

Abstract
Aim
Characterization of the biliary disposition of GSK1325756, using a non-invasive bile sampling technique and spectrometric analyses, to inform the major routes of metabolic elimination and to enable an assessment of victim drug interaction risk.
Method
Sixteen healthy, elderly subjects underwent non-invasive bile capture using a peroral string device (Entero-Test®) prior to and following a single oral dose of GSK1325756 (100 mg). The device was swallowed by each subject and once the weighted string was judged to have reached the duodenum, gallbladder contraction was stimulated in order to release bile. The string was then retrieved via the mouth and bile samples were analyzed for drug-related material using spectrometric and spectroscopic techniques following solvent extraction.
Results
Nuclear magnetic resonance spectroscopy (NMR) indicated that the O-glucuronide metabolite was the major metabolite of GSK1325756, representing approximately 80% of drug-related material in bile. As bile is the major clearance route for GSK1325756 (only 4% of the administered dose was excreted in human urine), this result indicates that uridine 5'-diphospho-glucuronosyltransferases (UGTs) are the major drug metabolizing enzymes responsible for drug clearance. The relatively minor contribution made by oxidative routes reduces the concern of CYP-mediated victim drug interactions.
Conclusion
The results from this study demonstrate the utility of deploying the Entero-Test® in early human studies to provide information on the biliary disposition of drugs and their metabolites. This technique can be readily applied in early clinical development studies to provide information on the risk of interactions for drugs that are metabolized and eliminated in bile.
Keywords: glucuronidation, non-invasive bile sampling, victim drug interactions
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Inhibition of a drug clearance mechanism by another co-administered drug can result in a victim drug interaction. Understanding the enzymes and transporters involved in the clearance of a drug, generally derived from in vitro systems, can help inform the drug interaction risk.
Knowledge of the drug and metabolite excretion profiles is required in human subjects to put mechanistic enzyme and transporter information into clinical context and this is generally limited to metabolic investigations of urine and faeces following a dose of radiolabelled drug during late clinical development.
What does this Study Add?
This clinical study demonstrates that human bile samples collected for drug and metabolite analysis can be used to inform the risk of victim drug interactions for drugs in early clinical development.
Using non-invasive bile sampling technology the biliary disposition of GSK1325756, a drug being developed for the treatment of chronic obstructive pulmonary disease (COPD), was evaluated in healthy elderly subjects. This showed that an O-glucuronide was the major metabolite in bile, confirming that clearance of GSK1325756 is mediated predominately by UGT enzymes.
The relatively low levels of oxidative metabolites in human bile indicate a lesser contribution of CYP enzymes to the elimination of GSK1325756. The risk of victim drug interactions with co-medications in COPD patients is therefore low, thus facilitating recruitment in forthcoming clinical trials.
Introduction
Biliary secretion is often a major route of elimination of drugs and their metabolites from the body. Knowledge of the biliary disposition of a drug is essential to understanding the relative importance of different routes of metabolism and their relationship to overall clearance [1]. As the clearance of a drug can be modulated by co-administered therapies resulting in clinically significant drug interactions, understanding the drug-related material in bile can help to assess this risk [2].
There is a wealth of published information illustrating how drug interactions can be attributed to modulation of the clearance mechanism of drugs. The most significant drug interactions in terms of both magnitude and clinical impact are mediated by the cytochrome P450 enzymes (CYPs), i.e. when CYP mediated pathways of metabolism are inhibited by the co-administration of a CYP inhibitor resulting in elevated concentrations of circulating victim drug which may be associated with adverse drug reactions [3].
The risk of victim drug interactions can be inferred by combining a mechanistic understanding of the enzymes and transporters involved in drug clearance, as determined by in vitro studies, with knowledge of the relative proportion of drug and metabolites excreted in human urine and faeces. Where biliary elimination is a significant route of drug clearance, understanding the composition of drug-related material in the bile is essential to understanding the risk of a victim drug interaction. However, since collection of bile from human subjects is generally recognized to be a complex and invasive process, biliary disposition information is rarely available.
The drug under investigation (GSK1325756, molecular weight 441, partition coefficient [log P] 3.9, see Figure 1) in the current study is intended for the maintenance treatment of chronic obstructive pulmonary disease (COPD). Many COPD patients are elderly and are frequently exposed to many other concomitant medications including some known to cause clinically relevant drug interactions by inhibiting drug metabolizing enzymes and transporters. An understanding of the risks of drug interactions is therefore important in the medical management of COPD patients. An analysis of co-medications has been conducted in the GSK-funded ECLIPSE study (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints [4]) which had over 2000 enroled COPD patients and captured patient-reported medication use. This analysis confirmed that CYP3A4 inhibitors are of most concern in this patient population. For example at the time of screening for the study 9% and 6% of patients reported using the CYP3A4 inhibitors, atorvastatin and amlodipine, respectively. Diltiazem and verapamil have been shown to result in the greatest drug interaction due to CYP3A4 inhibition, where an increased plasma exposure of approximately 5-fold has been observed for CYP3A4 metabolized drugs like simvastatin [5, 6]. Understanding the enzymes involved in the metabolic clearance of GSK1325756 is therefore critical to assess the risk of victim drug interactions with co-administered CYP inhibitors.
Figure 1.

Structure of GSK1325756 and metabolites identified in human bile captured using the Entero-Test®. Percentage of observed drug related material in pooled bile shown in parenthesis (estimated by semi-quantitative NMR)
Preclinical excretion and metabolism studies (unpublished data) in the rat indicated that bile is a major elimination route for GSK1325756. In rat bile and urine the major components were parent drug and the O-glucuronidated metabolite (M11, see Figure 1). The metabolism pathways were well predicted using in vitro methodologies in this species i.e. O-glucuronidation was also identified as a major metabolite in rat hepatocytes. The contribution of drug transporters to the elimination of GSK1325756 and its metabolites is currently unknown. Although O-glucuronidation was considered a likely pathway of metabolism in humans, the turnover in human hepatocytes was too low to substantiate this. Furthermore, other human data [unpublished] indicated a potential for oxidative metabolism as GSK1325756 was metabolically unstable when incubated with CYP1A2, 2C9, 2D6 and 3A4 recombinant enzymes and the only observed drug-related material in human blood (in addition to unchanged parent drug) was an oxidative metabolite i.e. there was no evidence of the O-glucuronide metabolite. In human urine, parent drug represented approximately 50% observed drug-related material and the predominant metabolite was the O-glucuronide (M11), which represented approximately 30% of observed drug-related material, with the remaining drug-related material consisting of oxidative metabolites. However, as the total drug-related material in urine accounted for merely 4% of the total administered dose, only a small proportion of the total elimination of GSK1325756 was understood, therefore prompting this investigation of biliary elimination in human subjects.
A simple method for the collection of duodenal bile in human subjects has been previously reported by Guiney et al. [7] and has demonstrated that the Entero-Test®, a commercially available device, could be used reliably to collect human duodenal bile in order to determine metabolites of the lipid lowering drug simvastatin. The work described herein demonstrates a further application of the Entero-Test® technique to aid the assessment of victim drug interaction risk by characterization of the biliary disposition of GSK1325756.
Methods
Materials
Entero-Test® devices (adult version, 140 cm) were purchased from HDC Corporation (Milpitas, USA). GSK1325756 (N-[4-chloro-2-hydroxy-3-(3-piperidinylsulfonyl)phenyl]-N’-(3-fluoro-2-methylphenyl)urea) was synthesized in GlaxoSmithKline, Stevenage, UK. High performance liquid chromatography (HPLC) grade acetonitrile and acetic acid were obtained from Fisher Scientific (Loughborough, UK). Analytical grade ammonium acetate was purchased from BDH (Poole, UK). Formic acid was purchased from Sigma-Aldrich (Gillingham, UK). De-ionized water was generated in the laboratory using a Millipore Mill-Q water filter unit (Molsheim, France). Deuterium oxide was purchased from GOSS Scientific Ltd (Essex, UK).
Entero-Test® procedure
The Entero-Test® is a commercially available diagnostic tool consisting of a gelatin capsule containing 140 cm of a highly absorbent nylon string which is capable of absorbing approximately 15 μl of fluid per 1 cm of line. The capsule is swallowed and one end of the string is taped to the corner of the mouth. The capsule dissolves in the stomach and the string, which is weighted at its distal end is allowed to pass into the duodenum over a period of 3.5 to 4 h, as recommended by the device manufacturer. Following a period of approximately 5 h, the string and any adsorbed gastro-intestinal fluid is withdrawn through the mouth. During withdrawal the small steel weight which is attached to the distal end of the string detaches once it encounters resistance at the pyloric sphincter and is eliminated in the stool.
Study design
The results reported here were from a study designed to assess the pharmacokinetics and pharmacodynamics of GSK1325756 (100 mg twice daily) in healthy male and female volunteers (registered at http://www.clinicaltrials.gov, Id NCT01267006, GSK study number CX3114922). Only the elderly cohort was administered the Entero-Test® and therefore the results in this report are from these volunteers only. The clinical study was conducted at Hammersmith Medicines Research Ltd, London, UK in accordance with Good Clinical Practice and the principles of the Declaration of Helsinki. The protocol was reviewed and approved by the Edinburgh Independent Ethics Committee for Medical Research (Edinburgh, Scotland, UK). Written informed consent was obtained from all subjects prior to any protocol-specific procedures. A total of sixteen subjects (eight males and eight females) with an age range of 65–80 years participated in this study.
The subjects were admitted to the study unit on the evening prior to dosing and each subject swallowed an Entero-Test® capsule with the proximal string taped to the face. Ingestion was assisted by drinking approximately 200 ml water. The subjects retired to bed with the devices left in situ in order to facilitate overnight collection of control bile. The following morning (approximately 12 h later) the Entero-Test® string was withdrawn through the mouth and processed as described below (see Sample processing). The purpose of this sample was to provide a baseline bile sample in the absence of any administered drug. Each subject was subsequently dosed with a single oral administration of GSK1325756 (100 mg tablet) with 240 ml water within 30 min following a light breakfast. Two hours post dosing, when the oral dose was judged to have transitioned from the stomach to the duodenum, each subject swallowed a second Entero-Test® as described above and was permitted up to 500 ml water over the subsequent period prior to string removal. Four hours later each subject was given a food stimulus (a sausage sandwich) in order to stimulate bile release from the gallbladder. One hour later, the strings were withdrawn from each subject and processed as described below (see Sample processing). The timings (summarized in Figure 2) were designed to ensure sufficient time for transit of the string from the stomach into the duodenum and adequate exposure to released bile, prior to withdrawal.
Figure 2.
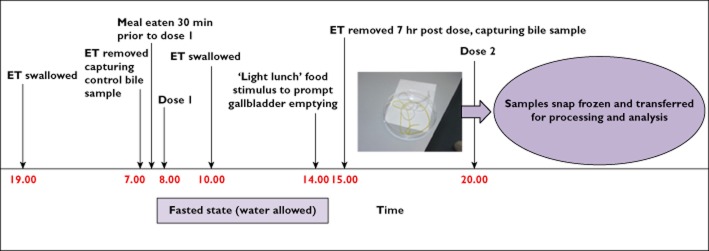
Time and events schedule for collection of Entero-Test® samples in the GSK1325756 clinical study. ET, enterotest
Sample processing
Following removal from each subject, all Entero-Test® strings (pre and post dose) were placed immediately into individually labelled 50 ml Falcon tubes. A record was made of those strings which had obvious bile staining and those which had little or no obvious colouration. The strings were frozen over solid carbon dioxide and then stored at −80°C until use. On the day of processing, the strings were allowed to defrost at room temperature. Each bile-soaked string was individually placed into the barrel of a 10 ml hypodermic plastic syringe before the plunger was reinserted and hand pressure applied in order to squeeze the sample from the string into a labelled glass vial. Acetonitrile (approximately 2 ml) was drawn into the syringe containing the string which was then agitated by hand before being eluted into the glass vial as above. The sample was eluted with two further washes of acetonitrile. The final string wash was used to rinse the appropriate 50 ml Falcon tube which had originally contained the string before being combined with the other washes.
The extracted samples were concentrated to near dryness using a Genevac DD4-X solvent evaporator (Genevac, Ipswich, Suffolk, UK) at 37°C. The concentrated extracts were individually reconstituted to a volume of 1 ml with 0.2 m ammonium acetate. Aliquots (approximately 75 μl) were taken for individual analysis by ultra performance liquid chromatography-mass spectrometry (UPLC-MS) to compare qualitative (due to the lack of available authentic standards) metabolite profiles across the subjects. Quantitative information was obtained using the remaining extracts from strings with obvious bile staining (approximately 900 μl) which were pooled for fractionation by preparative HPLC prior to analysis by nuclear magnetic resonance spectroscopy (NMR). An aliquot of pooled bile (approximately 75 μl) was also taken for analysis by UPLC-MS to aid with identification of the metabolites.
In addition, control (pre dose) bile samples collected from each subject were processed as per the post dose samples, pooled and analyzed by UPLC-MS to assist with discrimination of drug-related material from endogenous components in the bile.
UPLC-mass spectrometry analysis
UPLC separation was carried out on a Thermo Accela LC system (Thermo Scientific, San Jose, CA, USA) using an Acquity BEH C18 column (100 × 2.1 mm, 1.7 μm; Waters, Milford, USA). The column temperature was maintained at 40°C. The mobile phase consisted of 20 mm ammonium acetate adjusted to pH 3.0 with 10% formic acid (solvent A) and acetonitrile (solvent B). This was delivered at a constant flow of 0.3 ml min−1 with an initial gradient of 5% B increasing linearly to 20% B over 1 min followed by a linear increase to 50% B over the next 10 min, then a linear increase to 95% B over 1.5 min. Injections of individual and pooled bile samples (10 μl) together with an appropriate control sample were made.
The UPLC was coupled to a Thermo Accela photodiode array detector (Thermo Scientific, San Jose, CA, USA) and a Thermo LTQ Orbitrap XL (Thermo Scientific, San Jose, CA, USA) mass spectrometer controlled with Xcalibur™ version 2.07 software (Thermo Scientific, San Jose, CA, USA) with electrospray ionization in positive and negative ionization modes. Capillary voltages of 12 and −42 V (positive and negative ion), respectively, normalized collision energies of 20 eV, and source and HESI probe temperatures of 275°C and 150°C, respectively, were employed. Orbitrap mass resolution was set at 15000. A combination of accurate mass and MS/MS data on the molecular ions was used for structural identification of individual metabolites. Peak areas for metabolites of interest were automatically generated on the relevant ion traces using Xcalibur™ integration tools in order to give an estimate of the relative proportions of metabolite to parent drug across the individual samples.
Preparative HPLC
The pooled bile extract (approximately 5 ml) was separated by preparative HPLC using an Agilent series 1100 Preparative-LC system (Waldbronn, Germany). Separations were carried out on an Xbridge Prep-Phenyl HPLC column (250 × 10 mm i.d., Waters, Manchester, UK) at ambient temperature with a mobile phase of 20 mm ammonium acetate adjusted to pH 3.0 with formic acid (solvent A) and acetonitrile (solvent B) at a constant flow rate of 4 ml min−1 with an initial gradient of 5% B increasing linearly to 20% B at 4 min. This then increased linearly to 50% B at 35 min, and finally increasing linearly to 95% B at 37 min. The gradient was held at 95% B for a further 8 min before returning to initial conditions. HPLC eluent was collected into fractions, in a time-slice mode, into two 96 deep well plates using a frequency of 15 s per fraction. This resulted in 204 fractions, each containing 1 ml of column eluent. The HPLC flow was split 100:1 into a Micromass ZQ mass spectrometer (Waters, Manchester, UK) fitted with an electrospray source operated in both positive and negative ionization modes. System control was mediated through MassLynx™ and FractionLynx™ (Waters, Milford, USA). The fractions were taken to dryness under nitrogen at 37°C within the 96 deep well plates using a Micro DS96 dry down station (Porvair Scientific Ltd, Shepperton, UK) and then reconstituted in approximately 0.6 ml of deuterium oxide : acetonitrile (1:1) before being transferred to 5 mm NMR tubes.
Nuclear magnetic resonance spectroscopy
NMR spectroscopy was performed on all 204 fractions using a Bruker AVII+ spectrometer equipped with an inverse 5 mm TFI CryoProbe™ (1H/13C/19F) operating at 564.89 MHz (for 19F) under the control of TopSpin version 2.1 (Bruker, Rheinstetten, Germany). 19F NMR spectra were acquired using a standard 19F proton decoupled pulse sequence. In these experiments, 256 transients were acquired into 128 K data points over a spectral width of 113636 Hz (201 ppm) with an inter-scan delay of 2 s giving a pulse repetition time of 2.6 s. 1H NMR spectra were acquired using a standard NOESY presaturation pulse sequence for solvent suppression with time shared double pre-saturation of the water and acetonitrile frequencies. In these experiments, 256 transients were acquired into 48 K data points over a spectral width of 12019 Hz (20 ppm) with an inter-scan delay of 3 s giving a pulse repetition time of 5 s. Prior to Fourier transformation, an exponential line broadening function of 1.0 Hz was applied to each spectrum to improve the signal to noise ratio. Appropriate peaks in the 19F NMR spectra were quantified using the signal integration feature of TopSpin 2.1 software.
Results
Identification of metabolites
All sixteen elderly subjects underwent the bile sampling procedure twice, before and after dosing with GSK1325756. Based on colour assessment by visual inspection, nine out of the sixteen post dose Entero-Test® strings appeared to be soaked in duodenal bile. No observed drug-related material (oDRM) was detected by UPLC-MS on the strings that were not soaked in bile. Thirteen successful sampling occasions were noted for the pre-dose bile collection.
In the bile samples, a total of 13 metabolites were detected by UPLC-MS comprising oxidations, cyclizations and glucuronide conjugations of GSK1325756. 1H NMR data, accurate mass MS of the molecular ions and/or MS/MS data were used for definitive structural assignment of individual metabolites. For other metabolites, comparison of retention time, MS and MS/MS data with those metabolites previously reported for human blood and urine were used (unpublished data). No significant qualitative differences in metabolite profiles were observed by MS for individual bile strings which contained drug-related material.
Six drug-related components (shown in Figure 1) were detected by 19F NMR: GSK1325756 (P), M7 formed by dechlorination, cyclization and O-glucuronidation, a piperidinone O-glucuronide conjugate (M10), an O-glucuronide conjugate of GSK1325756 (M11), hydroxymethyl GSK1325756 (M14) and M16 formed by oxidation and glucuronidation. Several additional minor metabolites were detected by MS only and are not discussed in this manuscript as the remit of these investigations was to determine the major metabolites excreted in bile. The summed reconstructed ion chromatograms for GSK1325756 and the five major metabolites in pooled bile extract is shown in Figure 3 together with the pre dose bile for comparison.
Figure 3.
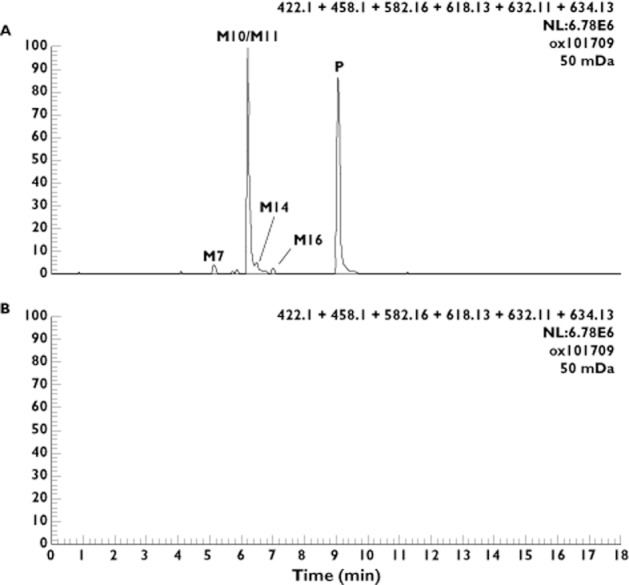
Reconstructed ion chromatograms for major human metabolites in (A) pooled bile from humans dosed orally with GSK1325756 at 100 mg and (B) pre dose pooled bile
Quantitative evaluation of major metabolites
Relative concentrations of the major metabolites in the pooled bile sample were estimated from the 19F NMR data by integration of the 19F signal using similar methodology to that described by Dear et al. [8]. The quantification limit by 19F NMR for an individual component was approximately 1% of total drug-related material which corresponded to approximately 100 ng of material.
The O-glucuronide conjugate (M11) was the major metabolite accounting for approximately 80% of the total observed drug-related material detected by NMR. The metabolite formed by oxidation to a piperidinone and O-glucuronidation (M10) accounted for approximately 6% oDRM. The remainder of oDRM was made up of unchanged parent drug, M14, M16 and M7 which individually contributed approximately 6%, 5%, 3% and 2% oDRM, respectively.
Safety and tolerability of the Entero-Test®
The procedure was very well tolerated in healthy elderly volunteers and no adverse events associated with the use of the Entero-Test® device were reported.
Discussion
Drug–drug interactions (DDIs) are an important consideration in the discovery, development and clinical use of new pharmaceuticals as there are several historical examples of serious harm leading to withdrawal from the market resulting from DDIs caused by inhibition of metabolic clearance (mibefradil [9] and terfenadine [10]). Understanding the clinical risk associated with drug interactions is therefore essential when developing new therapeutic agents in order to aid internal decision making and understand the potential impact on clinical safety in a target patient population. Ideally a drug should have minimal interaction liability as a victim otherwise a wide therapeutic index is required to minimize any clinical consequence of increasing drug concentrations. There is also a regulatory requirement to understand drug interaction risks and the FDA [11] and EMA [12] recommend that the underlying mechanisms for drug interactions are explored in advance of drug registration. Both regulatory agencies request an understanding of the enzymes involved in metabolic pathways contributing to >25% of a drug's elimination.
A large number of drug interactions have been attributed to the inhibition of CYPs which play a significant role in oxidative drug metabolism. In the CYP superfamily, the CYP3A4 enzyme is responsible for the metabolism of circa 50% of therapeutic drugs and is most frequently implicated in clinical DDIs [13]. Co-administration of a CYP3A4 inhibitor (perpetrator) with a drug whose clearance routes are predominately mediated by CYP3A4 can result in elevated and potentially toxic concentrations of the victim drug [14]. In comparison, drugs that are predominately metabolized by direct conjugation, for example glucuronidation (mediated by uridine 5'-diphospho-glucuronosyltransferase enzymes, UGT), are less sensitive to drug interactions. This can be attributed to the low affinity of substrates to UGT enzymes, the involvement of multiple UGT enzymes with over-lapping specificities and the limited potency of UGT inhibitors [15, 16]. Williams et al. [16] report that the exposure increases of a glucuronidation substrate rarely exceed 2-fold in the presence of a UGT inhibitor. This is a small magnitude of change compared with CYP3A4 victim drug interactions which can result in over 20-fold increases in the plasma concentration of drugs like midazolam [17].
In vitro methods to understand the major enzymes and transporters involved in drug elimination are widely implemented. However this information needs to be considered along with knowledge of the parent drug and metabolites eliminated in vivo to enable an accurate assessment of the clinical drug interaction risk [18]. A traditional approach to this involves dosing radiolabelled drug to humans and collecting urine and faeces to assess the routes of excretion and identify the major components. Owing to the considerable investment and enabling studies required to support a human radiolabel study [19, 20], these studies are often conducted too late in clinical development to impact on the co-medication strategy. Also bile is typically not collected in these studies thereby missing a potential key route of drug elimination [7]. Biliary elimination can sometimes be inferred by the drug-related material in faeces. However investigating this latter matrix is often technically challenging and may provide misleading information, e.g. for drugs that are poorly absorbed or those which undergo biliary secretion as conjugates (e.g. glucuronides) which are then subject to hydrolysis by gut microflora.
The human metabolism data obtained in the current study with duodenal bile collection, has significantly contributed to the assessment of the drug interaction risk for GSK1325756. The hypothesis of O-glucuronidation being the major elimination route (based on in vitro and preclinical data) was supported, as indicated by the metabolite M11 representing 80% of the observed drug-related material in bile. Therefore concerns of victim drug interactions are reduced in proposed patient studies. It can be speculated that CYP enzymes are responsible for the production of the oxidative metabolites observed in bile (<15% oDRM) and in urine (<16% oDRM). This is supported by the previous data obtained with CYP recombinant enzymes. However the current data put this oxidative metabolism into context as a likely minor elimination route in vivo. Using extrapolation techniques it can be estimated that contribution of CYP enzymes would need to comprise more than 50% total drug elimination to result in a 2-fold increase in drug exposure with a CYP inhibitor [21].
Understanding the relative proportion of metabolism and direct secretion of parent drug in bile (mediated by drug transporters [22]) may further mitigate CYP mediated victim drug interaction risks. This has minimal impact for GSK1325756 as the proportion of parent drug in bile represented only 5% observed drug-related material. The potential for contamination by unabsorbed parent drug on the bile string should be acknowledged. However the estimated ratios of metabolite to parent drug in bile were similar across subjects in this study (data not shown) which tends to support biliary secretion of parent drug rather than contamination by unabsorbed drug. This conclusion is supported by the observation that parent drug was not detected on the strings retrieved without a bile sample. Proving that clearance is predominately via secretion of unchanged parent drug may help mitigate victim drug interaction risk for other drugs, for example where metabolism is mediated primarily by CYP enzymes. Using the Entero-Test® to collect bile following an intravenous dose may be one approach to investigate this, i.e. any drug-related material detected on the string can be directly linked to biliary secretion of parent drug and/or metabolites.
The collection of bile samples was successfully conducted in this clinical study in healthy, elderly volunteers. There were no compliance or adverse events reported with use of the Entero-Test® device and the presence of food less than 3 h before administering the Entero-Test® did not preclude the collection of bile samples in over half the subjects, neither did food interfere with the analysis of the bile samples using spectrometric techniques. As reported previously [7] there appears to be wide variation in the percentage of gallbladder contraction between subjects and within an individual which may explain the differences in successful pre and post bile collection in this study.
Other potential caveats to the application of the Entero-Test® technique should be considered. It is unclear how changes in bile, pancreatic and intestinal flow may impact on the sample collected and it should be remembered that the bile sample collected represents a snapshot in time of total biliary output, in this instance 7 h post-dose. Therefore there is a risk that the metabolic profile indicated may not represent the complete excretion of drug via bile. To maximize the metabolic relevance of the sample observed in this study, the biliary collection time was optimized to coincide with the approximate half-life of the drug (i.e. 7 h). It is worth noting that this limitation is also common to other bile sampling techniques, e.g. duodenal intubation.
A further potential complication is the phenomenon of entero-hepatic recirculation which may impact on the proportion of drug cleared as glucuronide conjugates [23]. Drugs that are conjugated in the liver and secreted into bile will enter into the intestinal lumen where they may be hydrolyzed by gut microflora, e.g. β-glucuronidase, before de-conjugated drug is reabsorbed into the bloodstream and recycled to the liver. Entero-hepatic recirculation is eventually terminated by the formation of oxidative metabolites or by elimination of parent or metabolite in the faeces or in urine, via the systemic circulation. Taking a bile sample at any point during this process may partially interrupt recycling and the biliary composition may not accurately represent the contribution of a particular human metabolic pathway in the absence of sampling. This could be because less drug-related material may be available for re-conjugation and subsequent biliary secretion or, more significantly, the contribution of oxidative metabolism may be less due to limited recycling through the liver. That said, this situation is unlikely to present when sampling using the Entero-Test® given the very low volumes of bile that are sampled (≤1 ml).
For GSK1325756 the impact of entero-hepatic recirculation in human subjects is thought to be minimal due to the lack of notable and consistent secondary input peaks in the plasma concentration–time profiles, which are often associated with this phenomenon, and the relatively short apparent half-life of 7 h. Therefore the biliary profiles obtained in this study (at 7 h post dosing) are likely to be fairly reflective of the overall contribution of glucuronidation (estimated 80%) to the clearance of GSK1325756 in man and only major changes in this would alter the assessment of drug interaction risk. It can be estimated that the observed contribution of oxidative metabolism would need to increase by >3-fold before any notable impact on GSK1325756 exposure is observed following co-administration with a CYP inhibitor [21].
This study has demonstrated that for drugs with high biliary secretion, the Entero-Test® technique can be used to obtain bile samples for the evaluation of metabolic routes of clearance. For GSK1325756, which is predominately cleared by glucuronidation, this information has reduced the concern of victim drug interactions with CYP3A4 inhibitors. These data also prompt further mechanistic investigations of GSK1325756 to understand the UGT enzymes involved in glucuronidation and the drug transporters involved in elimination of drug and metabolites. Although further clinical investigations may be a regulatory expectation for drug registration, this study has provided confidence to co-administer GSK1325756 with CYP inhibitors thereby enabling the progression of clinical trials in a COPD patient population.
Acknowledgments
The authors would like to thank the staff of Hammersmith Medicines Research (London, UK) and most importantly, the volunteers for their participation in this clinical study which was funded by GlaxoSmithKline. We also thank Nick Locantore, Biomedical Data Sciences, GlaxoSmithKline for compiling the co-medication data from the ECLIPSE study.
Competing Interests
All authors are employees of GlaxoSmithKline and hold shares in the company.
References
- 1.Rollins DE, Klaassen CD. Biliary excretion of drugs in man. Clin Pharmacokinet. 1979;4:368–379. doi: 10.2165/00003088-197904050-00003. [DOI] [PubMed] [Google Scholar]
- 2.Ghibellini G, Leslie EM, Brouwer KLR. Methods to evaluate biliary excretion of drugs in human: an updated review. Mol Pharm. 2006;3:198–211. doi: 10.1021/mp060011k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guengerich FP. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol. 1997;43:7–35. doi: 10.1016/s1054-3589(08)60200-8. [DOI] [PubMed] [Google Scholar]
- 4.Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, Hagan G, Knobil K, Lomas DA, MacNee W, Silverman EK, Tal-Singer R, on behalf of the ECLIPSE investigators Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Eur Respir J. 2008;31:869–873. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 5.Mousa O, Brater DC, Sunblad KJ, Hall SD. The interaction of diltiazem with simvastatin. Clin Pharmacol Ther. 2000;67:267–274. doi: 10.1067/mcp.2000.104609. [DOI] [PubMed] [Google Scholar]
- 6.Kantola T, Kivistö KT, Neuvonen PJ. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther. 1998;64:177–182. doi: 10.1016/S0009-9236(98)90151-5. [DOI] [PubMed] [Google Scholar]
- 7.Guiney WJ, Beaumont C, Thomas SR, Robertson DC, McHugh SM, Koch A, Richards D. Use of Entero-test, a simple approach for non-invasive clinical evaluation of the biliary disposition of drugs. Br J Clin Pharmacol. 2011;72:133–142. doi: 10.1111/j.1365-2125.2011.03956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dear GJ, Roberts AD, Beaumont C, North SE. Evaluation of preparative high performance liquid chromatography and cryoprobe-nuclear magnetic resonance spectroscopy for the early quantitative estimation of drug metabolites in human plasma. J Chromatogr. 2008;876:182–190. doi: 10.1016/j.jchromb.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 9.Krayenbuhl JC, Vozeh S, Kondo-Oestreicher M, Dayer P. Drug-drug interactions of new active substances: mibefradil example. Eur J Clin Pharmacol. 1999;55:559–565. doi: 10.1007/s002280050673. [DOI] [PubMed] [Google Scholar]
- 10.Monahan BP, Ferguson CL, Killeavy ES, Lloyd BK, Troy J, Cantilena LR., Jr Torsades de pointes occurring in association with terfenadine use. JAMA. 1990;264:2788–2790. [PubMed] [Google Scholar]
- 11.US Food and Drug Administration Center for Drug Evaluation and Research. Draft Guidance for Industry: Drug Interaction Studies-Study Design, Data Analysis, and Implications for Dosing and Labeling. Rockville, MD: FDA Information Branch; 2006. [Google Scholar]
- 12.European Medicines Agency. Draft guideline on the investigation of drug interactions. April 2010.
- 13.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 14.Thummel KE. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 15.Kiang TKL, Ensom MHH, Chang TKH. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharma Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32:1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 17.Yuan R, Flockhart DA, Balian JD. Pharmacokinetic and pharmacodynamic consequences of Metabolism-Based Drug Interactions with alprazolam, midazolam, and triazolam. J Clin Pharmacol. 1999;39:1109–1125. [PubMed] [Google Scholar]
- 18.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PHRMA) perspective. Drug Metab Dispos. 2003;31:815–832. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 19.Penner N, Klunk LJ, Prakash C. Human radiolabeled mass balance studies: objectives, utilities and limitations. Biopharm Drug Dispos. 2009;30:185–203. doi: 10.1002/bdd.661. [DOI] [PubMed] [Google Scholar]
- 20.Obach RS, Nedderman AN, Smith DA. Radiolabelled mass-balance excretion and metabolism studies in laboratory animals: are they still necessary? Xenobiotica. 2012;42:46–56. doi: 10.3109/00498254.2011.621985. [DOI] [PubMed] [Google Scholar]
- 21.Ito K, Hallifax D, Obach RS, Houston JB. Impact of parallel pathways of drug elimination and multiple cytochrome P450 involvement on drug-drug interactions: CYP2D6 paradigm. Drug Metab Dispos. 2005;33:837–844. doi: 10.1124/dmd.104.003715. [DOI] [PubMed] [Google Scholar]
- 22.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation. Physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002;41:751–790. doi: 10.2165/00003088-200241100-00005. [DOI] [PubMed] [Google Scholar]