

Abstract
Aims
Two randomized, double-blind, placebo-controlled studies were performed to characterize the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of the investigational metastin analogue, TAK-683, in healthy men.
Methods
We first investigated a single subcutaneous (s.c.) dose of TAK-683 (0.01–2.0 mg) in 60 subjects (TAK-683, n = 42; placebo, n = 18). We then assessed a single s.c. bolus of 0.03–1.0 mg TAK-683 on day 1, followed by a 0.01–2.0 mg day−1 continuous infusion on days 2–13, to simulate a depot formulation, in 30 subjects (TAK-683, n = 25; placebo, n = 5) for 14 days.
Results
TAK-683 was well tolerated up to a dose of 2.0 mg day−1 by continuous s.c. infusion for 14 days. Adverse events were similar between TAK-683 and placebo subjects at all dose levels. TAK-683 plasma concentrations generally increased in proportion to dose with single and continuous dosing, with steady-state concentrations achieved by day 2 of continuous dosing. TAK-683 at 2.0 mg day−1 suppressed testosterone below castration level (<50 ng dl−1) in four of five subjects by day 7 of continuous dosing. Luteinizing hormone and follicle stimulating hormone concentrations were suppressed with TAK-683 continuous dosing compared with placebo by up to 70 and 43%, respectively, but this was not consistently dose-dependent.
Conclusions
In healthy men, s.c. administration of TAK-683 was well tolerated at all dose levels. The PK profile of TAK-683 was favourable, and TAK-683 suppressed testosterone profoundly during continuous dosing. Further investigation of metastin analogues is warranted for the treatment of castration-resistant prostate cancer.
Keywords: castration-resistant prostate cancer, metastin analogue, pharmacodynamics, pharmacokinetics, TAK-683
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Metastin is a peptide expressed by the KiSS1 gene and is a regulator of the gonadotropin-releasing hormone system.
TAK-683 is a metastin analogue that has demonstrated testosterone suppression in male rats.
Metastin analogues such as TAK-683 are being investigated for treatment of castration-resistant prostate cancer.
WHAT THIS STUDY ADDS
This paper reports first-in-human data from two phase 1 studies of TAK-683 in healthy men.
TAK-683 was well tolerated as a single subcutaneous dose and during continuous subcutaneous dosing.
Continuous dosing of TAK-683 suppressed testosterone concentrations to below castration level. This is the first demonstration in humans of this effect of a metastin analogue. Luteinizing hormone and follicle stimulating hormone concentrations were also suppressed, but that effect was not consistently dose dependent.
Introduction
Metastasis is a pivotal step in cancer progression. One endogenous mechanism for cell proliferation involves the product from the KiSS-1 gene, which has been shown to function as a tumour metastasis suppressor gene and to reduce the number of metastases in preclinical models of different cancers including human melanoma and breast cancer cells [1, 2]. Although the full mechanism by which KiSS-1 suppresses metastasis is not yet fully understood, expression of KiSS-1 is commonly reduced or completely absent in a variety of cancers [3–6]. Re-expression of KiSS-1 in tumour cell lines allows the precursor phases of metastasis to occur, but inhibits colonization of secondary sites. In addition, several clinical reports indicate that a loss or reduction of KiSS-1 expression in different human cancers inversely correlates with tumour progression, metastasis and survival [7].
KiSS-1 encodes a 145-amino acid peptide, which is further processed to a final 54-amino acid peptide known as metastin (or kisspeptin-54) [8]. Metastin is a ligand of the receptor OT7T175/GPR54 [8, 9], which is expressed in a variety of endocrine and gonadal tissues. Studies have shown that mutations of OT7T175/GPR54 are responsible for abnormal sexual maturation in mice [10] and are found in human hypogonadotrophic hypogonadism [11, 12]. Metastin and its analogues have an agonist effect on gonadotropin-releasing hormone (GnRH), luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in vivo [13]. Metastin stimulates GnRH secretion from the hypothalamus, leading to an increase in LH and FSH secretion and a corresponding rise in testosterone. Continuous infusion of metastin in adult male monkeys has been demonstrated to result in desensitization of GPR54 [14]. Intravenous administration of metastin to healthy male subjects up to a dose of 3.6 nmol kg−1 has been shown to increase plasma concentrations of LH, FSH and testosterone, and was well tolerated [15].
TAK-683 is an investigational peptide derivative of metastin that has been shown preclinically to be a potent metastin/GPR54 agonist both in vitro and in vivo [16]. Administration of TAK-683 to male rats for 4 or 8 weeks has been shown to deplete GnRH in the hypothalamus and to reduce plasma FSH, LH and testosterone concentrations [16]. It has been hypothesized that chronic administration of TAK-683 may result in depletion of hypothalamic GnRH, resulting in attenuation of its normal pulsatile release with consequent reductions in FSH, LH and testosterone concentrations [16]. The cornerstone of advanced prostate cancer management is androgen deprivation therapy [17, 18], but the andropausal state, achieved by the wide variety of medical therapies now available, results in significant long term adverse effects [19–21]. New approaches involving immunotherapy [22] or hormonal therapy [23] have had mixed results, indicating that alternative therapies are still needed as castration resistance develops. Therefore, TAK-683 might have additional clinical value in this disease setting by inhibiting metastasis while simultaneously suppressing circulating testosterone.
This paper reports the results of two first-in-human studies of TAK-683 to determine the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of single and continuous subcutaneous (s.c.) dosing in healthy male volunteers.
Methods
Objectives
The primary objective of these two studies (EudraCT nos. 2007-004101-94 and 2008-003158-14) was to determine the safety and tolerability of TAK-683 in healthy volunteers. The single dose study examined the safety and tolerability of single dose s.c. TAK-683, and the continuous dosing study examined an s.c. bolus of TAK-683 followed by a 13 day continuous s.c. infusion of TAK-683. The secondary objectives of both studies were to assess the PK of TAK-683 in plasma and urine, and to determine the PD effects of the study drug on serum hormone concentrations.
Study participants
The single dose study was conducted at two study sites in the UK and the continuous dosing study was conducted at a single site in the UK. Both studies used healthy male volunteers age ≥50 years. That age range was selected because it approximates to the age group of men with prostate cancer [5001]. The principal inclusion criteria were age ≥50 years, body mass index (BMI) ≥ 18.5 kg m−2 and <32.0 kg m−2, good overall health and results of clinical laboratory tests (haematology, clinical chemistry, liver function tests, urinalysis) and concentrations of LH, FSH and testosterone, either within the reference ranges or, if outside, not deemed relevant by the Investigator. Subjects were excluded if they had a history of any type of cancer, serum prostate-specific antigen concentrations >4 ng ml−1 (single dose study) or >5 ng ml−1 (continuous dosing study), a positive test for HIV, hepatitis B, or hepatitis C, any current or history of renal or hepatic impairment or any other illness that was considered to make them ineligible to participate. The study was conducted according to the principles founded in the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. Study protocols were approved by properly constituted Research Ethics Committees independent of the participating institutions, and were considered by the Medicines and Healthcare products Regulatory Agency. All patients gave written informed consent.
Study design
The single dose study was a phase 1, randomized, double-blind, placebo-controlled, ascending single dose study. Six cohorts of 10 subjects each were planned. In each cohort, seven subjects were randomized to receive TAK-683 and three to receive placebo. Each subject received a single dose of TAK-683 or placebo in the range 0.01–2 mg (Table 1), after a fast of ≥8 h. Study drug administration was conducted in three sequential groups, with subjects in each group being dosed in parallel. Initially, one subject received TAK-683 and one received placebo. After review of safety data over 24 h, a further two subjects were given study medication in the same way. After reviewing safety data for those subjects, the remaining six subjects were dosed, five with TAK-683 and one with placebo. Tests were done on the day of injection (day 1), and on days 2, 3, 4 and 14. A randomization schedule was generated by Takeda Global Research & Development Centre (Europe) Ltd personnel. All randomization information was secured and accessible only by authorized randomization personnel. Rationale for dose selection considered findings from toxicology and pharmacology studies in rats and dogs. The no observed adverse effect level for systemic toxicity in a single dose s.c. toxicity study in dogs was 0.06 mg kg−1 and this is equivalent to a human dose of 2.27 mg day−1. This provided a safety margin of approximately 230-fold higher than the starting dose (0.01 mg) used in this study.
Table 1.
Study medication (TAK-683 or placebo), dose and administration route (single and continuous dosing studies)
| Dose cohort | Study drug | Single dose | Continuous dosing | |||
|---|---|---|---|---|---|---|
| s.c. bolus (mg) | n | s.c. bolus day 1 (mg) | s.c. infusion days 2–14 (mg day−1) | n | ||
| 1 | TAK-683 | 0.01 | 7 | 0.03 | 0.01 | 5 |
| 2 | TAK-683 | 0.03 | 7 | 0.1 | 0.03 | 5 |
| 3 | TAK-683 | 0.1 | 7 | 0.3 | 0.1 | 5 |
| 4 | TAK-683 | 0.3 | 7 | 1.0 | 0.3 | 5 |
| 5 | TAK-683 | 1.0 | 7 | 1.0 | 2.0 | 5 |
| 6* | TAK-683 | 2.0 | 7 | – | – | – |
| All | Placebo | Placebo | 18 | Placebo | Placebo | 5 |
Single dose study only. There were no discontinuations in either study.
The continuous dosing study was also a phase 1, randomized double-blind, placebo-controlled study of ascending doses of TAK-683, but, in contrast with the single dose study, TAK-683 was administered in a protocol designed to mimic a depot formulation. TAK-683 was administered as a single s.c. dose on day 1, followed by a 13 day continuous infusion via an ambulatory pump from day 2 to day 14. Five cohorts, each of six subjects, were planned. In each cohort, five subjects received TAK-683 and one received placebo. In common with the single dose study, TAK-683 was administered in ascending daily doses in the range 0.01–2 mg (Table 1). Subjects remained at the clinic for the 14 days of dosing and for 3 days after completion of the course (day 17). Tests were done on days 1–17 and days 21, 28 and 35. Subjects in both studies who were sexually active with partners of child-bearing age were required to use adequate contraception throughout dosing and for 5 days afterwards.
Safety tests
Safety tests in both studies included adverse events (AEs), clinical laboratory tests, physical examination and 12-lead electrocardiogram (ECG). AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA version 11.0 for the single dose study and version 11.1 for the continuous dosing study), and were categorized as mild, moderate or severe. AEs were also categorized as either definitely, probably, possibly or not related to study drug. Serious adverse events (SAEs) were defined as AEs that resulted in death, or were life threatening, required in-patient hospitalization, were a congenital anomaly/birth defect or were considered to be an important medical event.
PK sampling and analyses
PK parameters were determined on days 1–4 in the single dose study and days 1–17 in the continuous dosing study. Concentrations of TAK-683 in human plasma and urine were measured by ultraperformance liquid chromatography with tandem mass spectroscopy assays with a lower limit of quantification (LLOQ) of 5.0 pg ml−1. Assay precision %CV was <6% and accuracy was ±2%. Concentrations below the LLOQ were set to zero for PK analysis.
For both studies, PK parameters included percent coefficient of variation (%CV =100 × √(exp(SDlog 2) − 1) where SDlog is the SD of log-transformed values); maximum observed plasma concentration (Cmax), time to reach Cmax (tmax), area under the plasma concentration–time curve from time 0 to infinity (AUC(0,∞), calculated using the linear trapezoidal rule), terminal elimination half-life (t1/2), the total amount of study drug excreted in urine from time 0 to time t (Ae(0,t)), the fraction of study drug excreted in urine (fe), and renal clearance (CLR). In addition, area under the plasma concentration–time curve during 24 h (AUC(0,24 h)) was assessed in the continuous dosing study. Area under the plasma concentration−time curve from time 0 to infinity was calculated as AUC(0, ∞) = AUC(0,tlqc) + lqc/λz, where lqc is the last quantifiable concentration. Cmax, tmax, AUC(0,∞) and t1/2 were determined on days 1–4 for the single dose study, and those parameters and AUC(0,24 h) were determined on days 1, 2 and 14 for the continuous dosing study. Ae(0,t), fe and CLR were determined up to 72 h post dose in the single dose study, and on days 1, 2 and 14 in the continuous dosing study.
PD sampling and analyses
In both studies, serum samples were analyzed for circulating concentrations of total testosterone, FSH and LH. Total testosterone was assessed by radioimmunoassay or chemiluminescent enzyme immunoassay with LOQ of 20 ng dl−1 for studies 1 and 2 (cohorts 1–4) and LOQ of 4 ng dl−1 for study 2 cohort 5. FSH was assessed by a chemiluminescent enzyme immunoassay with LOQ of 0.7 mIU ml−1 (study 1) and 0.1 mIU ml−1 (study 2). LH was assessed by a chemiluminescent enzyme immunoassay with LOQ of 0.2 mIU ml−1 (study 1) and 0.05 mIU ml−1 (study 2). PD parameters were determined for each dose level. In the continuous dosing study, the castration concentration of testosterone was defined as <50 ng dl−1 for all samples collected during a 24 h period on day 14. Analysis of those data included the number and percentage of subjects under the castration level at day 14.
Statistical analysis
In the single dose study, the sample size of 60 subjects was not based on any statistical analysis but was considered to be adequate to describe the PK, PD and safety characteristics, based on experience and results from other first-in-human studies. Based on the final safety results from the single dose study, 30 subjects were included in the continuous dosing study.
For each study, the safety dataset consisted of all subjects who received at least one dose of study medication. The PK and PD datasets contained all subjects in the safety set who had evaluable PK or PD data. In each study, data from all subjects receiving placebo were pooled for analysis.
For the dose proportionality evaluation, the power model was used. In the single-dose study, normalized AUC(0,∞) and normalized Cmax parameters were used in the model, with a and b as coefficients and Y the PK parameter (Y = a × [dose] × b × error). In the continuous dosing study, the same calculation was performed for log-transformed AUC(0,24 h), AUC(0,∞) and Cmax. Assessment of dose proportionality was reported by using the estimated slope of the linear regression line (b) and 95% confidence interval (CI). The Wilcoxon Rank-Sum test was performed on tmax. In the continuous dosing study, power calculations were performed on data from days 1, 2 and 14. In the single dose study, the calculation was performed on data gathered on day 1 only.
Statistical analyses were performed using SAS version 8.2 or later.
Results
Subject baseline characteristics
In the single dose study, 60 men were randomized and 42 received TAK-683. In the continuous dosing study, 30 men were randomized and 25 received TAK-683. Demographic characteristics were similar in the two studies and between the placebo and TAK-683 treatment groups in each study (data not shown). Median age (range) was 56.5 (50–79) and 66.5 (51–81) years in the single and continuous dosing studies, respectively; 93% of subjects were white.
Safety and tolerability
In the single dose study, 19 of 42 subjects (45%) experienced at least one AE considered to be associated with TAK-683, compared with seven of 18 subjects (39%) who received placebo. The overall frequency of AEs reported in the single dose study was similar in the TAK-683 and placebo groups (31% vs. 28%, respectively). The most common study drug-related AEs were related to the injection site and included injection site erythema (six subjects) and injection site pain (three subjects). All AEs were mild. No subjects discontinued for any reason including due to AEs.
Similarly, in the continuous dosing study, 12 of 25 subjects (48%) who received TAK-683 experienced at least one treatment-related AE, compared with three of five (60%) placebo-treated subjects. There was no consistent relationship between TAK-683 dose level and any type of AE, and all AEs were either mild or moderate in intensity. The most common AEs considered to be study drug-related were of a similar nature to those in the single dose study, with three subjects in the TAK-683 cohort 1 (0.1 mg) group reporting headache, and one subject in the cohort 5 (2.0 mg) group reporting injection site erythema. Hot flush was reported in one subject in the cohort 5 (2.0 mg) group.
In both studies, neither clinical laboratory variables, vital signs, physical examination, nor ECG measurements showed any clinically relevant difference between the TAK-683 and placebo groups.
PK
All subjects who received at least one dose of TAK-683 and had evaluable PK data were included in the PK analysis (in the single dose study, n = 42 and in the continuous dosing study, n = 25).
In the single dose study, plasma concentrations of TAK-683 were proportional to dose at all time points (Table 2). t1/2 was approximately 6–9 h, and tmax was 0.25–0.50 h. Although t1/2 appeared to be longer after higher doses, this was due to limitations in assay sensitivity: TAK-683 plasma concentrations could not be measured for long enough after the lower doses (LLOQ of 5.0 pg ml−1). Only small amounts of TAK-683 were excreted in urine at all dose levels (Table 2).
Table 2.
Single dose study: TAK-683 main PK parameters in plasma and urine, after a single bolus dose
| Geometric mean (%CV) | ||||||
|---|---|---|---|---|---|---|
| 0.01 mg (n = 7) | 0.03 mg (n = 7) | 0.1 mg (n = 7) | 0.3 mg (n = 7) | 1.0 mg (n = 7) | 2.0 mg (n = 7) | |
| Plasma | ||||||
| Cmax (pg ml−1) | 58.6 (4.17) | 125.4 (27.68) | 603.2 (56.97) | 1396.9 (43.74) | 5 404.8 (41.23) | 10 940.0 (26.08) |
| AUC(0,∞) (pg ml−1 h) | 173.0 (20.11) | 414.4 (21.64) | 2080.0 (21.04) | 5988.8 (23.04) | 20 027.7 (16.47) | 34 565.7 (21.12) |
| t1/2 (h) | 2.92 (52.96) | 3.07 (38.14) | 6.05 (18.87) | 7.41 (26.93) | 8.92 (31.75) | 8.81 (33.06) |
| tmax (h), median, (range) | 0.25 (0.08–0.50) | 0.25 (0.08–0.50) | 0.25 (0.25–0.50) | 0.25 (0.25–0.52) | 0.50 (0.25–0.50) | 0.50 (0.25–0.50) |
| Urine | ||||||
| CLR (l h−1) | 2.366 (312.23) | 0.072 (–) | 0.035 (209.14) | 0.036 (72.71) | 0.048 (224.65) | 0.065 (83.67) |
| Ae(0,t) (μg) | 0.039 (277.71) | 0.004 (–) | 0.077 (171.33) | 0.213 (81.57) | 0.960 (194.03) | 2.240 (72.76) |
| fe (%) | 3.913 (277.71) | 0.125 (–) | 0.077 (171.33) | 0.071 (81.57) | 0.096 (194.03) | 0.112 (72.76) |
AUC(0, ∞), area under the plasma concentration–time curve from time 0 to infinity; Ae(0,t), total amount of drug excreted in urine from time 0 to t; Cmax, maximum observed plasma concentration; CLR, renal clearance CLR = Ae(0,t)/AUC(0,t); fe, Fraction of drug excreted in urine, fe = (Ae(0,t)/dose) × 100; t1/2, terminal half-life, n(2)/λz; tmax, time to reach Cmax; %CV, percent coefficient of variation.
In the continuous dosing study, plasma concentrations of TAK-683 and the corresponding Cmax and AUC(0,∞) values generally increased in proportion to dose after the s.c. bolus dose on day 1 (Table 3; Figure 1A) and after continuous s.c. infusions on day 2 (Table 3; Figure 1B), after which the plasma concentrations generally remained constant until the end of the continuous infusions on day 14 (Figure 1C). Plasma concentrations of TAK-683 were slightly higher on day 1 than on day 14, as assessed by AUC(0,24 h) (Table 3). Statistical analysis of PK data following the s.c. bolus dose of TAK-683 on day 1 indicated dose proportionality. The amount of TAK-683 excreted in urine did not consistently increase with dose after the bolus dose on day 1, or when measured after the s.c. infusions on days 2 and 14. Mean fractional urinary excretion of TAK-683 was trivial at all dose levels, on both day 1 and day 14, ranging between 0.014–0.314% of the dose.
Table 3.
Continuous dosing study: TAK-683 plasma PK parameters for days 1, 2 and 14
| Geometric mean (%CV) | |||||
|---|---|---|---|---|---|
| Cohort 1 (n = 5) | Cohort 2 (n = 5) | Cohort 3 (n = 5) | Cohort 4 (n = 5) | Cohort 5 (n = 5) | |
| 0.01 mg | 0.03 mg | 0.1 mg | 0.3 mg | 2.0 mg | |
| Plasma | |||||
| Day 1 (s.c. bolus dose) | |||||
| Cmax (pg ml−1) | 173.7 (47.98) | 538.3 (18.12) | 1650 (11.34) | 7 824 (17.36) | 7 139 (23.30) |
| tmax (h), median, (range) | 0.25 (0.25–0.52) | 0.28 (0.25–0.52) | 0.50 (0.25–0.75) | 0.50 (0.28–0.75) | 0.50 (0.27–0.53) |
| AUC(0,24 h) (pg ml−1 h) | 495.6 (29.97) | 1579 (32.90) | 5760 (12.79) | 22 954 (2.30) | 19 364 (24.05) |
| AUC(0, ∞) (pg ml−1 h) | 558.2 (21.65) | 1617 (34.50) | 6059 (12.33) | 23 476 (2.63) | 20 272 (22.85) |
| t1/2 (h), median, (range) | 4.32 (3.43–9.36) | 5.32 (3.38–7.46) | 6.64 (5.68–8.27) | 5.45 (4.81–5.99) | 5.57 (4.08–10.40) |
| Day 2 (s.c. infusion) | |||||
| Cavg (pg ml−1) | 6.32 (20.31) | 19.54 (48.03) | 67.24 (25.68) | 224.9 (10.21) | 1 423 (23.62) |
| AUC(0,24 h) (pg ml−1 h) | 151.8 (20.29) | 469.0 (48.10) | 1614 (25.73) | 5 398 (10.21) | 34 163 (23.62) |
| Day 14 (s.c. infusion) | |||||
| Cavg (pg ml−1) | 4.14 (78.27) | 15.05 (38.89) | 55.10 (24.71) | 206.7 (22.83) | 677.5 (28.76) |
| AUC(0,24 h) (pg ml−1 h) | 99.83 (77.17) | 361.3 (38.76) | 1323 (24.68) | 4 960 (22.83) | 16 259 (28.76) |
| t1/2 (h), median, (range) | 3.44 (3.44–3.44) | 2.59 (1.18–4.00) | 3.67 (2.07–5.46) | 6.69 (4.11–10.80) | 14.26 (10.80–17.94) |
AUC(0, ∞), area under the plasma concentration–time curve from time 0 to infinity; AUC(0,24 h), area under the plasma concentration-time curve from time 0 to 24 h; Cmax, maximum observed plasma concentration; Cavg, Average observed plasma concentration over 24 h; t1/2, terminal half life, n(2)/λz; tmax, time to reach Cmax.
Figure 1.
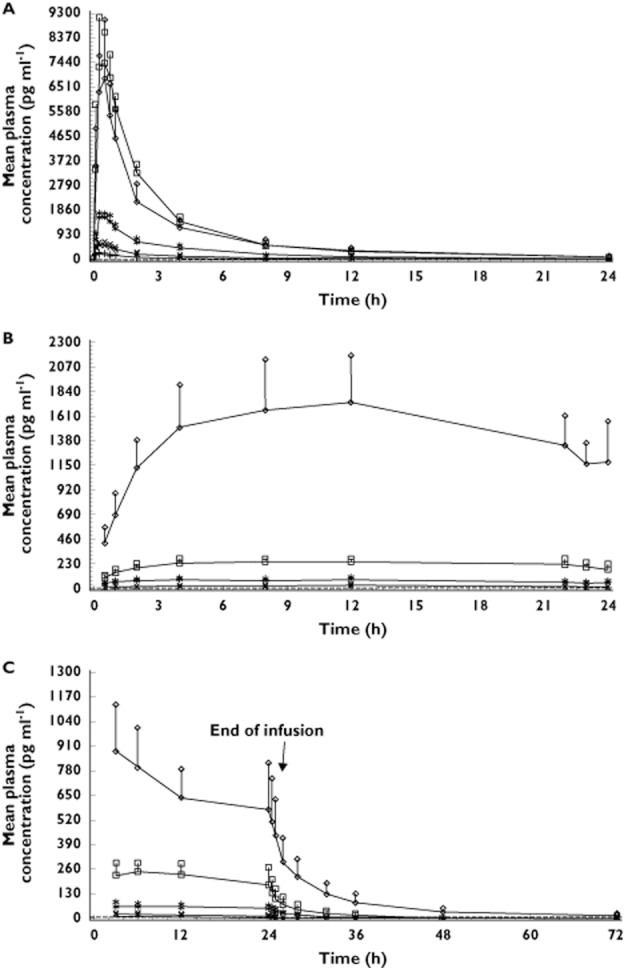
Continuous dosing study: mean plasma concentration of TAK-683 by treatment cohort on (A) day 1, (B) day 2, (C) and days 14–16. Values represent mean ± SD.  , TAK-683 0.01 mg day−1 (Cohort 1).
, TAK-683 0.01 mg day−1 (Cohort 1).  , TAK-683 0.03 mg day−1 (Cohort 2).
, TAK-683 0.03 mg day−1 (Cohort 2).  , TAK-683 0.1 mg day−1 (Cohort 3).
, TAK-683 0.1 mg day−1 (Cohort 3).  , TAK-683 0.3 mg day−1 (Cohort 4).
, TAK-683 0.3 mg day−1 (Cohort 4).  , TAK-683 2.0 mg day−1 (Cohort 5)
, TAK-683 2.0 mg day−1 (Cohort 5)
PD
All subjects were randomized and received at least one dose of TAK-683 and were included in the PD analysis. In the single dose study, TAK-683 administration resulted in a rapid, small increase in total plasma testosterone concentrations, which was not dose dependent, followed by a rapid decline within the first 8 h (Figure 2A). Between 16 h and 48 h after study drug administration, testosterone concentrations increased above baseline levels after TAK-683, but not after placebo. Concentrations of testosterone declined to near baseline values by 72 h and changes in testosterone concentrations were not dose dependent. Although placebo-treated subjects showed the same initial pattern of changes in testosterone concentration, the variations were smaller and values generally did not noticeably exceed baseline.
Figure 2.
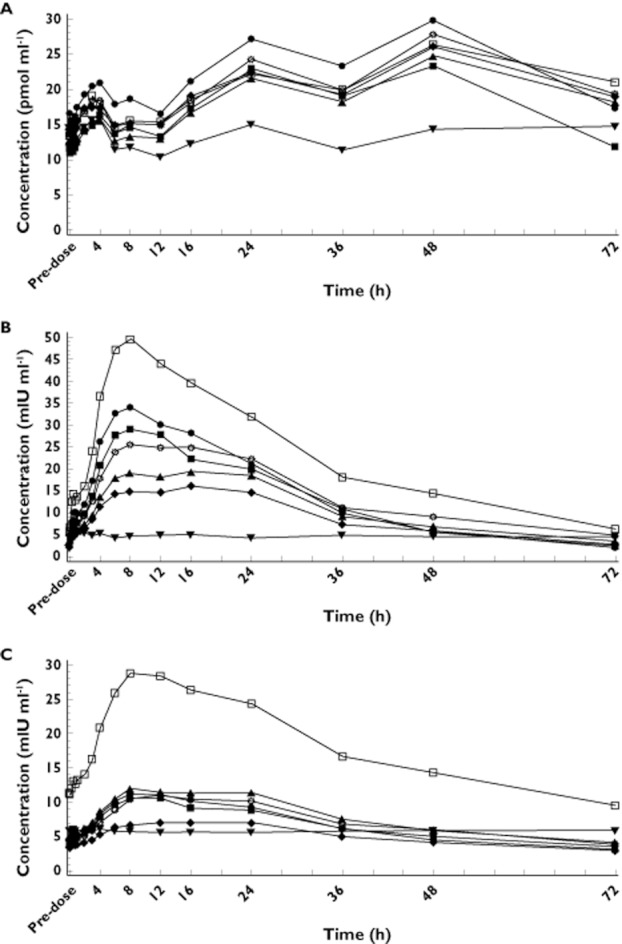
Single dose study: mean concentration–time profile (baseline to 72 h) for (A) total plasma testosterone, (B) LH and (C) FSH after administration of TAK-683 (0.01–2.0 mg) or placebo.  , placebo.
, placebo.  , TAK-683 0.01 mg.
, TAK-683 0.01 mg.  , TAK-683 0.03 mg.
, TAK-683 0.03 mg.  , TAK-683 0.1 mg.
, TAK-683 0.1 mg.  , TAK-683 0.3 mg.
, TAK-683 0.3 mg.  , TAK-683 1.0 mg.
, TAK-683 1.0 mg.  , TAK-683 2.0 mg
, TAK-683 2.0 mg
Plasma LH and FSH concentrations in the single dose study increased after TAK-683 administration, and decreased to baseline levels as plasma concentrations of the study drug decreased (Figure 2B,C). LH and FSH responses were not dose-dependent and placebo had no effect on the circulating concentrations of either hormone. Most subjects in the 0.1 mg TAK-683 group had substantially higher LH and FSH concentrations at all time points, including baseline, than did subjects in the other groups, leading to high mean LH and FSH concentrations in this group.
In common with the single dose study, LH and FSH concentrations in the continuous dosing study were all initially elevated in subjects treated with TAK-683. During days 1–4 of TAK-683 administration, the mean serum concentrations of both hormones were higher at all dose levels of TAK-683 than in the placebo group (Table 4). However, these increases were not dose-dependent. By day 14, the mean concentrations of LH and FSH in serum, measured as AUC(0,24 h) and Cavg (average concentration, calculated as AUC(0,24 h)/24), were notably lower after the 0.1 to 2.0 mg TAK-683 doses than after placebo (Table 4). At doses of TAK-683 below 0.1 mg, the reduction was either less or absent. Therefore, suppression of serum concentrations of LH and FSH by TAK-683 was not consistently dose-proportional.
Table 4.
Continuous-dosing study: PD parameters for total testosterone, LH and FSH for all cohorts on days 1 to 4 and day 14
| Analyte | Parameter | TAK-683 Mean (SD) | |||||
|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | |||
| Placebo | 0.01 mg day−1 | 0.03 mg day−1 | 0.1 mg day−1 | 0.3 mg day−1 | 2.0 mg day−1 | ||
| (n = 5) | (n = 5) | (n = 5) | (n = 5) | (n = 5) | (n = 5) | ||
| Days 1–4 | |||||||
| LH | AUC(0,96 h) | 89 | 654 | 927 | 605 | 617 | 1085 |
| (mIU ml−1 h) | (74) | (337) | (529) | (344) | (293) | (371) | |
| FSH | AUC(0,96 h) | 30 | 379 | 621 | 328 | 522 | 667 |
| (mIU ml−1 h) | (36) | (324) | (475) | (225) | (487) | (372) | |
| Testosterone* | AUC(0,96 h) | 1153 | 12023 | 6453 | 9941 | 6586 | 14629 |
| (ng dl−1 h) | (1957) | (8112) | (4041) | (5441) | (4558) | (6275) | |
| Day 14 | |||||||
| LH | AUC(0,24 h) | 107.0 | 69.9 | 105.2 | 58.5 | 52.4 | 32.4 |
| (mIU ml−1 h) | (52) | (35) | (54) | (27) | (23) | (8) | |
| Cavg | 4.5 | 2.9 | 4.4 | 2.4 | 2.2 | 1.4 | |
| (mIU ml−1) | (2.2) | (1.5) | (2.3) | (1.1) | (1.0) | (0.3) | |
| FSH | AUC(0,24 h) | 144.9 | 119.9 | 190.9 | 83.3 | 122.5 | 88.5 |
| (mIU ml−1 h) | (65) | (107) | (108) | (36) | (90) | (26) | |
| Cavg | 6.0 | 5.0 | 8.0 | 3.5 | 5.1 | 3.7 | |
| (mIU ml−1) | (2.7) | (4.5) | (4.5) | (1.5) | (3.8) | (1.1) | |
| Testosterone* | AUC(0,24 h) | 6889 | 7731 | 4095 | 3467 | 2798 | 805 |
| (ng dl−1 h) | (2109) | (2964) | (927) | (1768) | (985) | (602) | |
| Cavg | 287.0 | 322.1 | 170.6 | 144.5 | 116.6 | 33.5 | |
| (ng dl−1) | (88.9) | (123.5) | (38.6) | (73.7) | (41.0) | (25.1) | |
Total testosterone. FSH, follicle-stimulating hormone; LH, luteinizing hormone.
In the continuous dosing study, mean total testosterone concentrations were not suppressed by TAK-683 at the lowest dose level (0.01 mg) compared with placebo, but suppression increased as the TAK-683 dose level increased from 0.03 to 2.0 mg day−1 (Table 4, Figures 3 and 4). Using the definition of <50 ng dl−1 testosterone as the castration level, testosterone concentrations were suppressed to below that level in four of five subjects (80%) by day 7 in the TAK-683 2.0 mg group. Similarly, on day 14, testosterone suppression below castration level was evident in most samples from subjects receiving 2.0 mg daily (Figure 4). At that dose level, the mean TAK-683 concentration on day 14 was 677.5 pg ml−1 (Table 3), with a mean testosterone concentration of 33.5 ng dl−1 (Table 4).
Figure 3.
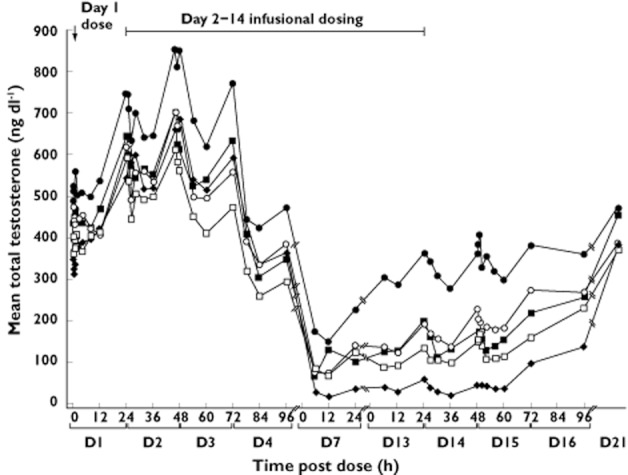
Continuous dosing study: mean serum concentrations of total testosterone with TAK-683 dosing by treatment cohort on days 1–4, 13 and 14. TAK-683 dose (mg kg−1).  , 0.01 mg kg–1.
, 0.01 mg kg–1.  , 0.03 mg kg–1.
, 0.03 mg kg–1.  , 0.10 mg kg–1.
, 0.10 mg kg–1.  , 0.30 mg kg–1.
, 0.30 mg kg–1.  , 2.00 mg kg–1
, 2.00 mg kg–1
Figure 4.
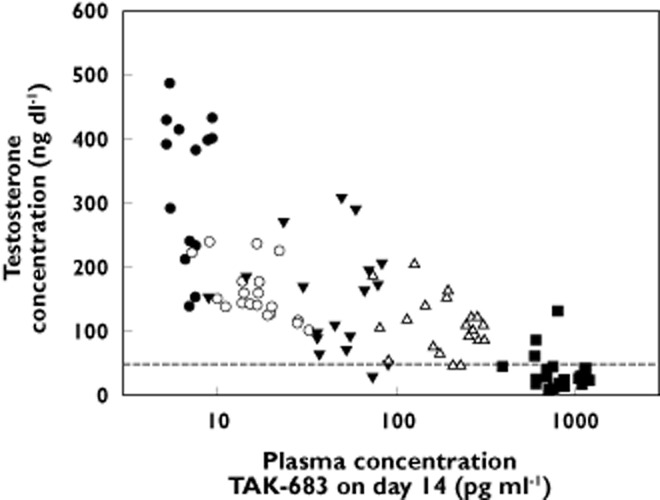
Continuous-dosing study testosterone concentration vs. TAK-683 plasma concentration on day 14, for subjects in cohorts 1 to 5; data are testosterone concentrations for all day 14 samples for all active subjects. •, 0.01 mg day−1. ◯, 0.03 mg day−1. ▾, 0.1 mg day−1. △, 0.3 mg day−1. ▪, 2.0 mg day−1
Discussion
In these two phase 1 studies including a total of 90 healthy male subjects, TAK-683 was well tolerated up to a dose of 2.0 mg administered as a single bolus and as an s.c. infusion for 13 days at doses up to 2.0 mg day−1. In both studies, AEs were mild or moderate, and there was no consistent relationship between increasing TAK-683 dose and occurrence of any type of study drug-related AE. PK analyses of plasma showed dose-proportional increases in TAK-683 after a single s.c. bolus dose and after continuous s.c. dosing of TAK-683 for 14 days. Steady-state plasma concentrations were reached by day 2 at all dose levels. The amount of TAK-683 excreted in urine was low.
TAK-683 administered as a single dose caused increases in LH, FSH and testosterone concentrations. However, those increases were not dose dependent, and the single dose study did not inform the selection of the range of doses of TAK-683 for the continuous dosing study. The single doses of TAK-683 were probably too low to cause the degree of testosterone suppression that had been observed in non-clinical studies. The increase in testosterone concentrations seen 16–48 h after TAK-683 administration, but not after placebo, was probably due to the absence of negative feedback on GnRH and LH concentrations, resulting from the decline in testosterone concentrations. Continuous dosing with TAK-683 suppressed concentrations of serum LH and FSH, but there was no clear relationship with the dose level. In contrast, suppression of testosterone by continuous TAK-683 administration at doses of 0.03–2.0 mg daily was clearly dose-dependent.
Testosterone concentrations were suppressed below castration levels (testosterone <50 ng dl−1) by day 7 in the majority of subjects receiving TAK-683 2.0 mg daily, and remained similarly suppressed for the remainder of the dosing period.
The suppression of testosterone achieved by continuous administration of a metastin analogue in the studies reported here suggests that metastin, optimally dosed, may be another form of medical castration therapy. Similar to GnRH agonist analogues, TAK-683 results in acute stimulation of FSH/LH concentrations followed by suppression during continuous exposure. Whether the onset of suppression is more rapid than the required dosing interval observed with GnRH agonists, i.e. 1 to 3 weeks, will require further study. The degree of testosterone suppression observed in this study of TAK-683 was not as profound as that observed with long term administration of GnRH analogues. Whether that is a function of dosing regimen or, alternatively, different biology of the metastin–GnRH pathway, will also require further study. In addition to androgen deprivation, other potential anti-tumour effects of TAK-683 as a potent metastin agonist may result in therapeutic benefit further to medical castration obtained with traditional GnRH-based androgen deprivation therapy. Further exploration of the therapeutic potential of the continuous administration of metastin analogues is supported by the good tolerability profile of TAK-683 in these studies.
In conclusion, in healthy male volunteers, continuous s.c. dosing with TAK-683 up to 2.0 mg daily for 14 days reduced plasma concentrations of LH, FSH and testosterone, with suppression of testosterone below castration level in 80% of subjects receiving the highest dose. However, suppression of LH and FSH was not consistently dose dependent. S.c. administration of TAK-683 was generally well tolerated at all dose levels and had a favourable PK profile. The data from these phase 1 studies demonstrate that the metastin analogue TAK-683 may be a promising candidate for further investigation in clinical trials in prostate cancer. A related metastin analogue, TAK-448, is also being evaluated in animal studies and in phase 1 clinical trials in prostate cancer [16, 25].
Acknowledgments
This research was funded by Takeda Pharmaceutical Company Limited. The authors would like to acknowledge the writing assistance of Catriona Scott and Stephen Mosley of FireKite during the development of this publication, which was funded by Millennium Pharmaceuticals, Inc.
Competing Interests
GS, IA, KH, DBM, CO and PW are employees of Takeda.
References
- 1.Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996;88:1731–1737. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 2.Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57:2384–2387. [PubMed] [Google Scholar]
- 3.Mitchell DC, Abdelrahim M, Weng J, Stafford LJ, Safe S, Bar-Eli M, Liu M. Regulation of KiSS-1 metastasis suppressor gene expression in breast cancer cells by direct interaction of transcription factors activator protein-2alpha and specificity protein-1. J Biol Chem. 2006;281:51–58. doi: 10.1074/jbc.M506245200. [DOI] [PubMed] [Google Scholar]
- 4.Masui T, Doi R, Mori T, Toyoda E, Koizumi M, Kami K, Ito D, Peiper SC, Broach JR, Oishi S, Niida A, Fujii N, Imamura M. Metastin and its variant forms suppress migration of pancreatic cancer cells. Biochem Biophys Res Commun. 2004;315:85–92. doi: 10.1016/j.bbrc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 5.Ikeguchi M, Yamaguchi K, Kaibara N. Clinical significance of the loss of KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:1379–1383. doi: 10.1158/1078-0432.ccr-1519-02. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Carbayo M, Capodieci P, Cordon-Cardo C. Tumor suppressor role of KiSS-1 in bladder cancer: loss of KiSS-1 expression is associated with bladder cancer progression and clinical outcome. Am J Pathol. 2003;162:609–617. doi: 10.1016/S0002-9440(10)63854-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck BH, Welch DR. The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. Eur J Cancer. 2010;46:1283–1289. doi: 10.1016/j.ejca.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, Ishibashi Y, Watanabe T, Asada M, Yamada T, Suenaga M, Kitada C, Usuki S, Kurokawa T, Onda H, Nishimura O, Fujino M. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 9.Gianetti E, Seminara S. Kisspeptin and KISS1R: a critical pathway in the reproductive system. Reproduction. 2008;136:295–301. doi: 10.1530/REP-08-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, Yang S, Monsma FJ, Gustafson EL. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun. 2003;312:1357–1363. doi: 10.1016/j.bbrc.2003.11.066. [DOI] [PubMed] [Google Scholar]
- 11.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF, Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 13.Matsui H, Takatsu Y, Kumano S, Matsumoto H, Ohtaki T. Peripheral administration of metastin induces marked gonadotropin release and ovulation in the rat. Biochem Biophys Res Commun. 2004;320:383–388. doi: 10.1016/j.bbrc.2004.05.185. [DOI] [PubMed] [Google Scholar]
- 14.Ramaswamy S, Seminara SB, Pohl CR, DiPietro MJ, Crowley WF, Jr, Plant TM. Effect of continuous intravenous administration of human metastin 45–54 on the neuroendocrine activity of the hypothalamic-pituitary-testicular axis in the adult male rhesus monkey (Macaca mulatta. Endocrinology. 2007;148:3364–3370. doi: 10.1210/en.2007-0207. [DOI] [PubMed] [Google Scholar]
- 15.Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, McGowan BM, Amber V, Patel S, Ghatei MA, Bloom SR. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. 2005;90:6609–6615. doi: 10.1210/jc.2005-1468. [DOI] [PubMed] [Google Scholar]
- 16.Matsui H, Takatsu Y, Tanaka A, Asami T, Nishizawa N, Kiba A, Kumano S, Suzuki A, Kusaka M, Ohtaki T. Potent and efficient testosterone suppression by chronic administration of novel metastin analogues, TAK-448 and TAK-683, in male rats. EJC Supplements. 2010;8:66. -(Abstract 251) [Google Scholar]
- 17.Kohli M, Tindall DJ. New developments in the medical management of prostate cancer. Mayo Clin Proc. 2010;85:77–86. doi: 10.4065/mcp.2009.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horwich A, Parker C, Bangma C, Kataja V. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl. 5):v129–v133. doi: 10.1093/annonc/mdq174. [DOI] [PubMed] [Google Scholar]
- 19.Keating NL, O'Malley AJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. J Clin Oncol. 2006;24:4448–4456. doi: 10.1200/JCO.2006.06.2497. [DOI] [PubMed] [Google Scholar]
- 20.Saigal CS, Gore JL, Krupski TL, Hanley J, Schonlau M, Litwin MS. Androgen deprivation therapy increases cardiovascular morbidity in men with prostate cancer. Cancer. 2007;110:1493–1500. doi: 10.1002/cncr.22933. [DOI] [PubMed] [Google Scholar]
- 21.Perlmutter MA, Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev Urol. 2007;9(Suppl. 1):S3–S8. [PMC free article] [PubMed] [Google Scholar]
- 22.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 23.Scher HI, Jia X, Chi K, de Wit R, Berry WR, Albers P, Henick B, Waterhouse D, Ruether DJ, Rosen PJ, Meluch AA, Nordquist LT, Venner PM, Heidenreich A, Chu L, Heller G. Randomized, open-label phase III trial of docetaxel plus high-dose calcitriol versus docetaxel plus prednisone for patients with castration-resistant prostate cancer. J Clin Oncol. 2011;29:2191–2198. doi: 10.1200/JCO.2010.32.8815. [DOI] [PubMed] [Google Scholar]
- 5001.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations), Bethesda, MD: National Cancer Institute. Based on November 2011 SEER data submission, posted to the SEER web site, April 2012. Available at http://seer.cancer.gov/csr/1975_2009_pops09/ (last accessed 10 August 2012)
- 25.Tanaka A, Matsui H, Asami T, Nishizawa N, Kitada C, Ohtaki T, MacLean D, Kusaka M. 2010. Suppression of testosterone release by chronic administration of investigational novel metastin analogues in male dogs and monkeys, and in healthy male volunteers. EJC Supplements (reference to be confirmed after ENA 2010 meeting)