

Abstract
Aim
Eribulin mesylate is a non-taxane microtubule dynamics inhibitor that was recently approved for treatment of metastatic breast cancer. The aim of this study was to determine the effect of rifampicin, a CYP3A4 inducer, on the plasma pharmacokinetics of eribulin in patients with solid tumours.
Methods
An open-label, non-randomized phase I study was carried out. Patients received intravenous 1.4 mg m−2 eribulin mesylate on days 1 and 15 and oral rifampicin 600 mg on days 9 to 20 of a 28 day cycle. Pharmacokinetic sampling for determination of eribulin plasma concentrations was performed up to 144 h following administration. AUC(0,∞) and Cmax for eribulin exposure without or with co-administration of rifampicin were subjected to an analysis of variance (anova) and corresponding 90% confidence intervals (CI) were calculated. Subsequently, patients were allowed to continue eribulin mesylate treatment with 1.4 mg m−2 eribulin mesylate on days 1 and 8 of a 21 day cycle. Also the adverse event profile and anti-tumour activity were assessed.
Results
Fourteen patients were included and 11 patients were evaluable for pharmacokinetic analysis. Co-administration of rifampicin had no effect on single dose exposure to eribulin (geometric least square means ratio: AUC(0,∞) = 1.10, 90% CI 0.91, 1.34 and Cmax = 0.97, 90% 0.81, 1.17). The most common treatment-related grade ≥3 adverse events were grade 3 neutropenia (4/14, 29%), leucopenia and fatigue (both 3/14, 21%).
Conclusions
These results indicate that eribulin mesylate may be safely co-administered with compounds that are CYP3A4 inducers.
Keywords: CYP3A4 induction, drug−drug interaction, eribulin mesylate, microtubule dynamics inhibitor, pharmacokinetics, rifampicin
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Eribulin mesylate is a non-taxane microtubule dynamics inhibitor that was recently approved by the European Medicines Agency as monotherapy indicated for the treatment of patients with locally advanced or metastatic breast cancer who have progressed after at least two chemotherapeutic regimens for advanced disease.
Preclinical data in human liver microsomes indicated that eribulin is predominantly cleared by hepatic metabolism in vitro and that CYP3A4 is the major enzyme involved.
The aim of this study was to explore the effect of repeated oral administration of rifampicin, a potent model CYP3A4 inducer, on the plasma pharmacokinetics of eribulin mesylate administered by intravenous infusion.
WHAT THIS STUDY ADDS
Co-administration of rifampicin had no effect on single-dose exposure to eribulin.
These results indicate that eribulin mesylate may be safely co-administered with compounds that are CYP3A4 inducers.
Introduction
Drug−drug interactions are estimated to be responsible for 20–30% of all adverse drug reactions and the risk of clinically significant drug interactions increases with the number of concomitantly prescribed drugs [1]. These concerns are especially relevant in anti-cancer therapy, where patients are often exposed to many other drugs for treatment-derived complications or concurrent diseases. Furthermore, anti-cancer drugs are usually dosed at the maximum tolerated dose. Next to this, patients may use over the counter drugs, or ‘complementary and alternative medicines’, such as St John's wort, that may interact with anti-cancer therapy and treating physicians may not be aware of this [2]. Therefore, it is important to gain insight into potentially clinically relevant drug−drug interactions early in the development of new drugs.
Eribulin mesylate is a non-taxane microtubule dynamics inhibitor with a distinct mechanism of action. Eribulin prevents microtubule growth and sequesters tubulin into non-functional aggregates. The overall effects lead to G2-M arrest and ultimately cell death. Eribulin is a synthetic analogue of halichondrin B, a product isolated from the marine sponge Halichondria okadai [3–6]. Eribulin pharmacokinetics exhibit a rapid distribution phase followed by a slow elimination phase [7, 8].
Eribulin mesylate is in development in phase I−III trials for the treatment of solid tumours, both as monotherapy as well as in combination therapy with capecitabine, trastuzumab, pemetrexed, erlotinib, gemcitabine, cisplatin and carboplatin [9, 10]. Recently, the results of the phase III trial (EMBRACE) demonstrated that eribulin mesylate treatment significantly improved median overall survival, by 2.47 months, of metastatic breast cancer patients who were previously treated with an anthracycline and a taxane compared with ‘treatment of physician's choice’ [11]. EMBRACE is the first phase III single agent study in heavily pre-treated patients with metastatic breast cancer to meet its primary end-point of prolonged overall survival. Following the results of this trial, eribulin mesylate was approved by the European Medicines Agency as monotherapy indicated for the treatment of patients with locally advanced or metastatic breast cancer who have progressed after at least two chemotherapeutic regimens for advanced disease. Prior therapy should have included an anthracycline and a taxane unless patients were not suitable for these treatments.
Human cytochrome P450 (CYP) 3A4 is involved in the metabolism of approximately 60% of currently prescribed drugs and plays a dominant role in many clinically relevant drug−drug interactions [12, 13]. Preclinical data in human liver microsomes indicated that eribulin is predominantly cleared by hepatic metabolism in vitro and that CYP3A4 is the major enzyme involved [14]. Animal studies showed though that metabolism was limited. After intravenous administration of radiolabelled eribulin mesylate in bile duct cannulated rats and dogs, unchanged drug was the major component found excreted in bile, faeces and urine. However, in humans, the contribution of CYP3A4 to the metabolism of eribulin is currently unknown and it is therefore potentially subject to interaction with compounds that are known to either inhibit or induce CYP3A4. This aspect is especially important for patients with cancer, who are likely to be exposed to other drugs, including CYP3A4 inhibitors or inducers, for treatment of concurrent diseases, treatment-derived complications or as ‘complementary and alternative medicine’.
The aim of this study was to explore the effect of repeated oral administration of rifampicin, a potent model CYP3A4 inducer recommended in regulatory guidelines [15, 16] on the plasma pharmacokinetics of eribulin mesylate administered by intravenous infusion. The secondary objectives were to determine the adverse event profile and tolerability of eribulin mesylate with and without co-administration of rifampicin.
Methods
Patient selection
Patients with histologically or cytologically confirmed progressive advanced solid tumours, for whom no standard treatment was available, were eligible. Other inclusion criteria were age ≥18 years, Eastern Cooperative Oncology Group (ECOG) [17] performance status ≤2, life expectancy of ≥3 months, resolution of all chemotherapy- or radiation-related toxicities to grade 1 severity or lower, except for stable sensory neuropathy ≤ grade 2 and alopecia, adequate renal and liver function (defined as: serum creatinine ≤176 μmol l−1 or calculated creatinine clearance ≥40 ml min−1 (ml min−1, Cockroft−Gault formula) [18], bilirubin ≤1.5 times the upper limit of normal (ULN), alkaline phosphatase (AP), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤3 times ULN, in the case of liver metastasis ≤5 times ULN, liver specific AP ≤3 times ULN), adequate bone marrow function [defined as neutrophil count (ANC) ≥1.5 × 109 l−1, haemoglobin ≥6.2 mmol l−1 and platelets ≥100 × 109 l−1] and adequate contraceptive protection. Exclusion criteria included: hypersensitivity to halichondrin B and/or like compounds or rifampicin, subjects with prior participation in an eribulin clinical trial, chemotherapy, radiation or biological therapy within 2 weeks, hormonal therapy within 1 week or any investigational drug within 4 weeks, any medication, dietary supplements or other compounds or substances known to induce or inhibit CYP3A4 activity at the time the study started, impaired intestinal absorption, significant cardiovascular impairment, clinically significant electrocardiogram (ECG) abnormality, brain or subdural metastases, unless stable and completed local therapy and discontinuation of corticosteroids for at least 4 weeks, meningeal carcinomatosis or any other significant disease or disorders that would exclude the patient from the study. The study protocol was approved by the Medical Ethics Committees (METC) of participating hospitals and all patients had to give written informed consent. The study was conducted in accordance with the requirements of the World Medical Association Declaration of Helsinki (2008), International Conference of Harmonization (ICH) and Good Clinical Practice (GCP) guidelines.
Study design, study procedures and treatment administration
This phase I study was a two centre, open label, non-randomized study in patients with solid tumours to investigate the effect of oral administration of rifampicin on the plasma pharmacokinetics of eribulin mesylate (E7389; Halaven® Eisai Inc) administered by intravenous infusion (EudraCT 2009-013430-24) [19]. Also the adverse event profile and tolerability of eribulin mesylate alone or when co-administered with rifampicin were assessed during cycle 1 (study treatment phase). Another objective was to explore further adverse effects and tolerability of eribulin when given alone on days 1 and 8 of a 21 day schedule in patients with solid tumours.
Up to a maximum of 14 patients were recruited in two centres: the University Medical Center Utrecht (UMCU), Utrecht and the Netherlands Cancer Institute (NKI), Amsterdam, both in the Netherlands.
In cycle 1, patients received intravenously 1.4 mg m−2 eribulin mesylate on days 1 and 15 and rifampicin 600 mg on days 9 to 20 of a 28 day cycle. A 15 day washout period was introduced to prevent the effects of the first treatment carrying over to the second treatment. Eribulin mesylate was provided in vials containing 1.0 mg eribulin mesylate in 2 ml solution (0.5 mg ml−1) with ethanol/water (5:95). A dose of 1.4 mg m−2 eribulin mesylate is equivalent to 1.23 mg m−2 eribulin in the ‘ready to use solution’ (0.44 mg ml−1) 20. Eribulin was administered as a 2–5 min intravenous bolus injection. Rifampicin (Sanofi-Aventis, UK; Rifadin™) [21] was orally administered once daily in two 300 mg capsules (total dose day−1: 600 mg). Rifampicin was taken at least 30 min before a meal or 2 h after a meal to ensure rapid and complete absorption. Patients were instructed to maintain a diary of rifampicin dosings, concomitant medication and side-effects from days 9 to 14. Patients were allowed to continue treatment with eribulin mesylate at a dose of 1.4 mg m−2 on day 1 and day 8 of a 21 day cycle.
Complete physical examinations, including vital signs, height, weight, ECOG performance status and ECG, and clinical laboratory tests (haematology, clinical chemistry and urinalysis) were performed at screening and at regular intervals during cycle 1, in any following cycles and at study termination.
Adverse effect assessment
Adverse events (AE) and concomitant medications were assessed throughout the study. The incidence and severity of AEs were evaluated and coded according to the National Cancer Institute Common Terminology Criteria of Adverse Events (CTCAE) version 3.0 [22]. Treatment emergent AEs (TEAEs) were defined as any AE with a start date/time beyond or equal to the day of initial dosing (cycle 1 day 1), up to 30 days after the last dose of study drug, and any AE which increased in severity during the study. Patients were discontinued from the study for any of the following reasons: progressive disease by clinical evaluation or as documented by RECIST 1.0 [23], loss of clinical benefit because of undue toxicity, patient refusal of further therapy or withdrawal of consent.
Anti-tumour activity assessment
Radiologic tumour assessments according to RECIST 1.0 were performed at baseline and at regular intervals. Best response was documented for each patient.
Pharmacokinetic sampling and analysis
All patients entering the study provided blood samples for pharmacokinetic analysis. Whole blood samples for the determination of free-base eribulin plasma concentrations were taken at day 1 and day 15 at pre-dose, at end of infusion and at 0.25, 0.5, 1, 2, 4, 6, 10, 24, 48, 72, 96, 120 and 144 h following the end of the administration. Whole blood samples were collected from the contralateral arm to the infusion. At each time point, 6 ml of whole blood was withdrawn into sodium heparinized collecting tubes (Vacutainer™) and blood and anti-coagulant were mixed, stored on ice and centrifuged within 60 min at 1500 g/3000 rev min−1 at 4°C for 10 min. Plasma was transferred into two separate polypropylene tubes and stored at −70°C until analysis. Plasma concentrations of free-base eribulin were quantified using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method [24]. The lower limit of quantitation (LLOQ) was 0.2 ng ml−1. The inter-assay accuracy ranged from −3.18 to 2.77% with an interassay precision of ≤13.0% [24]. Carry-over was defined as Ceribulin pre-dose, day 15 of >5% of Cmax, day 1.
Eribulin pharmacokinetic parameters were derived from plasma concentrations on days 1 to 7 and 15 to 21 by non-compartmental analysis using WinNonlin Professional® (version 5.1.1, Pharsight Corp, CA, USA). These included: area under the curve extrapolated to infinity (AUC(0,∞)), terminal half-life (t1/2), clearance (CL) and volume of distribution at steady state (Vss). Maximum observed plasma concentration (Cmax) and time to Cmax (tmax) were derived from the data directly. Statistical analysis was performed using SAS® (version 9.1).
Statistical analysis
Sample size calculations were made. The number of patients recruited was based on the number estimated to provide at least 90% power to detect a clinically significant decrease of 30% in AUC(0,∞) and Cmax between eribulin exposure in the presence and absence of rifampicin. The estimates of the within subject standard deviation for AUC(0,∞) and Cmax for the 1.4 mg m−2 dose ranged from 0.16 to 0.20, based on models with all data that were available at the time of study design, including subjects dosed at 1.4 mg m−2 at two time points as well as across all doses (0.25 mg m−2 to 1.4 mg m−2). Assuming a within subject standard deviation of 0.20 and a one-sided alpha level of 5%, 10 patients would provide at least 90% power to detect a 30% decrease in the AUC(0,∞) and Cmax by rifampicin.
Statistical analysis of AUC(0,∞) and Cmax used an estimation approach based on mean ratio [i.e. the ratio of the pharmacokinetic parameters for test (eribulin with rifampicin) over reference (eribulin alone) defining the magnitude of the interaction]. Natural logarithm (ln)-transformed AUC(0,∞) and Cmax were subjected to an analysis of variance (anova) and results were presented in terms of geometric means ratio with associated 90% confidence intervals (CI) and no P values. If the upper and lower bounds were within 0.7 to 1.43 for both AUC(0,∞) and Cmax, then the interaction was considered to have no effect. The estimated means were back-transformed to obtain an estimate for the ratio (eribulin + rifampicin) : eribulin. The anova model included terms for treatment and patient.
Results
Patient inclusion and demographics
A total of 14 patients were enrolled in the study. Patient demographics and characteristics are summarized in Table 1. The median age was 58.5 years (range 40.0–79.0 years) and the median weight was 70 kg (range 49–105 kg). The majority of the patients were male (57%) and Caucasian (93%). The most frequent primary tumour types were colorectal (29%) and ovarian cancer (21%).
Table 1.
Patient demographics and characteristics of the safety population
| Number (n) | 14 |
| Age (years) | |
| Median (range) | 58.5 (40.0–79.0) |
| Gender, n (%) | |
| Male | 8 (57) |
| Female | 6 (43) |
| Race, n (%) | |
| Caucasian | 13 (93) |
| Asian | 1 (7) |
| Weight (kg) | |
| Median (range) | 70 (49–105) |
| Height (cm) | |
| Median (range) | 172 (158–189) |
| Body surface area (m2) | |
| Median (range) | 1.83 (1.5–2.3) |
| Tumour type, n (%) | |
| Colorectal | 4 (29) |
| Ovary | 3 (21) |
| Lung | 2 (14) |
| Breast | 1 (7.1) |
| Other | 4 (29) |
Pharmacokinetic and statistical analysis
Eleven patients were evaluable for pharmacokinetics. Two patients discontinued before day 15 and one patient was excluded because of non-compliance with rifampicin intake. For all patients, AUC(0,∞) was evaluable for both days 1 and 15. In all pre-dose samples on day 15, the eribulin concentration was below the lower limit of quantitation. Therefore, no carry-over effect was observed.
Mean plasma eribulin concentration−time curves are presented in Figure 1. Pharmacokinetic data are summarized in Table 2 and shown in Figure 2 for AUC(0,∞) and Cmax. All calculated pharmacokinetic parameters were comparable for eribulin without and with rifampicin administration. Mean AUC(0,∞) (SD) for eribulin exposure without and with rifampicin was 757 (264) ng ml−1 h and 846 (326) ng ml−1 h, respectively (Figure 2B). Mean Cmax (SD) for eribulin exposure without and with rifampicin was 371 (164) ng ml−1 and 360 (168) ng ml−1, respectively (Figure 2D). Eribulin AUC(0,∞) and Cmax had moderate inter-individual variation, with CVs of 35% and 39%, and 44% and 47%, respectively, without and with rifampicin exposure (Figure 2A, C). Rifampicin had no effect on eribulin CL and t1/2.
Figure 1.
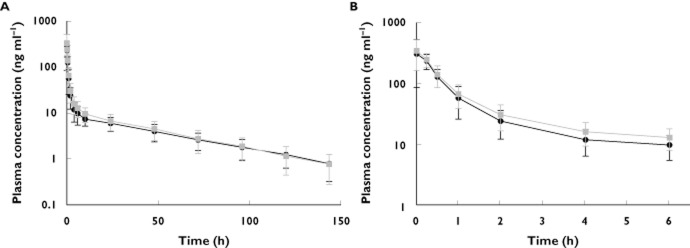
Mean (SD) plasma concentration (ng ml−1)−time (h) curve in log-linear scale following intravenous administration of eribulin mesylate alone (1.4 mg m−2; n = 11) or eribulin mesylate (1.4 mg m−2) with rifampicin (600 mg; n = 11 both groups). A) time scale up to 150 h, B) time scale up to 6 h.  , Eribulin only;
, Eribulin only;  , Eribulin plus rifampicin
, Eribulin plus rifampicin
Table 2.
Pharmacokinetic parameters for eribulin mesylate alone or co-administered with rifampicin: pharmacokinetic population
| Parameter Actual values | Eribulin mesylate | Eribulin mesylate with rifampicin |
|---|---|---|
| AUC(0,∞) (ng ml−1 h) | 757 (264) | 846 (326) |
| Cmax (ng ml−1) | 371 (164) | 360 (168) |
| tmax (h) | 0.17 (0.08–0.32) | 0.10 (0.05–0.37) |
| t1/2 (h) | 40.4 (9.67) | 36.6 (8.25) |
| CL (l h−1) | 3.41 (1.31) | 3.18 (1.38) |
| Vss (l) | 134 (42.7) | 109 (38.9) |
Eribulin mesylate dose was 1.4 mg m−2 (n = 11 both). Data are shown as mean (SD) except for tmax which are median (range) values. AUC(0,∞), area under the concentration−time curve from zero (pre-dose) extrapolated to infinity; CL, systemic clearance; Cmax, maximum observed plasma concentration; t1/2, terminal half-life; tmax, time to maximum observed plasma concentration; Vss, volume of distribution at steady state.
Figure 2.

Effect of rifampicin co-administration on AUC(0,∞) (A, B) and Cmax (C, D) of single dose intravenous administration of eribulin mesylate (1.4 mg m−2). Squares (without rifampicin) and triangles (with rifampicin) represent eribulin values for individual subjects. Left: intra-individual change in AUC(0,∞) (A) and Cmax (C; n = 11). Right: Comparative box-plots of eribulin AUC(0,∞) and Cmax (B,D; also summarized in Table 2) in the absence and presence of rifampicin. Box plot represents the 25th−75th percentiles, whiskers extend to 5th and 95th percentiles and median is indicated by a line within the box. Box-plots are flanked by scatter plots of individual data points, horizontal line represents the mean. Abbreviations: AUC(0,∞) = area under the concentration−time curve from zero (pre-dose) extrapolated to infinity; Cmax = maximum observed plasma concentration
For the primary pharmacokinetic endpoints of AUC(0,∞) and Cmax without or with co-administration of rifampicin, the geometric means, ratio and confidence intervals were calculated (Table 3). The confidence intervals of the ratios were within the pre-defined range of 0.7 to 1.43 (AUC(0,∞): geometric least square means ratio = 1.10, 90% CI 0.91, 1.34, and Cmax: geometric least square means ratio = 0.97, 90% 0.81, 1.17). Therefore, no difference was determined for AUC(0,∞) and Cmax of eribulin when administered alone or in co-administration with rifampicin.
Table 3.
Statistical analysis of the primary pharmacokinetic parameters
| Parameter | Geometric mean | Ratio of treatment means (eribulin plus rifampicin: eribulin) | 90% confidence interval | |
|---|---|---|---|---|
| Eribulin plus rifampicin | Eribulin | |||
| AUC(0,∞) (ng ml–1 h) | 789 | 716 | 1.10 | 0.91, 1.34 |
| Cmax (ng ml–1) | 333 | 342 | 0.97 | 0.81, 1.17 |
Model includes terms for treatment, and patient (n = 11 patients). AUC(0,∞), area under the concentration−time curve from zero (pre-dose) extrapolated to infinity; Cmax, maximum observed plasma concentration.
Adverse effects
All 14 patients were evaluable for the adverse event profile and tolerability of eribulin alone and when co-administered with rifampicin. Two patients discontinued during the first cycle, one due to an AE (leucocytopenia and febrile neutropenia, both grade 3) and one due to patient's choice. Ten patients (10/14, 71%) commenced the second cycle and the reason for discontinuation for these patients was progressive disease. The median number of treatment cycles was 2 (range 0–8).
All 14 patients experienced at least one AE. The most common treatment-related AEs were fatigue (9/14, 64%), alopecia (7/14, 50%), nausea (6/14, 43%) and pyrexia (5/14, 36%).
In total, 10/14 (71%) experienced a grade 3 or 4 treatment-related AE during the study. The incidence of all grade ≥3 treatment-related AEs that occurred during the study is summarized in Table 4. The most common treatment-related grade ≥3 AEs were grade 3 neutropenia (4/14, 29%), grade 3 leucopenia and fatigue (both 3/14, 21%) and grade 4 neutropenia (2/14, 14%). Other treatment-related grade ≥3 AEs included grade 4 leucopenia, grade 3 febrile neutropenia and grade 3 abdominal pain (all 1/14, 7.1%).
Table 4.
Incidence of all grade ≥3 treatment-related AEs that occurred during the study
| Term | CTCAE grade | Number of patients (%) | |
|---|---|---|---|
| Eribulin only (n = 14) | Total (n = 14) | ||
| Any TEAE | 3 | 6 (43) | 8 (57) |
| 4 | 1 (7.1) | 2 (14) | |
| Fatigue | 3 | 3 (21) | 3 (21) |
| Leucopenia | 3 | 4 (29) | 3 (21) |
| 4 | 0 | 1 (7.1) | |
| Neutropenia | 3 | 2 (14) | 4 (29) |
| 4 | 1 (7.1) | 2 (14) | |
| Febrile neutropenia | 3 | 1 (7.1) | 1 (7.1) |
| Abdominal pain | 3 | 0 | 1 (7.1) |
TEAE, treatment-emergent adverse event.
Six SAEs were reported to be related to the study drug and these included leucocytopenia, neutropenia, febrile neutropenia, pyrexia, abdominal pain and gastric haemorrhage. There were no deaths during the study or in the 30 days of follow-up after study discontinuation.
Anti-tumour activity
One patient (1/11, 9.1%) with breast cancer had a partial response and four patients had stable disease as best response (4/11, 36%). Four patients (4/11, 36%) had progressive disease and data were missing in two patients (2/11, 18%).
Discussion
In this study, the effect of rifampicin, a potent CYP3A4 inducer, was determined on the pharmacokinetics of a single intravenous dose of eribulin mesylate in patients with solid tumours. Rifampicin is a derivative of rifamycin B and is currently used as an oral antibiotic drug for treatment of tuberculosis and bacterial infections. Rifampicin is eliminated in the bile and has enterohepatic circulation [21]. Rifampicin is a potent inducer of the expression of CYP3A4 which is located in the liver and the epithelial layer of the intestine. In general, CYP3A4 induction by rifampicin can lead to clinically significant drug interactions by reducing the plasma concentrations and the effects of drugs that are metabolized by CYP3A4 [13]. Patients with advanced cancer may very well use co-medication, including a CYP3A4-inducer, during treatment. Examples of CYP3A4 substrates and inducers used in anti-cancer therapy include paclitaxel, cyclophosphamide and dexamethasone [25]. Another example of a CYP3A4 inducer is hyperforin, which is the active constituent of the herbal antidepressant St John's wort, a drug that is popular among cancer patients [26].
In pre-clinical in vitro studies [14] and in vivo studies in animals the likelihood of a clinically relevant drug−drug interaction with eribulin at the level of CYP3A4 has previously been determined, to predict whether such an interaction could occur in humans. These studies showed that CYP3A4 was the major enzyme involved in eribulin metabolism and that eribulin was excreted into the bile, faeces and urine. Zhang et al. attempted to make an in vivo prediction based upon in vitro data, and it was estimated that drug−drug interactions mediated by CYP3A4 were unlikely with eribulin (a Cmax,u/Ki, a predictive indicator for drug−drug interaction risk potential in the clinical setting [14]). This was calculated to lie in the range of 0.00211–0.0280, and was below the threshold of 0.1, indicating that drug−drug interactions mediated by CYP3A4 were unlikely with eribulin. However, these studies have not excluded the possibility of an interaction of eribulin with drugs that are metabolized by CYP3A4 in humans, especially in patients with advanced solid tumours. This could result in lower eribulin exposure, which in turn could lead to reduced anti-tumour activity. As eribulin mesylate is likely to be prescribed to patients with co-morbidity and co-medication, often as the third line of treatment or more, in this specific situation, it was considered useful to perform the present phase I study in a limited number of patients with advanced solid tumours. Therefore, rifampicin was chosen as a model CYP3A4-inducer to determine the impact on eribulin exposure and the potential for such unfavorable drug−drug interactions.
Co-administration of rifampicin had no effect on eribulin exposure as demonstrated by AUC(0,∞) and Cmax values. These results were consistent with results that became recently available from the eribulin-ketoconazole interaction study [27], in which inhibition of CYP3A4 was studied. Here, also, no effect on eribulin exposure was found for dose-normalized AUC(0,∞) and Cmax values. These results were also consistent with the results of a recent human metabolism and excretion study of radioactively-labelled eribulin [28]. The majority of eribulin was excreted unchanged in faeces (61.3%) except for a minor part in urine (8.1%). Plasma metabolite profiling showed that metabolites accounted for ≤0.6% of unchanged eribulin in plasma. These results indicate that, while CYP3A4 probably is the main enzyme for eribulin metabolism based on in vitro data, the contribution of metabolism to the elimination of eribulin is limited, and that elimination is mostly achieved by biliary excretion of unchanged drug. Therefore, the effects of CYP3A4 inducing and inhibiting drugs on eribulin exposure are not likely to have clinical significance in the treatment of cancer patients.
The study design was a non-randomized, one-treatment sequence study. In order to avoid any long term effects of enzyme induction by rifampicin, a randomized two-way crossover design was not employed, but a one-way design was chosen in which all patients first received eribulin mesylate monotherapy followed by eribulin mesylate co-administration with rifampicin. Carry-over effects of eribulin exposure from the first dose to day 15 were investigated, but were not observed, indicating that the wash-out period of 15 days was sufficient for the elimination of eribulin.
Rifampicin was administered at a dose of 600 mg daily during 12 days (days 9–20) according to FDA guidance [16]. Eribulin mesylate was dosed on the 7th day of rifampicin exposure (day 15) and rifampicin was continued for 5 additional days (up to day 20). Patient compliance was checked by a diary. Clinically relevant interactions have been reported using rifampicin 600 mg daily for at least 4 days [13]. In one study, actual hepatic CYP3A4 protein content was determined in liver biopsies and a 2.4 to 4.7 fold increase was found in patients exposed to rifampicin (600 mg daily for 4 days) compared with patients who were not exposed to rifampicin [29]. Thus, the rifampicin dosing schedule used was considered long enough and high enough to reach sufficient CYP3A4 induction in order to determine any potential clinical effect on eribulin exposure in this study.
Enzyme induction by rifampicin is mediated by the pregnane X receptor (PXR). Rifampicin binds and activates the PXR, leading to increased DNA transcription and subsequent protein synthesis of specific enzymes [30]. Next to CYP3A4, other proteins are also thought to be induced by rifampicin by modulation of the PXR receptor, such as the ATP-binding cassette drug transporter P-glycoprotein (P-gp, multidrug resistant (MDR) 1 gene, ABCB1) [31]. P-gp serves as an efflux transporter to form a permeation barrier in the gastrointestinal tract and the brain and to increase drug elimination in the liver and kidneys [32] and eribulin was shown to be a P-gp substrate [33]. Induction of P-gp by rifampicin could theoretically have resulted in increased elimination of eribulin, but such an effect was not observed in the present study.
In conclusion, analysis of AUC(0,∞) and Cmax indicated that co-administration of a potent CYP3A4 inducer, rifampicin, had no effect on single dose exposure to eribulin when administered to patients with solid tumours. These results indicate that eribulin mesylate may be safely co-administered with compounds that are CYP3A4 inducers.
Acknowledgments
This study was funded by Eisai Inc.
Competing Interests
J.W and W.C. were formerly employed by Eisai Ltd. G.E. was paid by Eisai Ltd for statistician consultancy services. K.L. is an employee of Eisai Ltd and L.R. and F.P. are employees of Eisai Inc. The institute of J.H.B. has received funds for research from Eisai. All remaining authors have declared no conflicts of interest.
References
- 1.Kohler GI, Bode-Boger SM, Busse R, Hoopmann M, Welte T, Boger RH. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38:504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- 2.Beijnen JH, Schellens JH. Drug interactions in oncology. Lancet Oncol. 2004;5:489–496. doi: 10.1016/S1470-2045(04)01528-1. [DOI] [PubMed] [Google Scholar]
- 3.Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsk BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013–1021. [PubMed] [Google Scholar]
- 4.Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4:1086–1095. doi: 10.1158/1535-7163.MCT-04-0345. [DOI] [PubMed] [Google Scholar]
- 5.Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA. Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Mol Cancer Ther. 2008;7:2003–2011. doi: 10.1158/1535-7163.MCT-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. 2010;49:1331–1337. doi: 10.1021/bi901810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goel S, Mita AC, Mita M, Rowinsky EK, Chu QS, Wong N, Desjardins C, Fang F, Jansen M, Shuster DE, Mani S, Takimoto CH. A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res. 2009;15:4207–4212. doi: 10.1158/1078-0432.CCR-08-2429. [DOI] [PubMed] [Google Scholar]
- 8.Tan AR, Rubin EH, Walton DC, Shuster DE, Wong YN, Fang F, Ashworth S, Rosen LS. Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clin Cancer Res. 2009;15:4213–4219. doi: 10.1158/1078-0432.CCR-09-0360. [DOI] [PubMed] [Google Scholar]
- 9.Twelves C, Cortes J, Vahdat LT, Wanders J, Akerele C, Kaufman PA. Phase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic breast cancer. Clin Breast Cancer. 2010;10:160–163. doi: 10.3816/CBC.2010.n.023. [DOI] [PubMed] [Google Scholar]
- 10.U.S.National Institute of Health. Available at http://www.clinicaltrials.gov (last accessed 26 April 2011)
- 11.Cortes J, O'Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Dieras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377:914–923. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- 12.Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38:41–57. doi: 10.2165/00003088-200038010-00003. [DOI] [PubMed] [Google Scholar]
- 13.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin : clinical relevance. Clin Pharmacokinet. 2003;42:819–850. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 14.Zhang ZY, King BM, Pelletier RD, Wong YN. Delineation of the interactions between the chemotherapeutic agent eribulin mesylate (E7389) and human CYP3A4. Cancer Chemother Pharmacol. 2008;62:707–716. doi: 10.1007/s00280-008-0755-1. [DOI] [PubMed] [Google Scholar]
- 15.European Medicines Agency Committee for Medicinal Products for Human Use (CHMP) Note for guidance on the investigation of drug interactions. 17-12-1997. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002966.pdf (last accessed 2 August 2012) [DOI] [PubMed]
- 16.U.S. Food and Drug Administration. Guidance for industry drug interaction studies – study design, data analysis, and implications for dosing and labeling. 2006. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM0292362.pdf (last accessed 2 August 2012)
- 17.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–655. [PubMed] [Google Scholar]
- 18.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 19. Summary of products characteristics for Halaven. 2011. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002084/WC500105112.pdf (last accessed 2 August 2012)
- 20.European Clinical Trials Database. Available at https://eudract.ema.europa.eu/ (last accessed 18 May 2012)
- 21. Summary of products characteristics for Rifadin. 2010. Available at http://www.medicines.org.uk/EMC/medicine/21223/SPC/Rifadin+300mg+Capsules/ (last accessed 2 August 2012)
- 22. Cancer Therapy Evaluation Program NCI Common Terminology Criteria AE Version 3.0 (NCI-CTCAE v3.0). 2006.
- 23.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 24.Dubbelman AC, Rosing H, Thijssen B, Lucas L, Copalu W, Wanders J, Schellens JH, Beijnen JH. Validation of high-performance liquid chromatography-tandem mass spectrometry assays for the quantification of eribulin (E7389) in various biological matrices. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:1149–1155. doi: 10.1016/j.jchromb.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 25.Harmsen S, Meijerman I, Beijnen JH, Schellens JH. The role of nuclear receptors in pharmacokinetic drug-drug interactions in oncology. Cancer Treat Rev. 2007;33:369–380. doi: 10.1016/j.ctrv.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Meijerman I, Beijnen JH, Schellens JH. Herb-drug interactions in oncology: focus on mechanisms of induction. Oncologist. 2006;11:742–752. doi: 10.1634/theoncologist.11-7-742. [DOI] [PubMed] [Google Scholar]
- 27.Devriese LA, Mergui-Roelvink M, Wanders J, Jenner A, Edwards G, Reyderman L, Copalu W, Peng F, Marchetti S, Beijnen JH, Schellens JHM. Eribulin mesylate pharmacokinetics in patients with solid tumors receiving repeated oral ketoconazole. Invest New Drugs. 2012 doi: 10.1007/s10637-012-9829-3. Epub May 5; doi: s10637-012-9829-3. [DOI] [PubMed] [Google Scholar]
- 28.Dubbelman AC, Rosing H, Jansen RS, Mergui-Roelvink M, Huitema AD, Koetz B, Lymboura M, Reyderman L, Lopez-Anaya A, Schellens JH, Beijnen JH. Mass balance study of 14C-eribulin in patients with advanced solid tumours. Drug Metab Dispos. 2012;40:313–321. doi: 10.1124/dmd.111.042762. [DOI] [PubMed] [Google Scholar]
- 29.Combalbert J, Fabre I, Fabre G, Dalet I, Derancourt J, Cano JP, Maurel P. Metabolism of cyclosporin A. IV. Purification and identification of the rifampicin-inducible human liver cytochrome P-450 (cyclosporin A oxidase) as a product of P450IIIA gene subfamily. Drug Metab Dispos. 1989;17:197–207. [PubMed] [Google Scholar]
- 30.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- 32.Lee CA, Cook JA, Reyner EL, Smith DA. P-glycoprotein related drug interactions: clinical importance and a consideration of disease states. Expert Opin Drug Metab Toxicol. 2010;6:603–619. doi: 10.1517/17425251003610640. [DOI] [PubMed] [Google Scholar]
- 33.Taur JS, Desjardins CS, Schuck EL, Wong YN. Interactions between the chemotherapeutic agent eribulin mesylate (E7389) and P-glycoprotein in CF-1 abcb1a-deficient mice and Caco-2 cells. Xenobiotica. 2010;41:320–326. doi: 10.3109/00498254.2010.542256. [DOI] [PubMed] [Google Scholar]