Janus kinase (JAK)/STAT signaling pathway. These included ligands, receptors, and modulators of the STAT3 signaling pathway, which is particularly interesting, because STAT3 has been documented to be an essential mediator of hESC decidualization (26).
We first confirmed the expression profile of several STAT3 pathway components, including the upstream signaling cytokine, IL-11, the receptors, PRL receptor 2 (PRLR2) and membrane glycoprotein 130 (GP130), and also STAT3 by q-PCR. As shown in Figure 5A, the STAT3 expression level was higher after knockdown of REA during hESC differentiation, being 2-fold elevated at 24 hours and 6-fold elevated at 96 hours, whereas STAT5A and STAT5B expressions were not changed (data not shown). Interestingly, the expression of several suppressors of the STAT3 signaling pathway, including SOCS1, SOCS3, and SOCS5, were reduced after REA knockdown (Figure 5B). The elevated expression of STAT3 and the much higher presence of phospho-STAT3 associated with REA knockdown was also confirmed at the protein level by immunofluorescence analysis (Figure 5C), where enhanced accumulation of STAT3 nuclear staining in differentiating hESC after REA knockdown was noted. By contrast, SOCS3 protein staining was detected in both the cytoplasm and nucleus of cells, and the proportion of positively stained hESC was significantly reduced with si-REA treatment (Figure 5C). These observations indicate that REA impacts expression of these important STAT3 pathway components and thus appears to elevate pathway signaling activity during hESC differentiation.
Figure 5.

The expression of STAT signaling pathway components was altered by REA knockdown during hESC differentiation. Expression of the STAT signaling pathway components, IL-11, STAT3, GP130, and PRLR2, was enhanced by knockdown of REA using siRNA during hESC decidualization (A), whereas the mRNA levels of several suppressors of cytokine signaling (SOCS1, SOCS3, and SOCS5) were reduced, as shown by q-PCR analysis (B). Elevated total and phosphorylated STAT3 protein, as well as reduced SOCS3 protein, in hESC after REA knockdown was observed by immunofluoresence in hESC at 96 hours of differentiation (C). Immunofluorescence of total STAT3, phospho-STAT3 (p-STAT3), or SOCS3 was merged with DAPI staining and presented under a higher magnification (×100) (C, right) to show cellular location of these proteins. Scale bar, 100 μm. *P < .05.
Stat3 signaling pathway activity is also influenced by REA level in the mouse uterus during decidualization in vivo
Because Stat3 is known to be required for mouse implantation and decidualization (27–29), we examined the influence of REA on Stat3 during decidualization in the mouse in vivo. As shown in Figure 6A, Stat3 mRNA was significantly higher in endometrial stromal cells isolated from REA+/− vs REA wild-type uteri. Moreover, stronger Stat3 and phospho-Stat3 immunostaining were also observed in REA+/− decidua at 72 hours after intrauterine oil stimulation (Figure 6, A and B), demonstrating more Stat3 and also more active Stat3 associated with the enhanced decidualization observed in the REA heterozygous mouse uterus in vivo. Also, the strong Socs3 staining observed in wild-type decidual cells was reduced in the REA+/− uterus. These findings demonstrate that activation of the Stat3 signaling pathway was also greatly affected by REA during mouse uterine decidualization in vivo.
Figure 6.
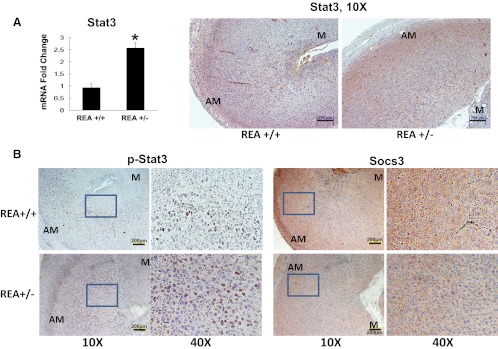
Enhanced Stat3 signaling in the REA heterozygous knockout (REA+/−) mouse uterus during decidualization in vivo. Increased Stat3 in REA+/− vs wild-type REA+/− mouse uteri at 72 hours after initiation of decidualization was seen by q-PCR (A) and immunohistochemistry (B). Scale bar, 200 μm. Levels of p-Stat3 and Socs3 protein were compared between wild-type (REA+/+) and REA+/− decidual uteri by immunohistochemistry. Magnification ×10 and ×40 (B). AM, antimesometrial; M, mesometrial. Scale bars: in panel A, 200 μm; in panel B p-Stat3, 200 μm; in panel B Socs3, 200 μm. *P < .05.
Discussion
Our findings highlight the pleiotropic ability of REA to modulate key aspects of decidualization, with its reduction accelerating and amplifying the decidualization process. The observations indicate that REA normally acts as a safeguard against premature and untimely endometrial decidualization, thereby maintaining optimal function of the endometrium for implantation. Our findings have important implications for understanding subfertility and deficiencies in embryo implantation and maintenance of early stages of pregnancy. The promotion of early decidualization in the face of reduced REA appears to result in asynchrony between the uterine endometrium and the implantation-ready embryo that may lead to the subfertility or complete infertility that we have previously reported in heterozygous or homozygous uterine conditional REA knockout animals, respectively (17). Of interest, Pabona et al have proposed involvement of a similar model, in which deletion of the transcriptional repressor Kruppel-like factor 9 changes REA levels in the endometrium, with temporal asymmetry between the maternal uterus and the embryo that resulted in reduced fertility (30) .
We have shown previously that reduction in REA in heterozygous conditional uterine-specific REA knockout animals and also in heterozygous conventional REA knockout animals resulted in a uterus that was hyperstimulated by E2 and showed enhanced cell proliferation (17, 19). These animals had fewer and smaller litters, indicating that the proper level of REA is required for optimal fertility (17). The current findings highlight that acceleration in time and enhancement in magnitude of the decidualization process observed in REA+/− uteri and endometrial stromal cells contribute to endometrial desynchronization, making it less suitable for effective implantation and maintenance of early pregnancy. We submit that an analogous embryo-uterine desynchronization occurs in human in vitro fertilization cycles, where advanced endometrial development has been attributed to suboptimal implantation rates (31–36).
Our initial studies identified REA as an estrogen receptor-interacting protein, and showed that REA reduced the transcriptional activity of estrogen receptor in human breast cancer cells and in the uterus and mammary glands of mice (37–39). In the studies reported here, we show that REA expression decreased as hESC differentiation progressed. Previous studies in transgenic animals, where levels of REA were reduced by conventional or conditional knockout, revealed that uterine development, receptive function, and fertility were all influenced by this important steroid receptor corepressor protein. Consistent with this hypothesis, we found that knockdown of REA in hESC increased the expression of several ESR1 target genes, such as PGR, P57, and TXN, confirming that REA suppresses estrogen actions in hESC during differentiation. In conditional murine models, REA knockdown impacted the entire spectrum of genes involved in STAT3-mediated uterine cell differentiation. Taken together, our data reveal that REA serves as a repressor-like restraint on endometrial stromal differentiation and that its level is critical for the decidualization response. The findings provide new understanding of REA function in female reproduction and underscore that steroid hormone receptors and their coregulators coordinate actions to provide precise control of the timing and magnitude of uterine decidualization that is essential for optimal fertility.
Implantation is a complex phenomenon requiring a succession of properly timed and quantitatively optimized biochemical and cellular interactions between the receptive uterus and the developing embryo. This process is driven by cascades of signaling events in the endometrium, regulated by the steroid hormones estrogen and P4, through their receptors and coregulators, which in turn regulate the production of key signaling molecules, such as the BMP2, indian hedgehog, leukemia inhibitory factor, and others (22, 40, 41). The proper synchronization of uterine events and concomitant development of the embryo are essential for successful implantation. From our unpublished work, we know from immunohistochemistry that REA is present in the uterus at days 1–6 during normal implantation and early pregnancy in wild-type mice and therefore is expressed in the periimplantation period before decidualization. Our current findings indicate that the corepressor REA restrains uterine decidualization and imply that alterations in its level or activity could disrupt endometrial differentiation, leading to early pregnancy loss and infertility.
In investigating the impact of REA on the expression of regulators of decidualization, we found that the levels of IGFBP-1, PGR, and STAT3 were significantly increased during stromal cell decidualization when REA was reduced. In the mouse, expression of Pgr in endometrial stromal cells is controlled by estrogen (42, 43), and Pgr knockout mice display a severe decidualization defect (7). It is also of interest that our studies, revealing accelerated decidualization with reduction in REA, showed that this was accompanied by increased IL-11 and STAT3, because these factors have been shown to be important in regulating hESC differentiation (26), and uterine STAT3 expression is known to be stimulated by E2 (29). Moreover, successful implantation in the mouse is dependent on STAT3 signaling pathways (27). Activation of STAT3 signaling in hESC by paracrine cytokine IL11 is also required for initiation and progression of hESC decidualization (26). Although further investigation is required to explore the mechanism by which stromal STAT3 expression is impacted by REA, PGR might serve as a mediator based on the observation that PGR is required for STAT3 expression in differentiating hESC (26). Collectively, these data demonstrate that as a corepressor of ESR1, REA controls stromal differentiation, at least partially via regulation of other master regulators, and suggest an emerging STAT3 signaling network during decidualization, in which REA serves as a central coregulator.
IL-11, which belongs to the IL-6 family of cytokines, works through receptor heteromultimers related to PRLR2 and GP130, ultimately activating the JAK/STAT pathway. Hence, it is notable that the acceleration of endometrial stromal cell differentiation that we observed was accompanied by early increases not only in IL-11 but also in the 2 receptors PRLR2 and GP130 and STAT3. The SOCS protein family (suppressors of cytokine signaling) regulates a negative feedback loop in the JAK/STAT circuitry to block the activities of IL-11 (42). Thus, it is noteworthy that the expression of several SOCS family members was reduced during decidualization when REA declined. The combination of increased STAT3 and reduction in suppressors of cytokine signaling may both contribute to promoting and progressing endometrial decidualization. The observations by others (26) that overexpression of SOCS3 in hESC retarded decidualization is consistent with the hypothesis that SOCS3 is an important inhibitor of decidualization and that its reduced expression when REA is low may contribute to the acceleration of decidualization. In other data, a STAT3 peptide inhibitor prevented implantation (27), supporting that perturbations in REA that impact STAT3 may cause infertility by affecting the decidualization process.
Our findings imply that an optimal level of REA is required for the precise control of decidualization and also suggest that a proper decidualization response under normal physiological estrogen range might be essential for successful pregnancy establishment and outcome. It has been suggested that one cause of the low rate of implantation after in vitro fertilization and embryo transfer technologies could be due to the high estrogen level resulting from ovarian hyperstimulation or as a direct effect of gonadotropins on the endometrium (9, 10, 36). In the mouse, Ma et al also reported that although estrogen within physiological concentrations can initiate implantation, the window of uterine receptivity for implanting of the embryo rapidly closes at higher levels of estrogen in the mouse (33). These studies suggest that estrogen action needs to be tightly controlled for normal uterine function. Indeed, in our previous study, REA conditional heterozygous female mice showed hyperresponsiveness to estrogen but also displayed a subfertility phenotype that included a lower frequency of pregnancies and smaller litter size (17). Reduced levels of REA in the uterus may lead to the altered stromal decidualization observed in this report and thus the impairment of fertility.
In summary, we show that decidualization in endometrial stromal cells occurs concomitantly with a decrease in REA. Reducing the level of REA experimentally leads to an acceleration of the timing and magnitude of the decidualization program, marked by higher expression of ESR1 target genes and several key mediators of decidualization, including PGR, IL-11, and STAT3. Our studies presented here highlight the role of REA as a restraint in controlling the stromal decidualization response, thus providing new insight into REA functions in uterine biology. Furthermore, this new understanding of REA's actions in regulating endometrial decidualization may help inform the improvement of fertility therapies in the clinic.
Acknowledgments
We thank Beth Papanek for excellent assistance in some of this research.
This work was supported by National Institutes of Health (NIH) Grant U54 HD055787 as part of the Eunice Kennedy Shriver National Institute of Child Health and Human Development/NIH Centers Program in Reproduction and Infertility Research.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Bmp2
- bone morphogenetic protein 2
- 8-Br-cAMP
- 8-bromo-cAMP
- Cx43
- connexin 43
- DAPI
- 4′,6-diamidino-2-phenylindole
- E2
- estradiol
- ESR1
- estrogen receptor α
- GP130
- membrane glycoprotein 130
- HBSS
- Hanks' balanced salt solution
- hESC
- human endometrial stromal cell
- IGFBP-1
- IGF binding protein 1
- JAK
- Janus kinase
- P4
- progesterone
- P57
- cyclin-dependent kinase inhibitor 1C
- PGR
- P4 receptor
- PRL
- prolactin
- Prl8a2
- PRL-related protein
- PRLR
- PRL receptor
- q-PCR
- real-time RT-PCR
- REA
- repressor of estrogen receptor activity
- si-GL3
- GL3 luciferase control siRNA
- si-REA
- REA siRNA
- siRNA
- small interfering RNA
- SOCS
- suppressor of cytokine signaling
- STAT
- signal transducer and activator of transcription
- TXN
- thioredoxin.
References
- 1. Wang H, Dey SK. Roadmap to embryo implantation: clues from mouse models. Nat Rev Genet. 2006;7:185–199 [DOI] [PubMed] [Google Scholar]
- 2. Bagchi IC, Li Q, Cheon YP. Role of steroid hormone-regulated genes in implantation. Ann NY Acad Sci. 2001;943:68–76 [DOI] [PubMed] [Google Scholar]
- 3. Carson DD, Bagchi I, Dey SK, et al. Embryo implantation. Dev Biol. 2000;223:217–237 [DOI] [PubMed] [Google Scholar]
- 4. Critchley HO, Saunders PT. Hormone receptor dynamics in a receptive human endometrium. Reprod Sci. 2009;16:191–199 [DOI] [PubMed] [Google Scholar]
- 5. Curtis SW, Clark J, Myers P, Korach KS. Disruption of estrogen signaling does not prevent progesterone action in the estrogen receptor α knockout mouse uterus. Proc Natl Acad Sci USA. 1999;96:3646–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gellersen B, Brosens J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: a decidualizing affair. J Endocrinol. 2003;178:357–372 [DOI] [PubMed] [Google Scholar]
- 7. Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278 [DOI] [PubMed] [Google Scholar]
- 8. Ramathal CY, Bagchi IC, Taylor RN, Bagchi MK. Endometrial decidualization: of mice and men. Semin Reprod Med. 2010;28:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. La Marca A, Carducci Artenisio A, Stabile G, Rivasi F, Volpe A. Evidence for cycle-dependent expression of follicle-stimulating hormone receptor in human endometrium. Gynecol Endocrinol. 2005;21:303–306 [DOI] [PubMed] [Google Scholar]
- 10. Lei ZM, Reshef E, Rao V. The expression of human chorionic gonadotropin/luteinizing hormone receptors in human endometrial and myometrial blood vessels. J Clin Endocrinol Metab. 1992;75:651–659 [DOI] [PubMed] [Google Scholar]
- 11. Gellersen B, Brosens IA, Brosens JJ. Decidualization of the human endometrium: mechanisms, functions, and clinical perspectives. Semin Reprod Med. 2007;25:445–453 [DOI] [PubMed] [Google Scholar]
- 12. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474 [DOI] [PubMed] [Google Scholar]
- 13. Simmen RC, Eason RR, McQuown JR, et al. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Kruppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem. 2004;279:29286–29294 [DOI] [PubMed] [Google Scholar]
- 14. Mukherjee A, Soyal SM, Fernandez-Valdivia R, et al. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol Cell Biol. 2006;26:6571–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai MJ, O'Malley BW. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science. 1998;279:1922–1925 [DOI] [PubMed] [Google Scholar]
- 16. Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci USA. 2000;97:6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S, Yoon S, Zhao Y, et al. Uterine development and fertility are dependent on gene dosage of the nuclear receptor coregulator REA. Endocrinology. 2012;153:3982–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park S, Zhao Y, Yoon S, et al. Repressor of estrogen receptor activity (REA) is essential for mammary gland morphogenesis and functional activities: studies in conditional knockout mice. Endocrinology. 2011;152:4336–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park SE, Xu J, Frolova A, Liao L, O'Malley BW, Katzenellenbogen BS. Genetic deletion of the repressor of estrogen receptor activity (REA) enhances the response to estrogen in target tissues in vivo. Mol Cell Biol. 2005;25:1989–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryan IP, Schriock ED, Taylor RN. Isolation, characterization, and comparison of human endometrial and endometriosis cells in vitro. J Clin Endocrinol Metab. 1994;78:642–649 [DOI] [PubMed] [Google Scholar]
- 21. Wang W, Li Q, Bagchi IC, Bagchi MK. The CCAAT/enhancer binding protein β is a critical regulator of steroid-induced mitotic expansion of uterine stromal cells during decidualization. Endocrinology. 2010;151:3929–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Q, Kannan A, Wang W, et al. Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J Biol Chem. 2007;282:31725–31732 [DOI] [PubMed] [Google Scholar]
- 23. Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor β on gene networks regulated by estrogen receptor α in breast cancer cells. Endocrinology. 2006;147:4831–4842 [DOI] [PubMed] [Google Scholar]
- 24. Chang EC, Charn TH, Park SH, et al. Estrogen Receptors α and β as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol Endocrinol. 2008;22:1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wetendorf M, DeMayo FJ. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol Cell Endocrinol. 2012;357:108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dimitriadis E, Stoikos C, Tan YL, Salamonsen LA. Interleukin 11 signaling components signal transducer and activator of transcription 3 (STAT3) and suppressor of cytokine signaling 3 (SOCS3) regulate human endometrial stromal cell differentiation. Endocrinology. 2006;147:3809–3817 [DOI] [PubMed] [Google Scholar]
- 27. Catalano RD, Johnson MH, Campbell EA, Charnock-Jones DS, Smith SK, Sharkey AM. Inhibition of Stat3 activation in the endometrium prevents implantation: a nonsteroidal approach to contraception. Proc Natl Acad Sci USA. 2005;102:8585–8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shuya LL, Menkhorst EM, Yap J, Li P, Lane N, Dimitriadis E. Leukemia inhibitory factor enhances endometrial stromal cell decidualization in humans and mice. PLoS ONE. 2011;6:e25288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teng CB, Diao HL, Ma XH, Xu LB, Yang ZM. Differential expression and activation of Stat3 during mouse embryo implantation and decidualization. Mol Reprod Dev. 2004;69:1–10 [DOI] [PubMed] [Google Scholar]
- 30. Pabona JM, Velarde MC, Zeng Z, Simmen FA, Simmen RC. Nuclear receptor co-regulator Kruppel-like factor 9 and prohibitin 2 expression in estrogen-induced epithelial cell proliferation in the mouse uterus. J Endocrinol. 2009;200:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casper RF. It's time to pay attention to the endometrium. Fertil Steril. 2011;96:519–521 [DOI] [PubMed] [Google Scholar]
- 32. Koot YE, Teklenburg G, Salker MS, Brosens JJ, Macklon NS. Molecular aspects of implantation failure. Biochim Biophys Acta. 2012;1822(12):1943–1950 [DOI] [PubMed] [Google Scholar]
- 33. Ma WG, Song H, Das SK, Paria BC, Dey SK. Estrogen is a critical determinant that specifies the duration of the window of uterine receptivity for implantation. Proc Natl Acad Sci USA. 2003;100:2963–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saadat P, Boostanfar R, Slater CC, Tourgeman DE, Stanczyk FZ, Paulson RJ. Accelerated endometrial maturation in the luteal phase of cycles utilizing controlled ovarian hyperstimulation: impact of gonadotropin-releasing hormone agonists versus antagonists. Fertil Steril. 2004;82:167–171 [DOI] [PubMed] [Google Scholar]
- 35. Toner JP, Hassiakos DK, Muasher SJ, Hsiu JG, Jones HW., Jr. Endometrial receptivities after leuprolide suppression and gonadotropin stimulation: histology, steroid receptor concentrations, and implantation rates. Ann NY Acad Sci. 1991;622:220–229 [DOI] [PubMed] [Google Scholar]
- 36. Valbuena D, Jasper M, Remohi J, Pellicer A, Simon C. Ovarian stimulation and endometrial receptivity. Human Reproduction. 1999;14(suppl 2):107–111 [DOI] [PubMed] [Google Scholar]
- 37. Delage-Mourroux R, Martini PG, Choi I, Kraichely DM, Hoeksema J, Katzenellenbogen BS. Analysis of estrogen receptor interaction with a repressor of estrogen receptor activity (REA) and the regulation of estrogen receptor transcriptional activity by REA. J Biol Chem. 2000;275:35848–35856 [DOI] [PubMed] [Google Scholar]
- 38. Montano MM, Ekena K, Delage-Mourroux R, Chang W, Martini P, Katzenellenbogen BS. An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens. Proc Natl Acad Sci USA. 1999;96:6947–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mussi P, Liao L, Park SE, et al. Haploinsufficiency of the corepressor of estrogen receptor activity (REA) enhances estrogen receptor function in the mammary gland. Proc Natl Acad Sci USA. 2006;103:16716–16721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee K, Jeong J, Kwak I, et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006;38:1204–1209 [DOI] [PubMed] [Google Scholar]
- 41. Dey SK, Lim H, Das SK, et al. Molecular cues to implantation. Endocr Rev. 2004;25:341–373 [DOI] [PubMed] [Google Scholar]
- 42. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283 [DOI] [PubMed] [Google Scholar]
- 43. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci USA. 2010;107:19272–19277 [DOI] [PMC free article] [PubMed] [Google Scholar]