

Abstract
Motivation: Statistical methods for comparing relative rates of synonymous and non-synonymous substitutions maintain a central role in detecting positive selection. To identify selection, researchers often estimate the ratio of these relative rates (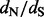 ) at individual alignment sites. Fitting a codon substitution model that captures heterogeneity in
) at individual alignment sites. Fitting a codon substitution model that captures heterogeneity in 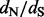 across sites provides a reliable way to perform such estimation, but it remains computationally prohibitive for massive datasets. By using crude estimates of the numbers of synonymous and non-synonymous substitutions at each site, counting approaches scale well to large datasets, but they fail to account for ancestral state reconstruction uncertainty and to provide site-specific
across sites provides a reliable way to perform such estimation, but it remains computationally prohibitive for massive datasets. By using crude estimates of the numbers of synonymous and non-synonymous substitutions at each site, counting approaches scale well to large datasets, but they fail to account for ancestral state reconstruction uncertainty and to provide site-specific 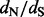 estimates.
estimates.
Results: We propose a hybrid solution that borrows the computational strength of counting methods, but augments these methods with empirical Bayes modeling to produce a relatively fast and reliable method capable of estimating site-specific 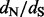 values in large datasets. Importantly, our hybrid approach, set in a Bayesian framework, integrates over the posterior distribution of phylogenies and ancestral reconstructions to quantify uncertainty about site-specific
values in large datasets. Importantly, our hybrid approach, set in a Bayesian framework, integrates over the posterior distribution of phylogenies and ancestral reconstructions to quantify uncertainty about site-specific 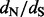 estimates. Simulations demonstrate that this method competes well with more-principled statistical procedures and, in some cases, even outperforms them. We illustrate the utility of our method using human immunodeficiency virus, feline panleukopenia and canine parvovirus evolution examples.
estimates. Simulations demonstrate that this method competes well with more-principled statistical procedures and, in some cases, even outperforms them. We illustrate the utility of our method using human immunodeficiency virus, feline panleukopenia and canine parvovirus evolution examples.
Availability: Renaissance counting is implemented in the development branch of BEAST, freely available at http://code.google.com/p/beast-mcmc/. The method will be made available in the next public release of the package, including support to set up analyses in BEAUti.
Contact: philippe.lemey@rega.kuleuven.be or msuchard@ucla.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Quantifying selective pressures on protein-coding genes is central to the goal of characterizing Darwinian processes in evolutionary biology. Among comparative and summary statistic approaches, the relative rate of silent and replacement substitutions represents one of the most popular measures to detect the molecular footprint of selection. Non-synonymous mutations that offer fitness advantages are expected to become fixed at a higher rate than synonymous mutations, implying that a non-synonymous/synonymous substitution rate ratio 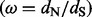 greater than one provides evidence for diversifying positive selection.
greater than one provides evidence for diversifying positive selection.
Although estimation of 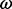 has led to the identification of positive selection in several systems (e.g. Hughes and Nei, 1988; Messier and Stewart, 1997), there are clear boundaries to the conditions under which it can reveal an unambiguous trace of molecular adaptation. First, an excess of non-synonymous substitutions over synonymous substitutions is almost invariably restricted to a handful of amino acid sites responsible for adaptive evolution. Therefore, sensible
has led to the identification of positive selection in several systems (e.g. Hughes and Nei, 1988; Messier and Stewart, 1997), there are clear boundaries to the conditions under which it can reveal an unambiguous trace of molecular adaptation. First, an excess of non-synonymous substitutions over synonymous substitutions is almost invariably restricted to a handful of amino acid sites responsible for adaptive evolution. Therefore, sensible 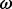 estimates need to take into account the variation in selection intensity across codon sites (Nielsen and Yang, 1998). Moreover, although divergent sequences can yield considerable information to estimate non-synonymous and synonymous substitution rates, the ratio of these rates may offer little insight when inferred from segregating polymorphisms within a single population (Kryazhimskiy and Plotkin, 2008). Finally, it is also important to distinguish between different selective regimes underlying molecular adaptation. Diversifying selection maintains amino-acid diversity at a given site and naturally results in elevated
estimates need to take into account the variation in selection intensity across codon sites (Nielsen and Yang, 1998). Moreover, although divergent sequences can yield considerable information to estimate non-synonymous and synonymous substitution rates, the ratio of these rates may offer little insight when inferred from segregating polymorphisms within a single population (Kryazhimskiy and Plotkin, 2008). Finally, it is also important to distinguish between different selective regimes underlying molecular adaptation. Diversifying selection maintains amino-acid diversity at a given site and naturally results in elevated 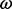 values, whereas directional selection may operate through a restricted number of amino-acid replacements, which has less impact on
values, whereas directional selection may operate through a restricted number of amino-acid replacements, which has less impact on 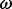 but can lead to rapid fixation of a new allele in the population. The former is ubiquitous in antagonistic systems such as pathogen-host interactions (Yang and Bielawski, 2000). Not surprisingly,
but can lead to rapid fixation of a new allele in the population. The former is ubiquitous in antagonistic systems such as pathogen-host interactions (Yang and Bielawski, 2000). Not surprisingly, 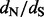 estimation methods have been frequently applied to viral gene sequences to detect escape from host immune responses or adaptation to novel hosts. Rapidly evolving viruses benefit from the ability to generate adaptive mutations de novo, whereas other organisms must rely on pre-existing variation maintained by population structure or balancing selection (Pybus and Rambaut, 2009). The impact of immune pressure on the genetic diversity of viruses can be highly dependent on how potent the immune responses are to different viral variants, a concept that has been put forward within the framework of phylodynamics (Grenfell et al., 2004). For this reason, methods to estimate
estimation methods have been frequently applied to viral gene sequences to detect escape from host immune responses or adaptation to novel hosts. Rapidly evolving viruses benefit from the ability to generate adaptive mutations de novo, whereas other organisms must rely on pre-existing variation maintained by population structure or balancing selection (Pybus and Rambaut, 2009). The impact of immune pressure on the genetic diversity of viruses can be highly dependent on how potent the immune responses are to different viral variants, a concept that has been put forward within the framework of phylodynamics (Grenfell et al., 2004). For this reason, methods to estimate 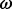 have become an integral part of the ‘phylodynamic toolbox’ (Pybus and Rambaut, 2009), which generally refers to recent statistical and computational developments to simultaneously estimate epidemic and spatiotemporal dynamics from viral genes sequences (Lemey et al., 2009, 2010).
have become an integral part of the ‘phylodynamic toolbox’ (Pybus and Rambaut, 2009), which generally refers to recent statistical and computational developments to simultaneously estimate epidemic and spatiotemporal dynamics from viral genes sequences (Lemey et al., 2009, 2010).
In this article, we are interested in site-specific estimation of 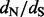 ratios. There are two main classes of methods designed for this task. The first class prescribes to build a model of codon evolution, with the
ratios. There are two main classes of methods designed for this task. The first class prescribes to build a model of codon evolution, with the 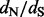 ratio as a model parameter that is allowed to vary across sites (Nielsen and Yang, 1998; Pond and Muse, 2005). Although we fully endorse these approaches and use them whenever possible, codon-based models become computationally prohibitive when analyzing large datasets that include hundreds, if not thousands, of molecular sequences (Lemey et al., 2007). Because of this computational limitation, researchers often turn to a second class of methods that are based on imputing (counting) unobserved synonymous and non-synonymous substitutions (Suzuki and Gojobori, 1999). These substitution counts are then used to construct a test statistic to assess the null hypothesis of neutrality (
ratio as a model parameter that is allowed to vary across sites (Nielsen and Yang, 1998; Pond and Muse, 2005). Although we fully endorse these approaches and use them whenever possible, codon-based models become computationally prohibitive when analyzing large datasets that include hundreds, if not thousands, of molecular sequences (Lemey et al., 2007). Because of this computational limitation, researchers often turn to a second class of methods that are based on imputing (counting) unobserved synonymous and non-synonymous substitutions (Suzuki and Gojobori, 1999). These substitution counts are then used to construct a test statistic to assess the null hypothesis of neutrality (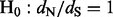 ). Such counting methods enjoy computational efficiency and often work well in practice (Kosakovsky Pond and Frost, 2005), but have two important shortcomings. First, counting approaches experience difficulties in handling uncertainty about the phylogenetic tree and about other nuisance parameters that play a role in counting unobserved synonymous and non-synonymous substitutions. Zhai et al. (2007) propose to overcome this problem by combining stochastic mapping, introduced by Nielsen (2002), with traditional counting methods’ test statistics as discrepancy measures in a posterior predictive diagnostics framework (Gelman et al., 1996). However, posterior predictive diagnostics, undoubtedly useful for visual exploration of model fit, are difficult to calibrate and to use in a semi-automatic fashion. The second limitation, shared by all counting methods, is their inability to provide reliable sites-specific
). Such counting methods enjoy computational efficiency and often work well in practice (Kosakovsky Pond and Frost, 2005), but have two important shortcomings. First, counting approaches experience difficulties in handling uncertainty about the phylogenetic tree and about other nuisance parameters that play a role in counting unobserved synonymous and non-synonymous substitutions. Zhai et al. (2007) propose to overcome this problem by combining stochastic mapping, introduced by Nielsen (2002), with traditional counting methods’ test statistics as discrepancy measures in a posterior predictive diagnostics framework (Gelman et al., 1996). However, posterior predictive diagnostics, undoubtedly useful for visual exploration of model fit, are difficult to calibrate and to use in a semi-automatic fashion. The second limitation, shared by all counting methods, is their inability to provide reliable sites-specific 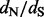 estimates and to quantify the relative strengths of selection across amino acid sites.
estimates and to quantify the relative strengths of selection across amino acid sites.
2 APPROACH
We propose a new counting method that overcomes both of the aforementioned shortcomings. Our solution arises in two steps. In the first step, we follow Nielsen (2002) and Zhai et al. (2007) and produce multiple stochastic mapping-based realizations of synonymous and non-synonymous counts while integrating over the posterior distribution of the nuissance parameters including the phylogenetics tree. However, to gain computational tractability, we exploit nucleotide-based codon partition models (Yang, 1996) in this step. These models can be fit to data in a fraction of the time it takes to fit even the simplest codon-based evolutionary models first introduced by Muse and Gaut (1994) and Goldman and Yang (1994). Although codon partition models do not account for selective pressures at amino acid sites (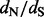 is one for all sites under these models), emerging probabilistic counting procedures have been shown to be robust to such gross model misspecification (Minin and Suchard, 2008b; Minin et al., 2011; O’Brien et al., 2009).
is one for all sites under these models), emerging probabilistic counting procedures have been shown to be robust to such gross model misspecification (Minin and Suchard, 2008b; Minin et al., 2011; O’Brien et al., 2009).
The second step of our approach is the main novelty of this article. We treat each random realization of synonymous and non-synonymous counts as pseudo-data and shrink these site-specific counts toward their means over all sites. We accomplish this regularization through an empirical Bayes procedure. We then use these regularized synonymous and non-synonymous counts to form site-specific 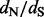 estimates. The end result is a posterior distribution of these ratios for all sites in the alignment. This distribution can be used for estimation [e.g. we report the posterior mean and 95% credible intervals (CIs) of site-specific
estimates. The end result is a posterior distribution of these ratios for all sites in the alignment. This distribution can be used for estimation [e.g. we report the posterior mean and 95% credible intervals (CIs) of site-specific 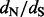 values] and for testing diversifying positive selection (e.g. a site is classified as positively selected if the posterior probability of
values] and for testing diversifying positive selection (e.g. a site is classified as positively selected if the posterior probability of 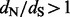 at this site is at least, say, 0.95). Empirical Bayes and, similar in spirit, full hierarchical Bayesian methods have been developed before for site-specific
at this site is at least, say, 0.95). Empirical Bayes and, similar in spirit, full hierarchical Bayesian methods have been developed before for site-specific 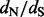 estimation and for identifying sites under diversifying positive selection (Huelsenbeck and Dyer, 2004; Yang et al., 2005). However, to our knowledge, we are the first to combine empirical Bayes philosophy and counting approaches, providing a revival of these simple methods to confront a formidable inferential problem. By performing the empirical Bayes regularization on the imputed count data, we avoid fitting computationally expensive codon-based models, making our ‘renaissance counting’ method practical for analyzing large datasets.
estimation and for identifying sites under diversifying positive selection (Huelsenbeck and Dyer, 2004; Yang et al., 2005). However, to our knowledge, we are the first to combine empirical Bayes philosophy and counting approaches, providing a revival of these simple methods to confront a formidable inferential problem. By performing the empirical Bayes regularization on the imputed count data, we avoid fitting computationally expensive codon-based models, making our ‘renaissance counting’ method practical for analyzing large datasets.
We assess our method using a simulation study almost identical to the one conducted and validated by Kosakovsky Pond and Frost (2005). The simulations demonstrate that our method’s site-specific 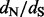 estimates and positively selected site identification are remarkably comparable with the estimates produced by the state-of-the-art codon-based models. We also apply our new renaissance approach to two empirical datasets. First, we reanalyze serially sampled feline panleukopenia and canine parvovirus VP2 capsid sequences. This analysis shows that renaissance counting produces sensible site-specific
estimates and positively selected site identification are remarkably comparable with the estimates produced by the state-of-the-art codon-based models. We also apply our new renaissance approach to two empirical datasets. First, we reanalyze serially sampled feline panleukopenia and canine parvovirus VP2 capsid sequences. This analysis shows that renaissance counting produces sensible site-specific 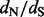 estimates, simultaneously accounting for uncertainty in all model parameters, and highlights important differences between our new and codon-based methods. We conclude by using our method in tandem with conventional approaches to examine differences in adaptive evolution in HIV pol genes in treated and drug naïve patient populations.
estimates, simultaneously accounting for uncertainty in all model parameters, and highlights important differences between our new and codon-based methods. We conclude by using our method in tandem with conventional approaches to examine differences in adaptive evolution in HIV pol genes in treated and drug naïve patient populations.
3 METHODS
3.1 Probabilistic counting under a codon partition model
Let 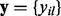 be a codon alignment matrix, where i spans n sequences, l runs over L sites in the alignment and each
be a codon alignment matrix, where i spans n sequences, l runs over L sites in the alignment and each 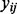 is one of 64 nucleotide triplets (codons). We assume that each site
is one of 64 nucleotide triplets (codons). We assume that each site 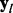 follows a codon partition model, meaning that each nucleotide position s = 1, 2, 3 within a codon evolves independently along a phylogenetic tree
follows a codon partition model, meaning that each nucleotide position s = 1, 2, 3 within a codon evolves independently along a phylogenetic tree  , according to one of the standard nucleotide substitution models (Yang, 1996). In all our examples and without loss of generality, we use a Hasegawa, Kishino and Yano (1985) model (HKY85) for each nucleotide position, with position-specific transition/transversion ratio
, according to one of the standard nucleotide substitution models (Yang, 1996). In all our examples and without loss of generality, we use a Hasegawa, Kishino and Yano (1985) model (HKY85) for each nucleotide position, with position-specific transition/transversion ratio 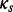 , substitution rate
, substitution rate 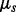 and stationary distribution
and stationary distribution 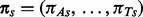 . Position rates are normalized for identifiability for contemporaneously sampled sequences, but remain free parameters in the case of serially sampled data.
. Position rates are normalized for identifiability for contemporaneously sampled sequences, but remain free parameters in the case of serially sampled data.
We fit the above codon partition model in a Bayesian framework and use Markov chain Monte Carlo (MCMC) integration to obtain a sample from the posterior distribution of model parameters 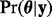 , where
, where 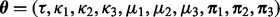 . Let
. Let 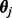 be parameter values at MCMC iteration j. At each iteration j, we use stochastic mapping to impute the full evolutionary history of each nucleotide position within each codon site in our alignment (Nielsen, 2002). We capitalize on a uniformization method, a modification of the original approach by Nielsen (2002), to draw realizations of the HKY continuous-time Markov chains (CTMCs) conditional on the codon data at the tips of the phylogeny
be parameter values at MCMC iteration j. At each iteration j, we use stochastic mapping to impute the full evolutionary history of each nucleotide position within each codon site in our alignment (Nielsen, 2002). We capitalize on a uniformization method, a modification of the original approach by Nielsen (2002), to draw realizations of the HKY continuous-time Markov chains (CTMCs) conditional on the codon data at the tips of the phylogeny 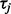 (Lartillot, 2006; Rodrigue et al., 2008). See Hobolth and Stone (2009) for an excellent review of different stochastic mapping algorithms. We also use simple forward CTMC simulation to draw the full evolutionary history of each site without conditioning on the data for normalization. For each stochastic mapping realization, conditional (C) and unconditional (U) on the data, we record the numbers of synonymous (S) substitutions
(Lartillot, 2006; Rodrigue et al., 2008). See Hobolth and Stone (2009) for an excellent review of different stochastic mapping algorithms. We also use simple forward CTMC simulation to draw the full evolutionary history of each site without conditioning on the data for normalization. For each stochastic mapping realization, conditional (C) and unconditional (U) on the data, we record the numbers of synonymous (S) substitutions 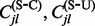 and non-synonymous (N) substitutions
and non-synonymous (N) substitutions 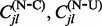 for each site
for each site 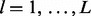 . Computing the ratio
. Computing the ratio 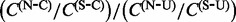 yields one straightforward way of converting these site-specific synonymous and non-synonymous counts into site-specific
yields one straightforward way of converting these site-specific synonymous and non-synonymous counts into site-specific 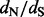 values. However, this naive method produces highly unstable estimates due to the high variance of site-specific substitution counts. Moreover, in many cases, some of the substitutions counts are zero, resulting in a
values. However, this naive method produces highly unstable estimates due to the high variance of site-specific substitution counts. Moreover, in many cases, some of the substitutions counts are zero, resulting in a 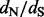 estimate of 0 or
estimate of 0 or 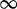 . To circumvent these problems, we apply an empirical Bayes regularization procedure to each of the four substitution counts, producing regularized rate estimates
. To circumvent these problems, we apply an empirical Bayes regularization procedure to each of the four substitution counts, producing regularized rate estimates 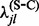 ,
, 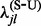 ,
, 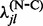 and
and 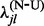 . With the regularized rate estimates at hand, we form our
. With the regularized rate estimates at hand, we form our 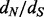 estimates
estimates
| (1) |
We explain our empirical Bayes regularization procedure in detail in the next section.
3.2 Regularized site-specific 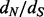 ratios
ratios
Within each MCMC iteration, we view the realized values of synonymous and non-synonymous substitutions as pseudo-data. As our empirical Bayes regularization procedure is identical for all four types of imputed counts, we use the generic notation 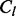 to denote one of the four possible count types,
to denote one of the four possible count types, 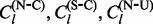 and
and 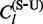 , at site l. Each of these counts represents the number of specifically labelled jumps of the codon model CTMC. In general, such random variables follow non-standard distributions that reduce to a Poisson distribution only in special cases (Minin and Suchard, 2008a). However, in the phylogenetic context, a Poisson distribution approximates these non-standard distributions very well (Siepel et al., 2006). Therefore, we assume that
, at site l. Each of these counts represents the number of specifically labelled jumps of the codon model CTMC. In general, such random variables follow non-standard distributions that reduce to a Poisson distribution only in special cases (Minin and Suchard, 2008a). However, in the phylogenetic context, a Poisson distribution approximates these non-standard distributions very well (Siepel et al., 2006). Therefore, we assume that
| (2) |
where 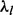 is an unknown site-specific Poisson rate. Next, we postulate a hierarchical model by assuming that
is an unknown site-specific Poisson rate. Next, we postulate a hierarchical model by assuming that
| (3) |
where the hierarchical prior carries unknown shape 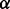 and rate
and rate 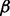 .
.
This second model level enables information sharing across sites so that sites with low counts can borrow information from more informative sites. This hierarchical model can be fitted in a fully Bayesian framework by approximating the posterior distribution 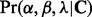 , where
, where 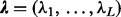 and
and 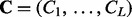 . Alternatively, one can first obtain estimates
. Alternatively, one can first obtain estimates 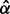 and
and 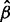 from the marginal probability density
from the marginal probability density 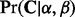 and then arrive at site-specific rate estimates in the form of the expectations
and then arrive at site-specific rate estimates in the form of the expectations 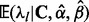 for
for 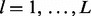 (Robbins, 1956). Maritz (1969) explores parametric and non-parametric hierarchical Poisson models and finds that even a simple gamma-Poisson model produces empirical Bayes estimates that enjoy good statistical properties.
(Robbins, 1956). Maritz (1969) explores parametric and non-parametric hierarchical Poisson models and finds that even a simple gamma-Poisson model produces empirical Bayes estimates that enjoy good statistical properties.
To execute our empirical Bayes approach, we start with sample means and variances
| (4) |
and match these empirical quantities with the theoretical mean and variance of a gamma-Poisson distribution with parameters 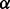 and
and 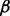 . This method of moments produces the following hyperparameter estimates
. This method of moments produces the following hyperparameter estimates
| (5) |
Given these estimated values, we have
| (6) |
from which we arrive at empirical Bayes estimates
| (7) |
In summary, to obtain a sample from the posterior distribution of the 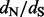 ratios, we
ratios, we
Draw M samples from the posterior distribution of model parameters
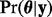 under the simple nucleotide-based codon partition model, and then
under the simple nucleotide-based codon partition model, and then- Post-process the MCMC output:
- For each iteration
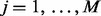 , impute the numbers of synonymous and non-synonymous substitutions,
, impute the numbers of synonymous and non-synonymous substitutions, 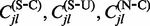 and
and 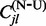 , with the help of stochastic mapping,
, with the help of stochastic mapping, - Use empirical Bayes regularization to arrive at rates of synonymous and non-synonymous subsitutions,
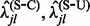 ,
, 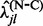 and
and 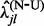 , and finally
, and finally
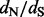
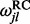
3.3 Computational considerations
Our site-specific 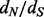 estimation method has two main steps. This first and the most time consuming step involves obtaining a posterior MCMC sample under the nucleotide-based codon partition model. An analogous step for the simplest codon models with no rate heterogeneity (Muse and Gaut, 1994; Goldman and Yang, 1994) takes at best 612/42/
estimation method has two main steps. This first and the most time consuming step involves obtaining a posterior MCMC sample under the nucleotide-based codon partition model. An analogous step for the simplest codon models with no rate heterogeneity (Muse and Gaut, 1994; Goldman and Yang, 1994) takes at best 612/42/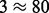 times as long, but in practice, performs worse, as the computational work of matrix exponentiation for the codon model becomes non-negligible. In the second step, we use stochastic mapping coupled with empirical Bayes regularization. Our empirical Bayes regularization of the imputed substitution counts is accomplished almost instantaneously because this estimation procedure does not involve computationally intensive calculations. The imputation step is potentially time consuming, but we draw stochastic mapping realizations for a small number of saved MCMC iterations (
times as long, but in practice, performs worse, as the computational work of matrix exponentiation for the codon model becomes non-negligible. In the second step, we use stochastic mapping coupled with empirical Bayes regularization. Our empirical Bayes regularization of the imputed substitution counts is accomplished almost instantaneously because this estimation procedure does not involve computationally intensive calculations. The imputation step is potentially time consuming, but we draw stochastic mapping realizations for a small number of saved MCMC iterations (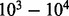 ), which is typically orders of magnitude smaller than the length of the entire Markov chain (
), which is typically orders of magnitude smaller than the length of the entire Markov chain ( ). Therefore, the running time of our two-step procedure is dominated by the first step, making our approach significantly faster than Bayesian site-specific
). Therefore, the running time of our two-step procedure is dominated by the first step, making our approach significantly faster than Bayesian site-specific 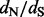 estimation under codon-based models.
estimation under codon-based models.
3.4 Identifying sites under selection
To classify sites as negatively selected, neutrally evolving or positively selected, we use for each site l the posterior sample  , to estimate the 95% CI
, to estimate the 95% CI 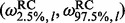 . Alternatively, we could use a 95% highest posterior density region. We declare site l to be under diversifying positive selection if
. Alternatively, we could use a 95% highest posterior density region. We declare site l to be under diversifying positive selection if 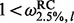 , neutral if
, neutral if 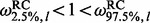 , and under negative selection if
, and under negative selection if 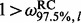 . Notice that we can not say anything about frequentist properties of this classification procedure. In other words, our method is not guaranteed to classify a site correctly 95% of the time during repeated replications of the evolutionary process. However, we point out that our new method offers more than merely a classification algorithm. In addition to classifying sites into the three categories above, we can assess the relative strengths of selection at individual sites by comparing site-specific CIs, including their relative rankings and the degree of overlap. To compare the use of a per-site
. Notice that we can not say anything about frequentist properties of this classification procedure. In other words, our method is not guaranteed to classify a site correctly 95% of the time during repeated replications of the evolutionary process. However, we point out that our new method offers more than merely a classification algorithm. In addition to classifying sites into the three categories above, we can assess the relative strengths of selection at individual sites by comparing site-specific CIs, including their relative rankings and the degree of overlap. To compare the use of a per-site 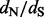 posterior distribution with a frequentist counting approach, we also explore a test procedure that only conditions on the expectations for the conditional and unconditional counts,
posterior distribution with a frequentist counting approach, we also explore a test procedure that only conditions on the expectations for the conditional and unconditional counts, 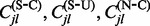 and
and 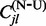 , for each site l to compute P-values using the extended binomial (EBin) distribution, as discussed in Kosakovsky Pond and Frost (2005).
, for each site l to compute P-values using the extended binomial (EBin) distribution, as discussed in Kosakovsky Pond and Frost (2005).
4 RESULTS
4.1 Performance
We conduct an extended simulation study to compare the relative performance of the renaissance approach to the state-of-the-art random effects likelihood codon-based model that accomodates both non-synonymous and synonymous rate variation across sites (DUAL REL) and the fixed effects likelihood (FEL) model approach implemented in HyPhy (Kosakovsky Pond et al., 2005). We reanalyze the datasets simulated by Kosakovsky Pond and Frost (2005). Briefly, the simulation procedure considers symmetric bifurcating trees with 8, 16, 32 and 64 tips and generates alignments encompassing 375 codons with a complex distribution of non-synonymous and synonymous substitution rates (resulting in 75 neutral, 335 negatively selected and 75 positively selected sites) (Kosakovsky Pond and Frost, 2005). We further extend this study to the 128-tip case and focus on the more realistic 16, 32, 64 and 128 range. Codon-based model substitution parameters were inspired by HIV polymerase sequence data, and 50 different replicates are generated for each collection of parameters.
We first focus on the binary classification problem of identifying sites under diversifying positive selection and map the proportion of misidentified sites (false positive rate) to the proportion of correctly identified sites (true positive rate) for various nominal 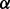 levels (P-values, empirical Bayes factors based on the ratio of posterior and prior odds of having
levels (P-values, empirical Bayes factors based on the ratio of posterior and prior odds of having 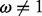 , or posterior quantiles) of the test procedures we compare. The resulting receiver operating characteristic curves for renaissance counting, DUAL REL and FEL are presented in Figure 1. For renaissance counting, we focus on the results for the CI coverage test approach (Supplementary Fig. S1 shows the EBin test has a very similar performance). Although the DUAL REL approach performs marginally better for a lower number of taxa, FEL benefits more from a large number of taxa. In general, the renaissance approach competes well with the codon model approaches with a roughly intermediate performance across the different simulation scenarios. Although the renaissance counting receiver operating characteristic curves are similar for CI coverage test and the EBin test (Supplementary Fig. S1), reasonably low false positive rates do not require very low EBin P-values, but they do require relative high posterior quantiles in the coverage approach (Supplementary Table S1). The EBin critical P-values also clearly decrease with the number of taxa for the same false positive rate, whereas a similar trend is not so obvious for the posterior quantiles. We anticipate that cut off values sensitive to the number of taxa will also be affected by sequence divergence, and only data-specific simulation provides an objective way to select such values.
, or posterior quantiles) of the test procedures we compare. The resulting receiver operating characteristic curves for renaissance counting, DUAL REL and FEL are presented in Figure 1. For renaissance counting, we focus on the results for the CI coverage test approach (Supplementary Fig. S1 shows the EBin test has a very similar performance). Although the DUAL REL approach performs marginally better for a lower number of taxa, FEL benefits more from a large number of taxa. In general, the renaissance approach competes well with the codon model approaches with a roughly intermediate performance across the different simulation scenarios. Although the renaissance counting receiver operating characteristic curves are similar for CI coverage test and the EBin test (Supplementary Fig. S1), reasonably low false positive rates do not require very low EBin P-values, but they do require relative high posterior quantiles in the coverage approach (Supplementary Table S1). The EBin critical P-values also clearly decrease with the number of taxa for the same false positive rate, whereas a similar trend is not so obvious for the posterior quantiles. We anticipate that cut off values sensitive to the number of taxa will also be affected by sequence divergence, and only data-specific simulation provides an objective way to select such values.
Fig. 1.
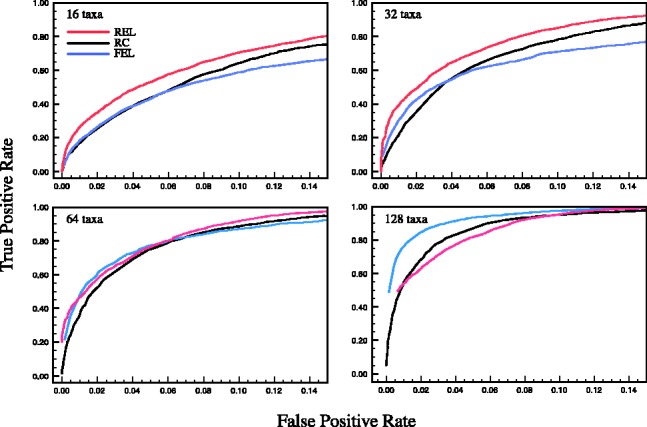
Receiver operating characteristic curves comparing classification performance of renaissance counting (RC), DUAL REL and FEL methods in identifying sites under diversifying positive selection. The curves plot true and false positive rates as functions of varying test-statistics. P-values, Bayes factors and posterior quantiles were used for FEL (blue line), DUAL REL (red line) and RC (black line), respectively
Next, we investigate the ability of the renaissance approach to estimate site-specific 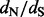 values. In Figure 2, we compare the mean relative errors of such site-specific estimation produced by our method, DUAL REL and FEL during the analysis of the 128-taxa simulated dataset. As the total number of simulations is fairly small, owing to the high computational costs of DUAL REL and FEL under phylogenetic tree uncertainty, we also report 95% Monte Carlo error (grey shaded areas). Although the renaissance approach tends to overestimate small
values. In Figure 2, we compare the mean relative errors of such site-specific estimation produced by our method, DUAL REL and FEL during the analysis of the 128-taxa simulated dataset. As the total number of simulations is fairly small, owing to the high computational costs of DUAL REL and FEL under phylogenetic tree uncertainty, we also report 95% Monte Carlo error (grey shaded areas). Although the renaissance approach tends to overestimate small 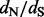 values, our method performs remarkably well when the true
values, our method performs remarkably well when the true 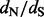 values are above or equal to one, the situation in which practitioners are most interested. This error pattern is reversed for the REL method, outperforming the renaissance method on small
values are above or equal to one, the situation in which practitioners are most interested. This error pattern is reversed for the REL method, outperforming the renaissance method on small 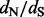 values, but loses to our approach on sites under positive selection. The relative errors of the FEL method are difficult to quantify owing to the fact that this method occasionally produces unreasonably high
values, but loses to our approach on sites under positive selection. The relative errors of the FEL method are difficult to quantify owing to the fact that this method occasionally produces unreasonably high 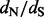 estimates. If we remove infinite
estimates. If we remove infinite 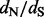 estimates during the relative error calculations, FEL still produces unreasonably high
estimates during the relative error calculations, FEL still produces unreasonably high 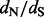 estimates on some of the remaining simulations (bottom left plot in Fig. 2). Only when we remove
estimates on some of the remaining simulations (bottom left plot in Fig. 2). Only when we remove 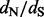 estimates that are above 10 000 do we arrive at reasonable (but now biased) relative errors for the FEL method. Although after this filtering, the FEL approach outperforms both the renaissance and REL methods, the FEL approach remains problematic because for some of the sites, it fails to provide reasonable
estimates that are above 10 000 do we arrive at reasonable (but now biased) relative errors for the FEL method. Although after this filtering, the FEL approach outperforms both the renaissance and REL methods, the FEL approach remains problematic because for some of the sites, it fails to provide reasonable 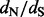 estimates in as many as 11 simulations (22% of all simulations).
estimates in as many as 11 simulations (22% of all simulations).
Fig. 2.
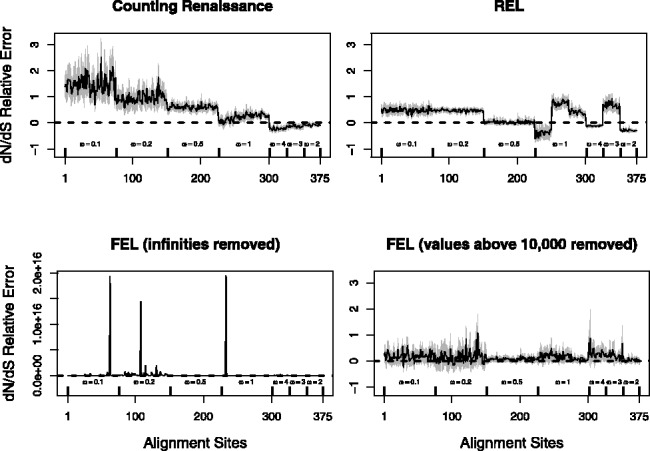
Relative errors of site-specific 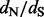 estimation. We show mean relative errors (solid black lines), produced by renaissance counting, DUAL REL and FEL for each site of simulated alignments with 128 taxa. The top facing tick marks in each plot demarcate changes in the true
estimation. We show mean relative errors (solid black lines), produced by renaissance counting, DUAL REL and FEL for each site of simulated alignments with 128 taxa. The top facing tick marks in each plot demarcate changes in the true 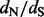 value. The true values are shown above the intervals produced by the tick marks. Gray shaded areas indicate 95% Monte Carlo error. We do not show the Monte Carlo error in the bottom left plot because outlier estimates, produced by the FEL method, violate normality assumptions of the Monte Carlo error calculations. In all plots, the horizontal dashed line corresponds to zero value of the relative error
value. The true values are shown above the intervals produced by the tick marks. Gray shaded areas indicate 95% Monte Carlo error. We do not show the Monte Carlo error in the bottom left plot because outlier estimates, produced by the FEL method, violate normality assumptions of the Monte Carlo error calculations. In all plots, the horizontal dashed line corresponds to zero value of the relative error
4.2 Empirical data
We analyze a feline and carnivore parvovirus dataset to compare renaissance counting implemented in BEAST (Drummond et al., 2012) to REL codon-based model approaches. This dataset includes 91 VP2 capsid sequences sampled from various geographical regions for a 42-year period and was previously used to demonstrate the rapid adaptation of feline panleukopenia parvovirus to canine hosts (Shackelton et al., 2005). Table 1 lists the positively selected sites identified using renaissance counting and REL approaches, and Supplementary Figure S2 plots the site-specific DUAL REL 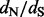 estimates against the renaissance counting
estimates against the renaissance counting 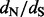 estimates and their CIs. The REL approaches include non-synonymous (NS REL) and both non-synonymous and synonymous (DUAL REL) rate variation across sites. To model this rate variation, we used generalized discrete distributions with three rate categories. The DUAL REL model provides a significantly better fit when compared with the NS REL model (P < 0.001), indicating significant variation in
estimates and their CIs. The REL approaches include non-synonymous (NS REL) and both non-synonymous and synonymous (DUAL REL) rate variation across sites. To model this rate variation, we used generalized discrete distributions with three rate categories. The DUAL REL model provides a significantly better fit when compared with the NS REL model (P < 0.001), indicating significant variation in 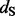 across sites. Shackelton et al. (2005) originally use a NS REL approach as implemented in PAML [Phylogenetic Analysis by Maximum Likelihood, Yang, (1997)] and relied on fairly conservative criteria to identify positive selected sites (underlined in Table 1). If we use the criterion of
across sites. Shackelton et al. (2005) originally use a NS REL approach as implemented in PAML [Phylogenetic Analysis by Maximum Likelihood, Yang, (1997)] and relied on fairly conservative criteria to identify positive selected sites (underlined in Table 1). If we use the criterion of 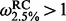 for renaissance counting and a log Bayes factor (for
for renaissance counting and a log Bayes factor (for 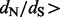 1) > 4.0 for the REL models, which is indeed the cut off that corresponds to a false positive rate of 5% according to data-specific simulations (data not shown), then these three approaches provide support for roughly the same sites as being positively selected with few exceptions. The FEL approach, however, does not yield support for any site under positive diversifying selection unless one is willing to accept high critical P-values as evidence. In Table 1, we also report results of a single-likelihood ancestor counting (SLAC) method (Kosakovsky Pond and Frost, 2005), a more advanced version of the original counting method of Suzuki and Gojobori (1999), which performs similarly to the FEL model. The same issues that emerged in the simulation analyses also complicate the site-specific dN/dS estimation by FEL for this dataset, and, for this reason, Kosakovsky Pond and Frost (2005) suggested to use scaled
1) > 4.0 for the REL models, which is indeed the cut off that corresponds to a false positive rate of 5% according to data-specific simulations (data not shown), then these three approaches provide support for roughly the same sites as being positively selected with few exceptions. The FEL approach, however, does not yield support for any site under positive diversifying selection unless one is willing to accept high critical P-values as evidence. In Table 1, we also report results of a single-likelihood ancestor counting (SLAC) method (Kosakovsky Pond and Frost, 2005), a more advanced version of the original counting method of Suzuki and Gojobori (1999), which performs similarly to the FEL model. The same issues that emerged in the simulation analyses also complicate the site-specific dN/dS estimation by FEL for this dataset, and, for this reason, Kosakovsky Pond and Frost (2005) suggested to use scaled 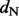 minus
minus 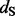 as a measure of selection to compare FEL with other methods. Similar to the FEL and SLAC P-values, the EBin P-values derived from the average counts for each site are of little use compared with a test approach that draws information from the posterior distribution of regularized site-specific
as a measure of selection to compare FEL with other methods. Similar to the FEL and SLAC P-values, the EBin P-values derived from the average counts for each site are of little use compared with a test approach that draws information from the posterior distribution of regularized site-specific 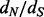 ratios. The poor performance of these approaches may be attributed to the relatively low diversity in this dataset (mean pairwise diversity of 0.0086 substitutions per site). Between the two clear exceptions to the consistency of renaissance counting and REL results, site 101 is only identified by NS REL to be under positive selection. When also considering rate variation for synonymous substitutions, this site falls under a class with high synonymous substitution rates in DUAL REL, resulting also in a considerably lower
ratios. The poor performance of these approaches may be attributed to the relatively low diversity in this dataset (mean pairwise diversity of 0.0086 substitutions per site). Between the two clear exceptions to the consistency of renaissance counting and REL results, site 101 is only identified by NS REL to be under positive selection. When also considering rate variation for synonymous substitutions, this site falls under a class with high synonymous substitution rates in DUAL REL, resulting also in a considerably lower 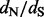 . So, it appears that despite conservative assignment, site 101 may be falsely identified as being positively selected when synonymous rate variation across sites is ignored. In this respect, renaissance counting is in better agreement with DUAL REL, and, in general, the renaissance estimates are in good agreement with the posterior
. So, it appears that despite conservative assignment, site 101 may be falsely identified as being positively selected when synonymous rate variation across sites is ignored. In this respect, renaissance counting is in better agreement with DUAL REL, and, in general, the renaissance estimates are in good agreement with the posterior 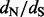 expectations estimated by DUAL REL. Both REL approaches find support for site 491 being under positive diversifying selection, despite the observation that the site also experiences a high synonymous substitution rate (S rate class = 3 in DUAL REL), whereas renaissance counting classifies the site as neutrally evolving. For this site, only two unrelated sequences have a different amino acid (histidine and arginine) compared with the wild-type amino acid (glutamine). These amino acid substitutions on external branches could therefore represent (slightly) deleterious substitutions that have subsequently experienced purifying selection (Lemey et al., 2007; Pond et al., 2006). For renaissance counting, similar to other counting and FEL approaches, we can restrict ourselves to summarizing the substitution history on internal branches only to avoid the impact of such transient mutations.
expectations estimated by DUAL REL. Both REL approaches find support for site 491 being under positive diversifying selection, despite the observation that the site also experiences a high synonymous substitution rate (S rate class = 3 in DUAL REL), whereas renaissance counting classifies the site as neutrally evolving. For this site, only two unrelated sequences have a different amino acid (histidine and arginine) compared with the wild-type amino acid (glutamine). These amino acid substitutions on external branches could therefore represent (slightly) deleterious substitutions that have subsequently experienced purifying selection (Lemey et al., 2007; Pond et al., 2006). For renaissance counting, similar to other counting and FEL approaches, we can restrict ourselves to summarizing the substitution history on internal branches only to avoid the impact of such transient mutations.
Table 1.
Comparison of sites under positive diversifying selection as identified using renaissance counting, two random effects likelihood (REL) models, the two-rate FEL approach and the SLAC method applied to the feline and carnivore parvovirus dataset
| Renaissance counting |
EBin | NS REL |
DUAL REL |
FEL |
SLAC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site | 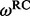 |
(95% CPD) | P-value | 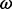 |
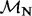 |
log(BF) | 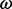 |
 |
 |
log(BF) |  |
P-value | P-value |
| 80 | 2.36 | (1.38–4.21)* | 0.54 | 1.35 | 3 | 3.89 | 2.27 | 1 | 3 | 4.00 |  |
0.44 | 0.52 |
| 85 | 2.28 | (1.35–3.96)* | 0.60 | 1.51 | 3 | 4.13 | 2.47 | 1 | 3 | 4.19 | 3.81E03 | 0.63 | 0.40 |
| 101 | 0.81 | (0.41–1.52) | 0.90 | 2.73 | 3 | 9.44 | 0.56 | 3 | 3 | 0.51 | 0.29 | 0.13 | 0.57 |
| 195 | 1.18 | (0.73–1.92) | 0.73 | 0.33 | 3 | 1.68 | 0.56 | 1 | 3 | 1.81 | 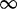 |
0.18 | 0.44 |
| 232 | 4.03 | (2.30–6.81)** | 0.32 | 2.52 | 3 | 6.40 | 4.09 | 1 | 3 | 6.24 | 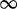 |
0.40 | 0.54 |
| 234 | 2.21 | (1.35–3.44)* | 0.58 | 1.66 | 3 | 4.38 | 2.73 | 1 | 3 | 4.42 | 3.78E03 | 0.63 | 0.54 |
| 300 | 5.68 | (3.36–9.19)** | 0.21 | 2.74 | 3 | 13.26 | 4.36 | 1 | 3 | 7.22 | 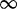 |
0.19 | 0.09 |
| 305 | 2.22 | (1.36–3.61)* | 0.57 | 1.66 | 3 | 4.37 | 2.70 | 1 | 3 | 4.40 | 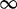 |
0.61 | 0.53 |
| 386 | 1.18 | (0.73–1.84) | 0.76 | 0.40 | 3 | 1.96 | 0.66 | 1 | 3 | 2.06 | 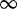 |
0.50 | 0.67 |
| 426 | 4.24 | (2.27–7.40)** | 0.34 | 2.72 | 3 | 9.21 | 4.24 | 1 | 3 | 6.66 | 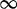 |
0.50 | 0.16 |
| 491 | 0.81 | (0.48–1.36) | 0.83 | 2.11 | 3 | 5.19 | 2.61 | 3 | 3 | 4.28 | 0.51 | 0.60 | 0.74 |
| 545 | 1.18 | (0.72–1.87) | 0.75 | 0.39 | 3 | 1.92 | 0.61 | 1 | 3 | 1.96 | 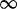 |
0.86 | 0.99 |
One REL model incorporates non-synonymous substitution rate variation (NS) and the other includes both synonymous and non-synonymous rate variation (DUAL). Underlined sites were identified as positively selected in the original study (Shackelton et al., 2005). 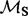 and
and 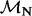 represent the rate class of the general discrete distribution with three rate classes to accommodate variation in
represent the rate class of the general discrete distribution with three rate classes to accommodate variation in 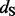 and
and 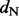 rates among sites, respectively. Bayes factors (BF) are reported on a logarithmic scale. * and ** indicate intervals correspond to the 95% and 99% cumulative posterior density (CPD) intervals, respectively, that exclude
rates among sites, respectively. Bayes factors (BF) are reported on a logarithmic scale. * and ** indicate intervals correspond to the 95% and 99% cumulative posterior density (CPD) intervals, respectively, that exclude 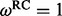 .
.
To appreciate the computational efficiency that renaissance counting affords, we first recall that direct run-time comparison with the existing implementation of the REL and FEL models is flawed because these implementations condition on a fixed tree. In this article, we overcome this implementation limitation by averaging results over an arbitrarily sized sample of trees from their posterior distribution, leading to almost arbitrarily long run-times. Alternatively, a slight underestimate of the computational cost to fit a full Bayesian REL model entails a full Bayesian fitting of the codon-based 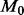 model. For this example, the codon-based model takes 92
model. For this example, the codon-based model takes 92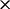 longer to fit than performing renaissance counting using an Intel Xeon E5620 CPU running at 2.4 GHz. Recently, Suchard and Rambaut (2009) introduce massive parallelization for fitting large state-space models through the BEAGLE library (Ayres et al., 2012). Even when exploiting a Tesla C2050 graphics processing unit for fitting
longer to fit than performing renaissance counting using an Intel Xeon E5620 CPU running at 2.4 GHz. Recently, Suchard and Rambaut (2009) introduce massive parallelization for fitting large state-space models through the BEAGLE library (Ayres et al., 2012). Even when exploiting a Tesla C2050 graphics processing unit for fitting 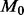 , renaissance counting still runs 3
, renaissance counting still runs 3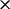 faster on the CPU.
faster on the CPU.
A second application of renaissance counting compares the site-specific selection patterns for partial HIV pol gene sequences from treated and drug naïve patients. Both the ‘treated’ and ‘untreated’ dataset encode amino acid position 1 to 220 in the reverse transcriptase and were previously used as examples of an intermediate size (81 taxa) and a large (297 taxa) dataset, respectively, for selection analyses (Kosakovsky Pond and Frost, 2005). In the treatment group, antiretroviral therapy consisted solely of the zidovudine (AZT) nucleoside reverse transcription inhibitor. Sites identified to be under positive diversifying selection by either renaissance counting, DUAL REL and FEL are listed in Tables 2 and 3 for the treated and untreated dataset, respectively. Supplementary Figure S2 provides a summary of mean 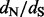 estimates with CIs for all sites in both datasets.
estimates with CIs for all sites in both datasets.
Table 2.
Positively selected sites in HIV-1 Reverse Transcriptase of AZT-treated patients identified by at least one of three methods (renaissance counting, DUAL REL and the two-rate FEL approach)
| Renaissance counting |
EBin | DUAL REL |
FEL |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Site | 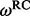 |
(95% CPD) | P-value | 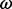 |
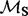 |
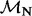 |
log(BF) | 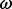 |
P-value |
| 20 | 2.16 | (1.37–3.36)** | 0.14 | 8.55 | 1 | 2 | 3.11 | 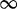 |
0.07 |
| 35 | 1.79 | (1.10–2.82)* | 0.08 | 2.3 | 2 | 3 | 7.25 | 2.62 | 0.17 |
| 39 | 1.55 | (0.99–2.43) | 0.25 | 7.05 | 1 | 2 | 3.68 | 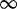 |
0.09 |
| 60 | 1.53 | (0.89–2.40) | 0.08 | 6.13 | 2 | 3 | 3.88 | 2.84 | 0.27 |
| 64 | 1.3 | (0.85–1.99) | 0.31 | 8.5 | 1 | 2 | 6.66 | 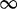 |
0.02 |
| 69 | 3.13 | (2.05–4.71)** | 0.06 | 34.89 | 1 | 3 | 10.00 | 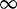 |
0.01 |
| 83 | 1.24 | (0.67–2.09) | 0.18 | 2.02 | 2 | 3 | 6.02 | 1.58 | 0.55 |
| 102 | 0.68 | (0.44–1.09) | 0.56 | 8.28 | 1 | 2 | 6.12 | 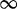 |
0.10 |
| 123 | 1.97 | (1.11–3.31)* | 0.19 | 1.69 | 2 | 3 | 4.47 | 0.83 | 0.76 |
| 135 | 2.27 | (1.30–3.80)* | 0.11 | 1.88 | 2 | 3 | 4.39 | 1.48 | 0.61 |
| 178 | 2.59 | (1.31–4.59)* | 0.12 | 9.96 | 1 | 3 | 3.06 | 3.71 | 0.70 |
| 200 | 4.63 | (2.92–7.27)** | 0.02 | 34.57 | 1 | 3 | 11.14 | 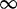 |
 0.01 0.01 |
| 207 | 1.92 | (1.15–3.15)* | 0.19 | 1.72 | 2 | 3 | 4.50 | 3.18 | 0.08 |
| 211 | 1.16 | (0.72–1.85) | 0.59 | 1.8 | 2 | 3 | 7.98 | 2.54 | 0.04 |
| 215 | 3.44 | (2.17–5.32)** | 0.05 | 39.75 | 1 | 3 | 15.28 |  |
 0.01 0.01 |
The sites indicated in bold are identified as positively selected by all three methods. EBin P-values were computed using the EBin distribution. 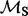 and
and 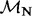 represent the rate class of the general discrete distribution with three rate classes to accommodate variation in
represent the rate class of the general discrete distribution with three rate classes to accommodate variation in 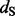 and
and 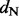 rates among sites, respectively. * and ** indicate intervals correspond to the 95% and 99% highest posterior density intervals, respectively, that exclude
rates among sites, respectively. * and ** indicate intervals correspond to the 95% and 99% highest posterior density intervals, respectively, that exclude 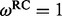 .
.
Table 3.
Positively selected sites in HIV-1 Reverse Transcriptase of drug naïve patients identified by at least one of three methods (renaissance counting, DUAL REL and the two-rate FEL approach)
| Renaissance counting |
EBin | DUAL REL |
FEL |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Site | 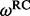 |
(95% CPD) | P-value | 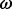 |
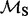 |
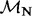 |
log(BF) | 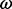 |
P-value |
| 35 | 1.98 | (1.30–3.00)** | 0.06 | 2.94 | 1 | 3 | 17.28 | 3.49 | 0.03 |
| 68 | 1.78 | (1.21–2.51)* | 0.13 | 0.72 | 1 | 2 |
 1 1 |
1.43 | 0.72 |
| 83 | 0.7 | (0.49–0.97) | 0.87 | 1.14 | 2 | 3 | 7.42 | 0.71 | 0.37 |
| 102 | 1.68 | (1.20–2.33)* | 0.14 | 0.75 | 1 | 2 |
 1 1 |
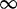 |
 0.01 0.01 |
| 123 | 0.91 | (0.65–1.25) | 0.68 | 1.11 | 2 | 3 | 6.34 | 0.88 | 0.70 |
| 135 | 2.31 | (1.64–3.20)** | 0.01 | 1.26 | 2 | 3 | 6.63 | 1.45 | 0.39 |
| 142 | 0.64 | (0.47–0.86) | 1 | 1.11 | 2 | 3 | 6.62 | 0.56 | 0.12 |
| 177 | 0.81 | (0.60–1.15) | 0.77 | 1.16 | 2 | 3 | 9.1 | 1 | 1.00 |
| 178 | 4.92 | (2.79–7.63)** |
 0.01 0.01 |
2.48 | 1 | 3 | 9.11 | 1.98 | 0.42 |
| 200 | 2.64 | (1.83–3.72)** | 0.01 | 2.91 | 1 | 3 | 17.85 | 3.67 |
 0.01 0.01 |
| 202 | 4.68 | (3.30–6.37)** |
 0.01 0.01 |
0.72 | 1 | 2 |
 1 1 |
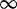 |
0.13 |
| 211 | 1.06 | (0.77–1.42) | 0.47 | 2.48 | 1 | 3 | 25.83 | 3.63 |
 0.01 0.01 |
See Table 2 for table details.
When considering 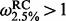 for renaissance counting, a log Bayes factor
for renaissance counting, a log Bayes factor  4.0 for DUAL REL and a P
4.0 for DUAL REL and a P
 0.1 for FEL as evidence for positive diversifying selection [cfr. (Kosakovsky Pond et al., 2005)], all methods identify about 8 to 11 positively selected sites in the treated dataset (Table 2). With a 5-fold higher diversity (a mean pairwise diversity of 0.0426 and 0.0413 substitutions per site for the treated and untreated dataset, respectively), the FEL approach does now result in convincing P-values for a number of sites. All three methods provide significant support for four sites under adaptation (site 69, 200, 207 and 215). Of those, amino acid substitutions at site 69 and 215 are known to confer resistance to AZT (Larder and Kemp, 1989), albeit mostly in combination with other substitutions for site 69 (Fitzgibbon et al., 1991; Winters and Merigan, 2001). The presence of specific substitutions at position 207 has also been correlated with reduced AZT susceptibility in biologically cloned HIV-1 isolates [Q207D and Q207E, (Stoeckli et al., 2002)], and in vitro evidence confirmed decreased AZT susceptibility owing to Q207D while this substitution also increased the relative fitness of AZT-resistant HIV-1 (Lu et al., 2005). Site 200 also undergoes adaptive evolution in the untreated dataset and most likely reflects selection from the cellular immune response, rather than antiviral therapy (see below). Substitutions at other sites listed in Table 2 may also bear some relation to nucleoside analogue resistance and AZT resistance in particular [e.g. V35M (Cane et al., 2007), T39A (Saracino et al., 2006), V60I (Huigen et al., 2006), R83K (Svicher et al., 2006), T200A (De Luca et al., 2006) and R211K (Kemp et al., 1998)], but these are generally considered to be ‘accessory’ substitutions that are supported by indirect evidence with little or no in vitro confirmation.
0.1 for FEL as evidence for positive diversifying selection [cfr. (Kosakovsky Pond et al., 2005)], all methods identify about 8 to 11 positively selected sites in the treated dataset (Table 2). With a 5-fold higher diversity (a mean pairwise diversity of 0.0426 and 0.0413 substitutions per site for the treated and untreated dataset, respectively), the FEL approach does now result in convincing P-values for a number of sites. All three methods provide significant support for four sites under adaptation (site 69, 200, 207 and 215). Of those, amino acid substitutions at site 69 and 215 are known to confer resistance to AZT (Larder and Kemp, 1989), albeit mostly in combination with other substitutions for site 69 (Fitzgibbon et al., 1991; Winters and Merigan, 2001). The presence of specific substitutions at position 207 has also been correlated with reduced AZT susceptibility in biologically cloned HIV-1 isolates [Q207D and Q207E, (Stoeckli et al., 2002)], and in vitro evidence confirmed decreased AZT susceptibility owing to Q207D while this substitution also increased the relative fitness of AZT-resistant HIV-1 (Lu et al., 2005). Site 200 also undergoes adaptive evolution in the untreated dataset and most likely reflects selection from the cellular immune response, rather than antiviral therapy (see below). Substitutions at other sites listed in Table 2 may also bear some relation to nucleoside analogue resistance and AZT resistance in particular [e.g. V35M (Cane et al., 2007), T39A (Saracino et al., 2006), V60I (Huigen et al., 2006), R83K (Svicher et al., 2006), T200A (De Luca et al., 2006) and R211K (Kemp et al., 1998)], but these are generally considered to be ‘accessory’ substitutions that are supported by indirect evidence with little or no in vitro confirmation.
Only two sites are identified as positively selected by all three methods in the drug naïve dataset using the same significance criteria as mentioned above (site 35 and 200). Twenty-three records can be retrieved from the Los Alamos HIV cytotoxic T-lymphocyte (CTL)/CD8+ T-Cell epitope database for epitopes that span site 35. Importantly, when patients elicit CTL responses against this epitope, they are generally directed against amino acid variation at site 35 (Karlsson et al., 2007), which makes this an example of virus evolution to the consensus B sequence (35V). Site 200 is also located in five epitopes listed in the CTL/CD8+ T-Cell epitope database, but we could not retrieve any information concerning specific amino acid variation at site 200 that induces different CTL responses. Without such information, it remains difficult to establish a clear role for particular positions in immune evasion. In fact, the collection of reported epitopes covers the majority of Reverse Transcriptase amino acid positions, and it is therefore not surprising that many sites listed in Table 3 can be mapped to known HIV-1 epitopes.
5 DISCUSSION
We present a novel and efficient Bayesian method for detecting positive diversifying selection in molecular sequences. Our method combines the computational speed of counting methods and statistical efficiency of empirical Bayes approaches. A further strength of the method is its simplicity of implementation, thanks to recent algorithmic advances in stochastic mapping (Nielsen, 2002; Lartillot, 2006). Stochastic mapping has been used before to speed up MCMC-based Bayesian estimation of codon-based models (Rodrigue et al., 2008), but this and subsequent approaches still operate within the codon-based modeling framework. In contrast, our method uses stochastic mapping to quickly approximate codon-based models that account for rate variation of 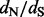 over sites. Our analysis of simulated and real data demonstrates that the proposed approach competes well with more computationally demanding methods in recovering site-specific
over sites. Our analysis of simulated and real data demonstrates that the proposed approach competes well with more computationally demanding methods in recovering site-specific 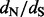 ratios and in accurately identifying sites under positive diversifying selection.
ratios and in accurately identifying sites under positive diversifying selection.
The main distinction between renaissance counting and previous attempts to use stochastic mapping to detect diversifying positive selection (Zhai et al., 2007) is the ability of our renaissance method to directly estimate 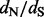 ratios and to quantify uncertainty in these estimates. This advance is important because all counting methods, including stochastic mapping-based ones, can only produce site-specific P-values, resulting from testing a null hypothesis of neutrality. However, it is well known that using P-values has limitations. In particular, as P-values are always calculated by conditioning on the null hypothesis, it would be inappropriate to use the magnitude of P-values as evidence in favor of the null (Goodman, 1999). The task of comparing P-values becomes even more challenging in the method of Zhai et al. (2007) because these authors use notoriously hard-to-calibrate posterior predictive P-values (Hjort et al., 2006). Therefore, it is difficult to use previously developed counting methods to quantify relative strength of selection at amino acid sites. Although our method cannot possibly resolve the long-standing statistical controversy of quantifying evidence in hypothesis testing, renaissance counting offers a straightforward estimation alternative by producing site-specific posterior distributions of
ratios and to quantify uncertainty in these estimates. This advance is important because all counting methods, including stochastic mapping-based ones, can only produce site-specific P-values, resulting from testing a null hypothesis of neutrality. However, it is well known that using P-values has limitations. In particular, as P-values are always calculated by conditioning on the null hypothesis, it would be inappropriate to use the magnitude of P-values as evidence in favor of the null (Goodman, 1999). The task of comparing P-values becomes even more challenging in the method of Zhai et al. (2007) because these authors use notoriously hard-to-calibrate posterior predictive P-values (Hjort et al., 2006). Therefore, it is difficult to use previously developed counting methods to quantify relative strength of selection at amino acid sites. Although our method cannot possibly resolve the long-standing statistical controversy of quantifying evidence in hypothesis testing, renaissance counting offers a straightforward estimation alternative by producing site-specific posterior distributions of 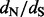 ratios. Importantly, this uniquely positions renaissance counting among other positive selection detection methods as a method that also appropriately quantifies the relative strengths of selection at individual sites.
ratios. Importantly, this uniquely positions renaissance counting among other positive selection detection methods as a method that also appropriately quantifies the relative strengths of selection at individual sites.
When imputing/counting the unobserved synonymous and non-synonymous substitutions, we produce substitution counts on each branch of the phylogenetic tree and then sum them up for each site. If a scientifically meaningful partition of branches into groups exists, we can easily sum the substitutions within each group separately and apply our empirical Bayes procedure to the site-specific counts in each group. For applications to within-host HIV evolution for example, we can consider synonymous and non-synonymous substitutions only on internal branches of the phylogeny. This partition of branch lengths is motivated by a widely accepted hypothesis that many terminal branches of intrahost HIV phylogenies represent lineages that are weeded out by selection (Pond et al., 2006). Therefore, it is reasonable, if not advisable, to avoid the mutational load on these branches and their impact on substitution rates (Lemey et al., 2007) by excluding them when identifying positive diversifying selection.
Concerning the identification site-specific selection patterns, the development of renaissance counting was clearly not to replace, but to complement existing methodology. Even after a comprehensive evaluation of several counting-based and maximum likelihood methods, Kosakovsky Pond and Frost (2005) found it difficult to make definitive recommendations on which methods to use in particular cases. In fact, they concluded that a consensus of several methods, but each accepting relatively high nominal 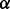 levels, would be a reasonable approach to rule out spurious results. In many ways, renaissance counting presents an interesting candidate method to include in such a consensus approach. Embedded in a Bayesian inference framework, renaissance counting accounts for phylogenetic error when estimating
levels, would be a reasonable approach to rule out spurious results. In many ways, renaissance counting presents an interesting candidate method to include in such a consensus approach. Embedded in a Bayesian inference framework, renaissance counting accounts for phylogenetic error when estimating 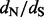 ratios. Perhaps more importantly, our approach uses a test statistic that is very different from both previous counting methods and maximum likelihood methods. For small or less divergent datasets, for example the test used in previous counting methods is notoriously conservative (Kosakovsky Pond and Frost, 2005), which is also confirmed by our comparison of the EBin test to the CI coverage test. Empirical Bayesian analysis of random-effects models on the other hand can suffer from large Type I error rates owing to excessive errors in the parameter estimates used (Kosakovsky Pond and Frost, 2005). Renaissance counting avoids the estimation of codon model parameters, and estimation uncertainty will be taken into account by the CI coverage test approach. The nucleotide-model approximation also offers a significant speed-up compared with fitting a codon model, but considerable computation time may still need to be invested in integrating over the posterior distribution of phylogenies. In this respect, we emphasize that renaissance counting is implemented in a Bayesian inference framework [BEAST (Drummond et al., 2012)] that provides access to an array of flexible models for estimating genealogical history, divergence times, flexible semi-parametric demographic and phylogeographic histories (Lemey et al., 2009, 2010). Although site-specific selection patterns may not be of prime interest when engaging in such analyses, renaissance counting readily delivers
ratios. Perhaps more importantly, our approach uses a test statistic that is very different from both previous counting methods and maximum likelihood methods. For small or less divergent datasets, for example the test used in previous counting methods is notoriously conservative (Kosakovsky Pond and Frost, 2005), which is also confirmed by our comparison of the EBin test to the CI coverage test. Empirical Bayesian analysis of random-effects models on the other hand can suffer from large Type I error rates owing to excessive errors in the parameter estimates used (Kosakovsky Pond and Frost, 2005). Renaissance counting avoids the estimation of codon model parameters, and estimation uncertainty will be taken into account by the CI coverage test approach. The nucleotide-model approximation also offers a significant speed-up compared with fitting a codon model, but considerable computation time may still need to be invested in integrating over the posterior distribution of phylogenies. In this respect, we emphasize that renaissance counting is implemented in a Bayesian inference framework [BEAST (Drummond et al., 2012)] that provides access to an array of flexible models for estimating genealogical history, divergence times, flexible semi-parametric demographic and phylogeographic histories (Lemey et al., 2009, 2010). Although site-specific selection patterns may not be of prime interest when engaging in such analyses, renaissance counting readily delivers 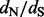 ratios for any coding sequence alignment, which may offer a starting point for in-depth characterization of selection pressures involving different methods.
ratios for any coding sequence alignment, which may offer a starting point for in-depth characterization of selection pressures involving different methods.
Funding: The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013) under Grant Agreement no. 278433-PREDEMICS and ERC Grant agreement no. 260864, the National Science Foundation (DMS 0856099) and the National Institutes of Health (R01 GM086887 and R01 HG006139). This research has also been supported by the Bioinformatics, Statistical Analysis and Evolutionary Core of the UCSD Center for AIDS Research (5P30AI36214). We acknowledge the support of the National Evolutionary Synthesis Center (NESCent) through a working group (Software for Bayesian Evolutionary Analysis). Further, research was partially completed while the authors were visiting the Institute for Mathematical Sciences, National University of Singapore in 2011.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ayres D., et al. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012;61:170–173. doi: 10.1093/sysbio/syr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane P.A., et al. Identification of accessory mutations associated with high-level resistance in HIV-1 reverse transcriptase. AIDS. 2007;21:447–455. doi: 10.1097/QAD.0b013e3280129964. [DOI] [PubMed] [Google Scholar]
- De Luca A., et al. Polymorphisms in the viral reverse transcriptase predict the evolution towards distinct thymidine analogue mutational patterns: a longitudinal analysis. Antivir. Ther. 2006;11:157. [Google Scholar]
- Drummond A., et al. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgibbon J.E., et al. In vivo prevalence of azidothymidine (AZT) resistance mutations in an aids patient before and after AZT therapy. AIDS Res. Hum. Retroviruses. 1991;7:265–269. doi: 10.1089/aid.1991.7.265. [DOI] [PubMed] [Google Scholar]
- Gelman A., et al. Posterior predictive assessment of model fitness via realized discrepancies. Stat. Sin. 1996;6:733–807. [Google Scholar]
- Goldman N., Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Goodman S. Toward evidence-based medical statistics. 1: The P value fallacy. Ann. Intern. Med. 1999;130:995–1004. doi: 10.7326/0003-4819-130-12-199906150-00008. [DOI] [PubMed] [Google Scholar]
- Grenfell B.T., et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–32. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., et al. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hjort N., et al. Post-processing posterior predictive p values. J Am. Stat. Assoc. 2006;101:1157–1174. [Google Scholar]
- Hobolth A., Stone E. Simulation from endpoint-conditioned, continuous-time Markov chains on a finite state space, with applications to molecular evolution. Ann. Appl. Stat. 2009;3:1204–1231. doi: 10.1214/09-AOAS247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J., Dyer K. Bayesian estimation of positively selected sites. J. Mol. Evol. 2004;58:661–672. doi: 10.1007/s00239-004-2588-9. [DOI] [PubMed] [Google Scholar]
- Hughes A.L., Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–70. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- Huigen M.C., et al. Compensatory fixation explains long term persistence of the m41l in HIV-1 reverse transcriptase in a large transmission cluster. Antivir. Ther. 2006;11:113. [Google Scholar]
- Karlsson A.C., et al. Sequential broadening of CTL responses in early HIV-1 infection is associated with viral escape. PLoS One. 2007;2:e225. doi: 10.1371/journal.pone.0000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S.D., et al. A novel polymorphism at codon 333 of human immunodeficiency virus type 1 reverse transcriptase can facilitate dual resistance to zidovudine and L-2’,3’-dideoxy-3’-thiacytidine. J. Virol. 1998;72:5093–5098. doi: 10.1128/jvi.72.6.5093-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Frost S.D.W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., et al. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S., Plotkin J.B. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larder B.A., Kemp S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- Lartillot N. Conjugate Gibbs sampling for Bayesian phylogenetic models. J. Comput. Biol. 2006;13:1701–1722. doi: 10.1089/cmb.2006.13.1701. [DOI] [PubMed] [Google Scholar]
- Lemey P., et al. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput. Biol. 2007;3:e29. doi: 10.1371/journal.pcbi.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., et al. Bayesian phylogeography finds its root. PLoS Comput. Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., et al. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., et al. Effect of the Q207D mutation in HIV type 1 reverse transcriptase on zidovudine susceptibility and replicative fitness. J. Acquir. Immune Defic. Syndr. 2005;40:20–23. doi: 10.1097/01.qai.0000172369.82456.36. [DOI] [PubMed] [Google Scholar]
- Maritz J. Empirical Bayes estimation for the Poisson distribution. Biometrika. 1969;56:349–359. [Google Scholar]
- Messier W., Stewart C.B. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Minin V., Suchard M. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 2008a;56:391–412. doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- Minin V., et al. Imputation estimators partially correct for model misspecification. Stat. Appl. Genet. Mol. Biol. 2011;10:17. [Google Scholar]
- Minin V.N., Suchard M.A. Fast, accurate and simulation-free stochastic mapping of discrete traits. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2008b;363:3985–3995. doi: 10.1098/rstb.2008.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse S., Gaut B. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Mapping mutations on phylogenies. Syst. Biol. 2002;51:729–739. doi: 10.1080/10635150290102393. [DOI] [PubMed] [Google Scholar]
- Nielsen R., Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J., et al. Learning to count: robust estimates for labeled distances between molecular sequences. Mol. Biol. Evol. 2009;26:801–814. doi: 10.1093/molbev/msp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond S., Muse S. Site-to-site variation of synonymous substitution rates. Mol. Biol. Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Pond S.L.K., et al. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2006;2:e62. doi: 10.1371/journal.pcbi.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pybus O.G., Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009;10:540–550. doi: 10.1038/nrg2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins H. Proceedings of the Third Berkeley Symposium on Mathematical Statistics and Probability, Volume 1: Contributions to the Theory of Statistics. Berkeley, California: 1956. An empirical Bayes approach to statistics; pp. 157–163. [Google Scholar]
- Rodrigue N., et al. Uniformization for sampling realizations of Markov processes: applications to Bayesian implementations of codon substitution models. Bioinformatics. 2008;24:56–62. doi: 10.1093/bioinformatics/btm532. [DOI] [PubMed] [Google Scholar]
- Saracino A., et al. Impact of unreported HIV-1 reverse transcriptase mutations on phenotypic resistance to nucleoside and non-nucleoside inhibitors. J. Med. Virol. 2006;78:9–17. doi: 10.1002/jmv.20500. [DOI] [PubMed] [Google Scholar]
- Shackelton L.A., et al. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl Acad. Sci. USA. 2005;102:379–384. doi: 10.1073/pnas.0406765102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., et al. Proceedings of the 10th International Conference on Research in Computational Molecular Biology. Venice, Italy: 2006. New methods for detecting lineage-specific selection; pp. 190–205. [Google Scholar]
- Stoeckli T.C., et al. Phenotypic and genotypic analysis of biologically cloned human immunodeficiency virus type 1 isolates from patients treated with zidovudine and lamivudine. Antimicrob. Agents Chemother. 2002;46:4000–4003. doi: 10.1128/AAC.46.12.4000-4003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M., Rambaut A. Many-core algorithms for statistical phylogenetics. Bioinformatics. 2009;25:1370–1376. doi: 10.1093/bioinformatics/btp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Gojobori T. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- Svicher V., et al. Involvement of novel human immunodeficiency virus type 1 reverse transcriptase mutations in the regulation of resistance to nucleoside inhibitors. J. Virol. 2006;80:7186–7198. doi: 10.1128/JVI.02084-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters M.A., Merigan T.C. Variants other than aspartic acid at codon 69 of the human immunodeficiency virus type 1 reverse transcriptase gene affect susceptibility to nuleoside analogs. Antimicrob. Agents Chemother. 2001;45:2276–2279. doi: 10.1128/AAC.45.8.2276-2279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Maximum-likelihood models for combined analyses of multiple sequence data. J. Mol. Evol. 1996;42:587–596. doi: 10.1007/BF02352289. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosc. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z., et al. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005;22:1107. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
-
Zhai W., et al. Exploring variation in the
 . ratio among sites and lineages using mutational mappings: applications to the influenza virus. J. Mol. Evol. 2007;65:340–348. doi: 10.1007/s00239-007-9019-7. [DOI] [PubMed] [Google Scholar]
. ratio among sites and lineages using mutational mappings: applications to the influenza virus. J. Mol. Evol. 2007;65:340–348. doi: 10.1007/s00239-007-9019-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.