

Abstract
The enzymatic activity of peptidases must be tightly regulated to prevent uncontrolled hydrolysis of peptide bonds, which could have devastating effects on biological systems. Peptidases are often generated as inactive propeptidases, secreted with endogenous inhibitors, or they are compartmentalized. Propeptidases become active after proteolytic removal of N-terminal activation peptides by other peptidases. Some peptidases only become active towards substrates only at certain pHs, thus confining activity to specific compartments or conditions. This review discusses the different roles proteolysis plays in regulating GPCRs. At the cell-surface, certain GPCRs are regulated by the hydrolytic inactivation of bioactive peptides by membrane-anchored peptidases, which prevent signalling. Conversely, cell-surface peptidases can also generate bioactive peptides, which directly activate GPCRs. Alternatively, cell-surface peptidases activated by GPCRs, can generate bioactive peptides to cause transactivation of receptor tyrosine kinases, thereby promoting signalling. Certain peptidases can signal directly to cells, by cleaving GPCR to initiate intracellular signalling cascades. Intracellular peptidases also regulate GPCRs; lysosomal peptidases destroy GPCRs in lysosomes to permanently terminate signalling and mediate down-regulation; endosomal peptidases cleave internalized peptide agonists to regulate GPCR recycling, resensitization and signalling; and soluble intracellular peptidases also participate in GPCR function by regulating the ubiquitination state of GPCRs, thereby altering GPCR signalling and fate. Although the use of peptidase inhibitors has already brought success in the treatment of diseases such as hypertension, the discovery of new regulatory mechanisms involving proteolysis that control GPCRs may provide additional targets to modulate dysregulated GPCR signalling in disease.
Keywords: peptidase, neuropeptide, GPCR, trafficking, signalling, proteolysis, endosome, cell-surface, ubiquitin, β-arrestins
Nomenclature
For receptors, the British Journal of Pharmacology's Guide to Receptors and Channels was used (Alexander et al., 2011). For peptidases, the MEROPS database was used (Rawlings et al., 2012).
The traditional notion of proteolysis
Proteolysis is the hydrolytic breakdown of a peptide bond, C(O)-NH found between amino acids in peptides, polypeptides and protein structures. Peptide bonds can spontaneously break in the presence of water, but do so, at an extremely slow rate. Therefore, in biological systems, enzymes are required to facilitate the breakage of these bonds. These enzymes or more specifically, peptidases (also known as proteases or proteinases) are themselves made of amino acids and are currently classified into six groups according to the critical residue in their catalytic site: (i) serine peptidases; (ii) cysteine peptidases; (iii) aspartic peptidases; (iv) threonine peptidases; (v) metallopeptidases; and (vi) glutamic peptidases (Rawlings et al., 2012). Traditionally, peptidases are mainly thought of as enzymes of digestion, breaking down food in the stomach and in the intestine. However, it is now clear that peptidases can contribute to the regulation of cell function by controlling levels of bioactive peptides and by cleaving cell-surface receptors and ion channels to regulate signalling pathways.
Cell-surface peptidases regulate the availability of GPCR ligands
The role of cell-surface peptides in the regulation of GPCR signalling is well documented. These peptidases regulate levels of circulating bioactive peptides, which function to initiate GPCR-mediated signalling. ACE compound peptidase (EC 3.4.15.1) is a zinc-dependent metallopeptidase that converts the inactive angiotensin I to angiotensin II by releasing the C-terminal dipeptide, His9-Leu10 (Figure 1A). Angiotensin II is a vasoconstrictor and exerts its effect through two types of angiotensin II receptors, AT1 and AT2. The AT1 receptor mediates most of the physiological and pathophysiological actions of angiotensin II and is the predominant receptor subtype expressed in the cardiovascular system. Interaction of the AT1 receptor with angiotensin II activates Gq/11, Gi, G12 and G13 proteins, leading to the mobilization of intracellular calcium, generation of reactive oxygen species and activation of numerous PKs and mitogenic signalling pathways [reviewed in Mogi et al. (2009)]. AT2 receptors couple to G proteins to activate PLC, promoting the mobilization of intracellular calcium and activation of PKC [reviewed in Porrello et al. (2009)]. Angiotensin II causes a plethora of effects, including tissue remodelling, leukocyte infiltration, inflammation, atherosclerosis, endothelial dysfunction, myocardial infarction, stroke, and heart and renal failure [reviewed in Cheng et al. (2005)]. Similarly, endothelin-converting enzyme-1 (ECE-1, EC 3.4.24.71) is responsible for the production of the vasoconstrictor endothelin-1 from big endothelin. Endothelin-1 can activate both endothelin A (ETA) and ETB receptors to elicit a broad range of signalling responses [reviewed in Khimji and Rockey (2010)]. Activation of ETA receptors by endothelin-1 promotes mobilization of intracellular calcium, indicating that ETA receptors are coupled to Gq/11 proteins. ETA receptors primarily mediate the vasoactive and proliferative effects of endothelin-1. ETB receptors have been proposed to act as endothelin-1 scavengers and thus, reduce circulating endothelin-1 levels. Thus, proteolysis acts to promote activation of GPCRs. In contrast, proteolysis can also act to prevent activation of GPCRs. For example, neprilysin (NEP, neutral endopeptidase 24.11, EC 3.4.24.11) cleaves and inactivates both bradykinin (Gly4↓Phe5 and Pro7↓Phe8) and substance P (SP, Gln6↓Phe↓Phe-Gly↓Leu10) (Matsas et al., 1984), thus preventing activation of their respective GPCRs, the bradykinin 2 receptor (B2 receptor) and the neurokinin 1 receptor (NK1 receptor) (Figure 1A). Peptidases can therefore play a major role in the production of vasoactive peptides and, therefore, regulate vascular functions and dysregulation can lead to vascular diseases. A great deal of time and effort has been spent on the development of peptidase inhibitors for the treatment of hypertension. It is over 30 years since the first ACE inhibitor, captopril, was designed (Ondetti et al., 1977). Now, however, other ACE inhibitors with improved pharmacokinetics and pharmacodynamics have since been developed and include enalaprilat (MK-421) (Gross et al., 1981) and imidaprilat (Ikeo et al., 1992). ACE inhibitors are effective treatments for hypertension and congestive heart failure (CONSENSUS, 1987; SOLVD, 1991) and are also beneficial for patients with atherosclerosis (Yusuf et al., 2000). With the success of ACE inhibitors for the treatment of hypertension, it was thought that the development of compounds able to inhibit multiple peptidases, thereby potentiating the effects of dilator peptides such as bradykinin, while reducing the availability of constrictors such as angiotensin II, may offer an even better way of treating diseases such as hypertension. Indeed, dual or triple vasoactive peptidase inhibitors have been developed. These compounds inhibit the proteolytic activities of ACE, ECE-1 and NEP in various combinations. The first described dual inhibitors were alatriopril and glycoprilat. Both compounds inhibit the activities of both ACE and NEP (Gros et al., 1991). Alatriopril was shown to be more effective than captopril alone in reducing cardiac hypertrophy in rats with myocardial infarction (Bralet et al., 1994). Later, omapatrilat (BMS-186716) was developed and was shown to be more effective in reducing blood pressure in humans than either placebo or ACE inhibitors (Neal et al., 2002; Regamey et al., 2002). However, omapatrilat failed in phase III clinical trials and was discontinued due to an increased incidence of angioedema as an unwanted side effect. CGS 35601, a triple vasopeptidase inhibitor, prevents the activities of ACE, NEP and ECE-1 (Trapani et al., 2004) and significantly reduced both systolic and diastolic blood pressure in a number of preclinical rodent models of hypertension (Daull et al., 2005; 2006a). Further, a preclinical safety profile assessment of CGS 35601 showed it to have no effect on either hepatic or liver toxicities (Daull et al., 2006b). Although these triple inhibitors may represent the future of peptidase inhibitors for the treatment of disease (Table 1), no clinical trials using triple peptidase inhibitors have yet been conducted. So, although these dual and triple peptidase inhibitor compounds are promising in humans and animal models of hypertension, none have yet been approved for the treatment of human disease. Thus, ACE inhibitors remain the compounds of choice for the treatment of hypertension, often in combination with angiotensin or β-adrenoceptor antagonists or diuretics. NEP also plays a major role in the catabolism of endogenous opioid peptides such as the enkephalins and dynorphins. Thus, together with aminopeptidase N (EC 3.4.11.2), NEP represents a major target for the development of drugs for the treatment of acute and chronic pain [reviewed in Thanawala et al. (2008)].
Figure 1.
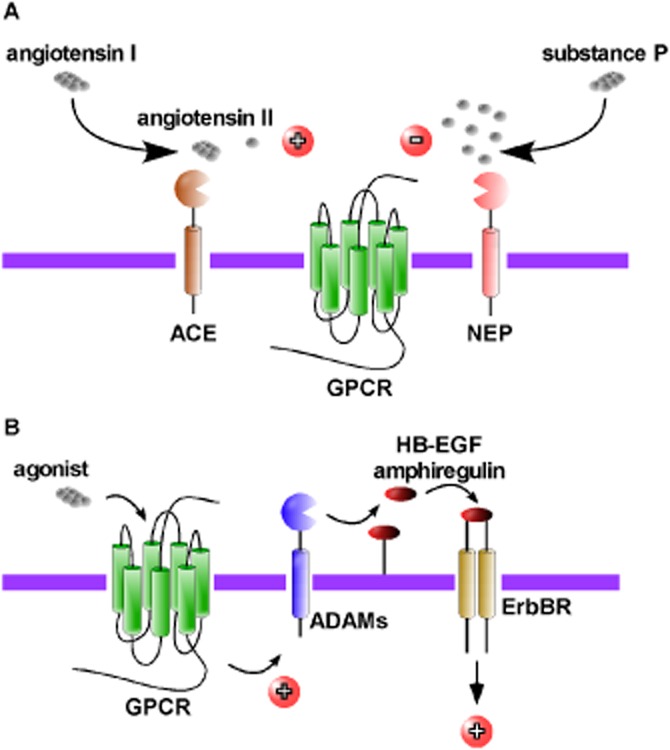
Cell-surface peptidases regulate GPCR-mediated signalling. (A) ACE compound peptidase cleaves angiotensin I to generate angiotensin II to promote activation of angiotensin II receptors. Conversely, NEP hydrolyses SP to prevent activation of SP of neurokinin 1 receptors. (B) Activation of certain GPCRs promotes the ADAMs that generate EGF-like ligands (e.g. heparin-binding EGF-like factor, amphiregulin). In turn, these ligands transactivate ErbB receptors to activate intracellular signalling pathways.
Table 1.
Future and current uses for peptidase inhibitors in the treatment of disease
| Peptidase | Drug perspectives | Targeted GPCRs | References/reviews |
|---|---|---|---|
| Cell-surface | |||
| ACE, ECE-1, NEPa | Hypertension | AT1, AT2, ETA, ETB, B2 | Dive et al. (2009) |
| NEP, aminopeptidase N | Pain | Opioid receptors | Thanawala et al. (2008) |
| ADAMs | Cancer progression | GPCRs causing ErbB receptor transactivation | Paolillo and Schinelli (2008) |
| Extracellular | |||
| Thrombin, Coagulation Factor Xa | Anticoagulants, thrombosis | PAR1, PAR3, PAR4 | Showkathali and Natarajan (2012) |
| Trypsin, Tryptase | Inflammatory bowel diseases | PAR2 | Rothmeier and Ruf (2012; Yoshida and Yoshikawa (2008) |
| Lysosomal | |||
| Cathepsins B, C, D, E, G and L, Carboxypeptidase A | No known functional consequences of preventing GPCR degradation | –b | N/A |
| Endosomal | |||
| ECE-1 | Modulation of GPCR trafficking and signalling | NK1, CLR•RAMP1, sst2, NT1, CRF1 | Murphy et al. (2009; 2011) |
| USPs | Modulation of GPCR trafficking and signalling | GPCR function affected by de-ubiquitination of β-arrestins | Murphy et al. (2009; Shenoy et al. (2009) |
Dual and triple ACE, ECE-1 and NEP inhibitors are in development.
Too numerous to list in table. N/A, not applicable.
GPCRs promote proteolysis to transactivate epidermal growth factor receptors
Certain GPCRs can activate intracellular signalling pathways via transactivation of cell-surface receptor tyrosine kinases such as PDGF (Linseman et al., 1995), insulin-like growth factor-1 receptor (Rao et al., 1995), ErbB receptor (Daub et al., 1996) and Trk neurotrophin receptors (Lee and Chao, 2001). Interestingly, only transactivation of ErbB receptors requires the activity of peptidases (Figure 1B). The GPCR agonists, endothelin-1, lysophosphatic acid and thrombin, were shown to induce phosphorylation of the ErbB receptor in Rat-1 cells (Daub et al., 1996). Specific inhibition of ErbB receptors using the selective tyrphostin AG1478 or expression of a dominant-negative ErbB receptor mutant that is unable to signal, suppressed the GPCR-induced activation of ERK1/2 (Daub et al., 1996). Later studies revealed that transactivation of ErbB receptors, involves the processing of membrane-anchored heparin-binding EGF-like growth factor by a metallopeptidase activity that is rapidly induced following GPCR activation. Inhibition of the peptidase activity using the metalloproteinase inhibitor batimastat prevented ErbB receptor transactivation and downstream signals (Prenzel et al., 1999). Additional work, indicated that this GPCR-mediated transactivation not only lead to the initiation of signalling cascades, but also ErbB receptor dimerization, tyrosine autophosphorylation and internalization (Maudsley et al., 2000). The identity of the metallopeptidase responsible for the processing of the membrane-anchored ligand remained elusive until a study conducted in cardiomyocytes showed that the metallopeptidase inhibitor, KB-R7785 and a dominant-negative of a disintigrin and metallopeptidase (ADAM) 12 peptidase attenuated GPCR-induced signalling (Asakura et al., 2002). The same study also showed that KB-R7785 inhibited the shedding of heparin-binding EGF-like growth factor and attenuated thoracic aortic constriction-induced thickening of the heart muscle in intact mice. A subsequent study suggested that ADAM10 peptidase (EC 3.4.24.81) is also implicated in bombesin-mediated ErbB receptor transactivation (Yan et al., 2002). As more than one peptidase is involved in ErbB receptor transactivation, it came as no surprise that additional ligands were also involved. In squamous cell carcinoma cells, the GPCR agonists, lysophosphatic acid and carbachol, which activate lysophospholipid and muscarinic receptors, respectively, specifically activate metallopeptidase-dependent release of amphiregulin by another ADAM peptidase, ADAM17 peptidase (TNF-α-converting enzyme, TACE; EC 3.4.24.86) to regulate proliferation and motility (Gschwind et al., 2003). However, in TccSup cancer cells, lysophosphatic acid-induced transactivation is mediated by ADAM15 peptidase and promotes cell survival (Schafer et al., 2004).
Following on from the fact that GPCRs can promote the activity of many different metallopeptidases to cause ErbB receptor transactivation, evidence exists for the involvement of numerous GPCR-induced pathways in the generation of EGF-like ligands [reviewed in Ohtsu et al. (2006)]. The β2-adrenoceptor agonist, isoprenaline causes ErbB receptor transactivation via a mechanism that requires Gβγ subunits and c-Src activity (Pierce et al., 2001). Similarly, glucagon-like peptide-1-induced transactivation also requires c-Src activity (Buteau et al., 2003). The involvement of G proteins has also been observed in other studies. Lysophosphatic acid-mediated transactivation by ADAM17 was partially blocked by pertussis toxin in squamous cell carcinoma-9 cells (Gschwind et al., 2003) and in MDA-MB-231 cells, lysophosphatic acid- and sphingosine-1 phosphate-dependent ErbB receptor transactivation involving ADAM15, was also prevented by pertussis toxin (Hart et al., 2005). A requirement for the activity of phospholipase C has also been observed for angiotensin II-induced, ADAM17-dependent ErbB receptor transactivation by the AT1 receptor (Mifune et al., 2005). The elevation of intracellular calcium levels by bradykinin (Zwick et al., 1999) and angiotensin II (Eguchi et al., 2003) has also been implicated in the transactivation of ErbB receptors. It is assumed that many of these GPCR-induced pathways lead to the phosphorylation of the metallopeptidases, enhancing the proteolytic activity of the peptidase. Although it is well established that numerous PKs can phosphorylate metallopeptidases to promote proteolytic activity [reviewed in Huovila et al. (2005)], none of the present studies have presented evidence of a direct GPCR-mediated effect on metallopeptidases.
The generation of reactive oxygen species by GPCRs has also been proposed to play an important role in promoting metallopeptidase activity and includes ErbB receptor transactivation mediated by 5-HT2B receptors and α1D-adrenoceptors (Pietri et al., 2005), AT1 receptor (Eguchi and Inagami, 2000) and purine P2Y receptors (Myers et al., 2009). The reactive oxygen species are thought to induce oxidation of a cysteine residue that lies within an inhibitory motif, thereby activating the peptidase (Zhang et al., 2001). A more recent development in elucidating the mechanism by which metallopeptidases become active following GPCR activation to promote ErbB receptor activation is the involvement of integrins (Gooz et al., 2012). 5-HT induces kidney mesangial cell proliferation through ADAM17 activation and ErbB receptor transactivation. In unstimulated cells, ADAM17 binds to α5β1 integrin, an interaction that prevents the proteolytic activity of ADAM17. However, following application of 5-HT, this interaction is disrupted, presumably promoting peptidase activity (Gooz et al., 2012).
A further consequence of the processing of EGF-like ligands from cell-surface precursors, is the generation of C-terminal fragments. It is now established that these C-terminal fragments also have a biological role [reviewed in Tanida et al. (2010)]. IL-8 induces cell proliferation and migration of the colon cancer cell lines, HT-29 and Caco2 by an ADAM-dependent intranuclear translocation pathway of HB-EGF-C-terminal fragment (Itoh et al., 2005). Thus, understanding how GPCRs activate cell-surface peptidases to generate the multiple ligands responsible for receptor transactivation is essential to develop new pharmacological interventions. The importance of metallopeptidases in generating EGF-like ligands that regulate cell proliferation and differentiation, has lead these peptidases to be considered as useful molecular targets in the treatment of cancer. Specific peptidase inhibitors may also be used to modulate GPCR-induced signalling and this may be an effective treatment for GPCR-driven diseases.
Peptidases can act as positive or negative regulators of GPCR signalling
It was long recognized that peptidases such as thrombin (EC 3.4.21.5) and trypsin could signal directly to cells (Burger, 1970; Sefton and Rubin, 1970; Chen and Buchanan, 1975; Carney and Cunningham, 1977, 1978). However, the mechanism through which these peptidases could activate intracellular signalling cascades remained unknown until the cloning of the thrombin receptor, now called proteinase-activated receptor 1 (PAR1) (Vu et al., 1991). PAR1 is a GPCR and thrombin activates PAR1 by direct cleavage of the receptor in a two-step process. Firstly, thrombin binds to PAR1 either side of the proteolytic cleavage site. One of these sites (D51KYEPF56) is similar to that of hirudin, an anticoagulant found in the saliva of leech (Vu et al., 1991). This hirudin-like binding domain increases the affinity of thrombin for PAR1. Following binding, thrombin cleaves between Arg41 and Ser42 to expose the new N-terminus starting with S42FLLRN47 (Vu et al., 1991). This tethered ligand domain then interacts with residues on the second extracellular loop of the receptor and presumably induces a conformational change, which activates the receptor. PAR1 activation by thrombin is the most potent known trigger for platelet aggregation (Sambrano et al., 2001) and as such, thrombin signalling and PAR1 are key targets for the prevention of thrombosis.
PAR2 is the second member of this receptor family and is activated by trypsin (Figure 2 A) (Nystedt et al., 1994; 1995a,b; Bohm et al., 1996). Cleavage of PAR2 occurs between Arg36 and Ser37 to reveal the tethered ligand and new amino terminus of S37LIGKV42. The use of PAR2-specific agonistic and antagonistic peptides and studies in PAR2-deficient mice, have helped to identify critical roles for PAR2 in inflammation, development, angiogenesis and immune responses [reviewed in Rothmeier and Ruf (2012)]. Subsequently, two other members of this subfamily of GPCRs have been cloned, PAR3 (Ishihara et al., 1997; Scase et al., 1997) and PAR4 (Kahn et al., 1998; Xu et al., 1998), both of which are activated by thrombin. PAR4 can also be activated by trypsin (Xu et al., 1998). Due to their sensitivity to thrombin, both PAR3 and PAR4 are important in platelet function (Ishihara et al., 1998; Kahn et al., 1998).
Figure 2.
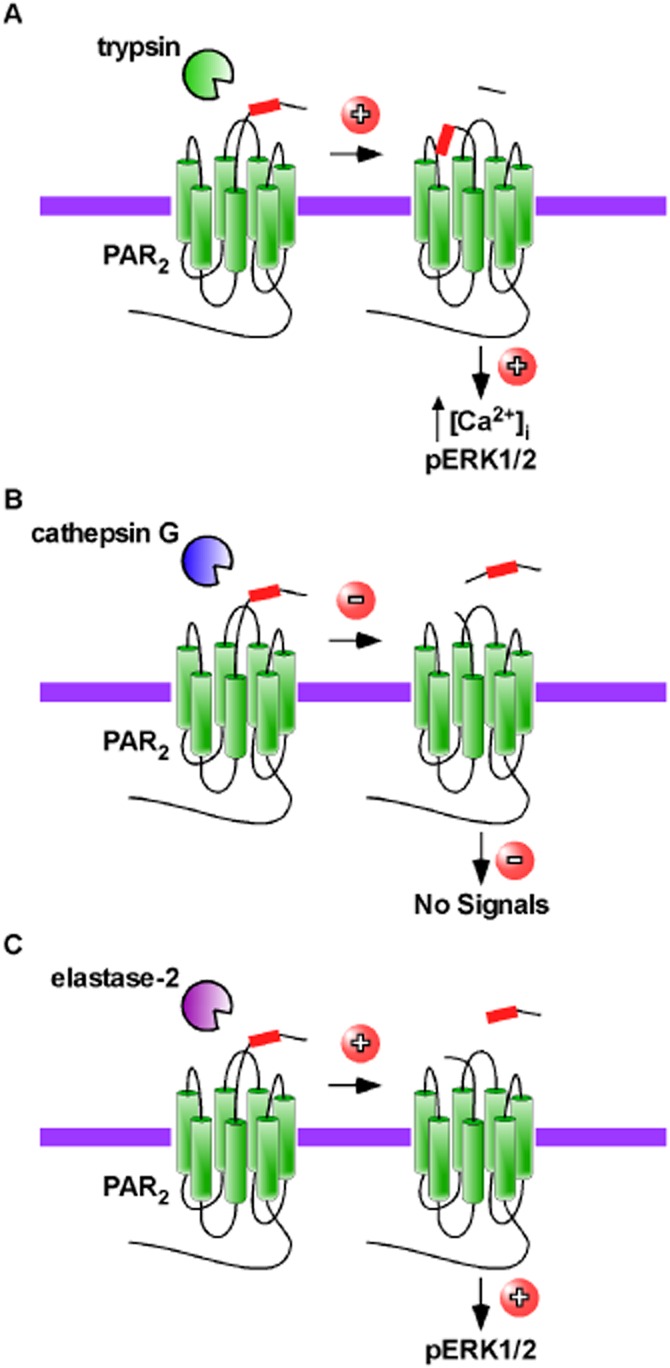
Peptidases act as biased agonists at GPCRs. (A) Trypsin cleaves PAR2 to create a new N-terminus that activates PAR2 mobilizing intracellular calcium and promoting phosphorylation (p) of ERK1/2. (B) Cathepsin G cleaves PAR2, but does not elicit any known signalling and prevents activation by other peptidases such as trypsin. (C) Other peptidases such as elastase-2, cleave PAR2 at a site distinct from trypsin. The action of this peptidase does not mobilize intracellular calcium, but does activate ERK1/2.
PARs are promiscuous receptors and may be activated by multiple peptidases. For example, PAR1 can also be activated by coagulation factor Xa (EC 3.4.21.6) (Camerer et al., 2000), activated protein C (EC 3.4.21.69) (Riewald et al., 2002), matrix metallopeptidase-1 (EC 3.4.24.7) (Boire et al., 2005) and plasmin (EC 3.4.21.7) (Mannaioni et al., 2008). Similarly, PAR2 is also cleaved by multiple peptidases, including mast cell tryptases (Molino et al., 1997), coagulation factor VIIa (EC 3.4.21.21) in complex with tissue factor and coagulation factor Xa (Camerer et al., 2000), matriptase-1 (membrane-type serine peptidase 1) (Takeuchi et al., 2000) and trypsin IV (Cottrell et al., 2004). PAR4 is cleaved and activated by trypsin (Xu et al., 1998), cathepsin G (Sambrano et al., 2000), trypsin IV (Cottrell et al., 2004) and plasmin (Quinton et al., 2004). Peptidases that cleave PARs to disarm the receptor and prevent subsequent activation by removing the tethered ligand have also been identified. For example, cathepsin G (EC 3.4.21.20) (Dulon et al., 2003), pseudolysin (EC 3.4.24.26) (Dulon et al., 2005) and myeloblastin (neutrophil leukocyte proteinase 3, EC 3.4.21.76) (Ramachandran et al., 2011) cleave PAR2 downstream of the tryptic cleavage site inactivating the receptor (Figure 2B). Cathepsin G and elastase-2 (neutrophil elastase, EC 3.4.21.37) abolish signalling by thrombin in PAR3-transfected cells, and thus disarm PAR3 (Cumashi et al., 2001).
The concept of biased agonism at GPCRs is not a new one [reviewed in Urban et al. (2007)]. Given that PARs are activated by many different peptidases, it is not surprising that the generation of different N-termini can result in peptidase-specific signalling. It is these unique N-termini that act as intramolecular ligands causing specific GPCR conformations to elicit biased signalling responses. The earliest indications that PARs could be selectively activated came from observations comparing activation by synthetic ligands to peptidases. For PAR1, mutations in the extracellular regions resulted in differential signalling between synthetic peptide agonists and thrombin (Blackhart et al., 2000). For example, a PAR1 deletion mutant lacking amino acids 68–93 of the N-terminus, failed to mobilize intracellular calcium in response to the synthetic peptide, but retained the ability to respond to thrombin (Blackhart et al., 2000). A subsequent study using human endothelial cells showed that thrombin favours G12/13 signalling and induction of endothelial barrier permeability rather than intracellular calcium mobilization, whereas synthetic peptides preferentially caused intracellular calcium mobilization by triggering Gq signalling (McLaughlin et al., 2005). Studies have also shown that activation of PAR1 by different peptidases can have different functional consequences for endothelial barrier permeability (Feistritzer and Riewald, 2005; Finigan et al., 2005). Whereas thrombin-dependent cleavage of PAR1 disrupts endothelial barrier integrity, activated protein C-dependent activation of PAR1 enhances endothelial barrier function (Feistritzer and Riewald, 2005; Finigan et al., 2005). Challenging human lung epithelial cells with elastase-2 leads to PAR1-mediated apoptosis, similar to that observed with thrombin and a synthetic-activating peptide, but modified kinetics (Suzuki et al., 2005; 2009). Matrix metallopeptidase-1 cleaves PAR1 at a distinct site to thrombin and activates Rho-GTP and mitogenic pathways in a biased mechanism, to promote cell shape change and motility (Trivedi et al., 2009).
Agonist-biased signalling has also been observed for PAR2. Certain synthetic peptide agonists fail to cause PAR2-mediated calcium signalling, while still activating ERK1/2, whereas SLIGRL-NH2, which mimics the naturally unmasked tethered ligand, triggers both calcium and MAP kinase signalling (Ramachandran et al., 2009). Further evidence came from the observation that a novel peptidomimetic PAR2 antagonist, K-14585 activated ERK1/2 signalling, but failed to elicit and calcium responses (Goh et al., 2009). It was initially thought that elastase-2 also disarmed PAR2 (Dulon et al., 2003). However, recent evidence suggests that elastase-2 is in fact a biased agonist of PAR2 (Ramachandran et al., 2011) (Figure 2C). Although elastase-2 does not cause internalization of PAR2 or mobilization of intracellular calcium, it does initiate activation of the ERK1/2 pathway (Ramachandran et al., 2011). The exact mechanism by which peptidases can act as biased agonists has not yet been fully established. However, in the case of PAR2 it is known that trypsin and elastase-2 cleave PAR2 at different positions within the N-terminus. Therefore, it is highly probable that the different cleavages result in different conformations of PAR2, which leads to biased activation of signalling pathways. With regard to PAR1, although the peptidases may cleave at the same sites, the fact that thrombin actually binds to PAR1 may lead to slight conformation changes, which also lead to biased activation of signalling cascades. However, until the crystal structures of the activated GPCRs are solved, this is pure conjecture. It is not yet known if biased agonism of PAR3 and PAR4 occurs. However, with numerous peptidases able to cleave members of this subfamily of GPCRs and perhaps each with different functional consequences, there is still much to discover about how peptidases signal through GPCRs.
Lysosomal peptidases degrade GPCRs to irrevocably terminate GPCR signalling
Once activation of a GPCR has occurred, many GPCRs are removed from the cell-surface to prevent uncontrolled signalling. The receptors are then either recycled back to the cell-surface mediating resensitization to allow cells to respond to the same stimulus again, or they are trafficked to intracellular compartments, such as lysosomes for degradation, which results in permanent signal arrest. The peptidases present in lysosomes mainly belong to the aspartic, cysteine and serine peptidase classes, with few metallo- and threonine peptidases residing in lysosomes. They are also mainly endopeptidases, cleaving within polypeptide chains and not at the end or beginning of chains. Examples of peptidases localized to lysosomes include the serine peptidases, serine carboxypeptidase A (EC 3.4.16.5) and cathepsin G, aspartic peptidases, cathepsin D (EC 3.4.23.5) and E (EC 3.4.23.24) and cysteine peptidases, cathepsin B (EC 3.4.22.1) and L (EC 3.4.22.15). One mechanism by which many GPCRs are targeted for degradation is by the post-translational modification of ubiquitination, although this is not an absolute requirement for all GPCRs. Once activated, ubiquitin moieties are added to intracellular facing lysine residues by a process requiring three enzymes, an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2) and an ubiquitin ligase (E3). Before the GPCR is degraded by peptidases present in the lysosomes, the ubiquitin molecules are removed by metallo- or cysteine peptidases, referred to as de-ubiquitinating enzymes or ubiquitin-specific peptidases (USPs). In fact it is an absolute requirement that the ubiquitin molecules are removed before the GPCR can enter the lysosome. The first identified GPCR to be regulated by ubiquitination was the yeast GPCR, Ste2p. Ubiquitination of Ste2p is a requirement for internalization (Hicke and Riezman, 1996). Prolonged exposure of β2-adrenoceptors to agonists promotes sorting of receptors to lysosomes and degradation by lysosomal peptidases (Moore et al., 1999). A combination of the aspartic peptidase inhibitor, pepstatin A and the serine peptidase inhibitor, leupeptin prevented degradation (Moore et al., 1999). The β2-adrenoceptor was the first identified mammalian GPCR shown to be directed to lysosomes for degradation by ubiquitination (Shenoy et al., 2001), an observation closely followed by that for the C-X-C chemokine receptor type 4 (CXCR4) (Marchese and Benovic, 2001). PARs are sometimes termed one-shot receptors, because of the proteolytic nature of their activation. PAR2 is also targeted for degradation by lysosomal peptidases by ubiquitination (Jacob et al., 2005). N-CBZ-L-phenylalanyl-L-alanine-diazomethylketone, an inhibitor of lysosomal cysteine peptidases, cathepsins B and L prevents degradation of PAR2. However, there are GPCRs that are trafficked to lysosomes without the need for modification by ubiquitin moieties. Although, agonists induce ubiquitination of the δ-opioid receptor, this ubiquitination is not an absolute requirement for lysosomal targeting, as a lysine-less mutant of the δ-opioid receptor is still efficiently trafficked to lysosomes and degraded by lysosomal serine peptidases (Tsao and von Zastrow, 2000; Tanowitz and Von Zastrow, 2002). However, further studies have indicated that ubiquitination does have a subtle role in the regulation of δ-opioid receptors. Ubiquitination, although not affecting trafficking to lysosomes does alter the rate at which the δ-opioid receptor is degraded in lysosomes (Hislop et al., 2009). A similar role for ubiquitination is observed for the μ-opioid receptor. It is sequences in the C-terminal tail of the μ-opioid receptor that direct its trafficking to lysosomes, whereas ubiquitination of lysine residues in the first intracellular loop promote transfer of internalized μ-opioid receptors from the limiting endosome membrane to lumen thereby facilitating degradation (Hislop et al., 2011). The receptor for calcitonin gene-related peptide (CGRP) is an unusual heterodimeric GPCR, comprising the GPCR, calcitonin receptor-like receptor (CLR) and a single transmembrane protein, receptor activity-modifying protein 1 (RAMP1) (McLatchie et al., 1998). Following sustained activation with CGRP, CLR•RAMP1 traffics to lysosomes and is degraded by lysosomal peptidases (Kuwasako et al., 2000; Cottrell et al., 2007). The identity of the lysosomal peptidases degrading CLR and RAMP1 is less clear, as an inhibitor cocktail blocking serine, aspartic and cysteine peptidases was used. However, CGRP does not induce ubiquitination of CLR or RAMP1 (Cottrell et al., 2007). Although there are known roles for lysosomal peptidases in the immune system, in generating or destroying antigenic peptides and in trafficking of growth factor receptors [reviewed in Muller et al. (2012)], the functional consequences of preventing the degradation of GPCRs in lysosomes remain to be determined.
Proteolytic removal of ubiquitin regulates GPCR-dependent trafficking and signalling
The identity of many of the E3 ligases responsible for facilitating the addition of ubiquitin moieties to GPCRs are now known and are summarized elsewhere (Hislop and von Zastrow, 2011). However, unlike the E3 ligases, fewer USPs responsible for the de-ubiquitination of GPCRs have been identified. USPs play an important role in the regulation of GPCRs by ubiquitination, opposing the action of E3 ligases by removing ubiquitin molecules. This proteolysis is not only important for regulating GPCR signalling, but is also critical for maintaining the cellular pools of ubiquitin, which is critical for many other processes including regulation of transcription, cell-cycle control, DNA damage responses, apoptosis and the immune response [reviewed in Malynn and Ma (2010); Ramaekers and Wouters (2011); Vucic et al. (2011); Hammond-Martel et al. (2012); Starostina and Kipreos (2012)]. Studies have shown that under basal conditions PAR1 is ubiquitinated at the cell-surface and that this ubiquitination promotes cell-surface retention (Wolfe et al., 2007). Following proteolytic activation by thrombin, PAR1 is de-ubiquitinated by unidentified USPs, a process that facilitates internalization to endosomes (Wolfe et al., 2007). CXCR7, a recycling GPCR is also ubiquitinated under basal conditions. After activation induced by the stromal-derived factor CXCL12, CXCR7 is reversibly de-ubiquitinated by an unidentified USP, before trafficking back to the cell-surface (Canals et al., 2012). Unusually, κ-opioid receptors are polyubiquitinated following agonist stimulation. These polyubiquitin chains are subsequently removed by CylD protein, a USP that specifically cleaves Lys-63 ubiquitin chains. After removal of the ubiquitin chains, κ-opioid receptors are degraded in lysosomes (Li et al., 2008). However, other than down-regulation of the κ-opioid receptor, the functional consequences of this polyubiquitination have yet to be elucidated. β2-Adrenoceptors are regulated by USP33 and USP20, which act in a coordinated fashion to regulate the β2-adrenoceptor recycling and resensitization (Berthouze et al., 2009). Similarly, ubiquitinated adenosine A2A receptors are de-ubiquitinated by USP4, promoting trafficking back to the cell-surface (Milojevic et al., 2006). In contrast, the proteolytic activities of associated molecule with the SH3 domain of signal-transducing adaptor molecule de-ubiquitinating peptidase (AMSH) and USP8 are required to promote entry of PAR2 to lysosomes (Hasdemir et al., 2009). AMSH and USP8 also regulate the proteolytic down-regulation of the δ-opioid receptor, but in contrast to PAR2, their activities do not affect the trafficking δ-opioid receptors to lysosomes (Hislop et al., 2009). Expression of dominant negative mutants of AMSH and USP8 caused accumulation of PAR2 in endosomes, but had no effect on endosomal mitogenic signalling, indicating that de-ubiquitination does not regulate association with β-arrestins (Hasdemir et al., 2009). The effect of deubiquitination on the mitogenic signalling of other GPCRs has not been examined. However, ubiquitination of CXCR4 by atrophin-interacting protein 4 is reported to enhance CXCR4-mediated ERK1/2 activation (Malik et al., 2012). Although only a few USPs that regulate ubiquitin dynamics for GPCRs have been identified to date, it is clear that de-ubiquitination by peptidases plays a different role for each GPCR, in regulating trafficking and signalling.
The cytosolic proteins, β-arrestin 1 and 2 are key regulators of GPCR signalling and internalization [reviewed in Moore et al. (2007); Shenoy and Lefkowitz (2011)] and may be ubiquitinated following agonist stimulation. The first observation of β-arrestin ubiquitination occurred following activation of the β2-adrenoceptor and is regulated by the E3 ligase, murine double minute 2 (Mdm2) (Shenoy et al., 2001). A later study showed that de-ubiquitination of β-arrestin triggers its release from β2-adrenoceptors and the V2 vasopressin receptors (Shenoy and Lefkowitz, 2003). GPCRs may be classed on their association with β-arrestins. Class A receptors bind only β-arrestin 2 and with low affinity, whereas class B receptors bind both β-arrestin 1 and 2 with similar affinities and for prolonged periods (Oakley et al., 2000). When a β-arrestin-ubiquitin fusion protein was overexpressed in cells expressing the β2-adrenoceptor, the receptor associated with the β-arrestin–ubiquitin fusion protein for prolonged periods, changing the β2-adrenoceptor from a class A to a class B GPCR (Shenoy and Lefkowitz, 2003). Consistent with this observation and the fact that β-arrestins can mediate GPCR-induced ERK1/2 activation (Luttrell et al., 1999), sustained ubiquitination of β-arrestins results in prolonged activation of ERK1/2 by agonists of β2-adrenoceptors and V2 vasopressin receptors (Shenoy et al., 2007) (Figure 3, panel B). The ubiquitination state of β-arrestins is reciprocally regulated by Mdm2 and USP33, with the actions of USP33 promoting the disassembly of endosomal signalling complexes terminating ERK signalling (Shenoy et al., 2009).
Figure 3.
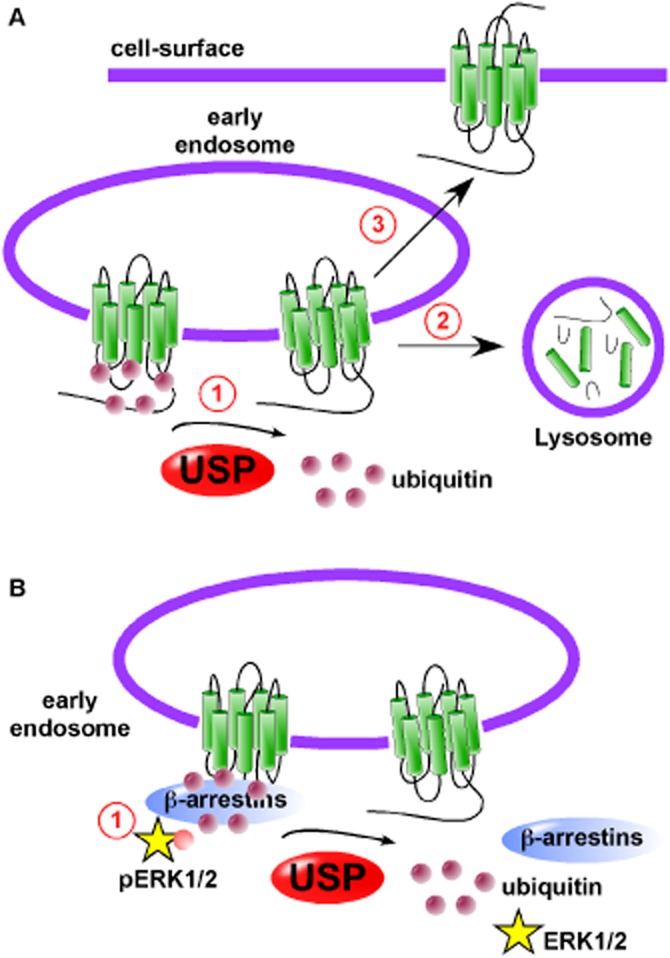
USPs promote lysosomal degradation and recycling of GPCRs. (A) (1) USPs cleave ubiquitin molecules from GPCRs to (2) promote entry in lysosomes and degradation by lysosomal peptidases or (3) recycling of GPCRs to the cell-surface mediating resensitization of signalling. (B) Ubiquitination of β-arrestins promotes GPCR-mediated phosphorylation of extracellular-regulated PK 1 and 2. (2) De-ubiquitination of β-arrestins by USPs destabilizes the GPCR•β-arrestin complex to terminate ERK1/2 activation.
There are additional ubiquitin-like modifying proteins, such as small ubiquitin-like modifier 1 (SUMO-1). Upon activation of the β2-adrenoceptor, β-arrestins are modified by the E2-modifying enzyme Ubc9, in a process termed sumoylation (Wyatt et al., 2011). Although it is known that this sumoylation promotes the internalization of the β2-adrenoceptor, the identity of the SUMO-specific peptidase and the functional consequences of the desumoylation are not yet known.
Endosomal proteolysis regulates the recycling and resensitization of GPCRs
SP and CGRP are neuropeptides expressed by primary sensory neurons (Lundberg et al., 1985). Noxious stimuli and trauma can lead to release of CGRP and SP from primary sensory neurons. The central release of CGRP and SP facilitates nociceptive transmission (Kuraishi et al., 1988; Kawamura et al., 1989; Pedersen-Bjergaard et al., 1991), whereas peripheral release mediates neurogenic inflammation, which is characterized by neutrophil infiltration, oedema and vasodilatation (McDonald, 1988; McDonald et al., 1988). CGRP, a potent vasodilator (Brain et al., 1985) is also implicated in the pathogenesis of migraine [reviewed in Raddant and Russo, (2011); Moore and Salvatore (2012)].
Following activation, many GPCRs internalize with β-arrestins to early endosomes. In order for a GPCR to recycle back to the cell-surface, β-arrestins must be released. Until recently, the molecular mechanisms that initiate the release of β-arrestins from neuropeptide GPCRs, such as the NK1 receptor and CLR•RAMP1 were poorly defined. NK1 receptors and CLR•RAMP1 are both class B GPCRs, having sustained interactions with β-arrestins (Oakley et al., 2000; Hilairet et al., 2001). Similar to the mechanisms that operate at the cell-surface, NK1 receptors and CLR•RAMP1 are regulated by the membrane-anchored peptidase, ECE-1 present in endosomes (Figure 4 ) (Padilla et al., 2007; Roosterman et al., 2007). SP and CGRP are substrates for the endosomal peptidase, ECE-1, but only at endosomal pH (Johnson et al., 1999; Padilla et al., 2007; Roosterman et al., 2007). As NK1 receptors and CLR•RAMP1 are trafficked through the endosomal system, vacuolar type H+-ATPases pump protons inside the vesicles lowering the pH of endosomes (Forgac et al., 1983; Cain et al., 1989). Acidification has two effects: firstly, the affinity of SP and CGRP for their respective receptors is lowered, and secondly, SP and CGRP become substrates for ECE-1 (Padilla et al., 2007; Roosterman et al., 2007). ECE-1 cleaves SP and CGRP into inactive metabolites that can no longer interact with their GPCRs. β-Arrestins are then released and NK1 receptors and CLR•RAMP1 are free to recycle back to the cell-surface (Padilla et al., 2007; Roosterman et al., 2007). Initial experiments were carried out using transfected cell lines, but further studies have shown that ECE-1-dependent cleavage of these neuropeptides regulates GPCR trafficking both in cells that endogenously express these receptors and in in vivo models. For example, ECE-1 regulates the resensitization of SP-induced plasma extravasation both in mice and rats, indicating the ECE-1 regulates NK1 receptors in endothelial cells (Roosterman et al., 2007; Cattaruzza et al., 2009). ECE-1 also regulates the trafficking of NK1 receptors in primary myenteric neurons (Pelayo et al., 2011). More recently, we have shown that ECE-1 regulates resensitization of CGRP-induced cAMP generation in primary mesenteric artery smooth muscle cells (McNeish et al., 2012). We also demonstrated that ECE-1 inhibition prevents the resensitization of CGRP-induced relaxation in rat mesenteric resistance arteries (McNeish et al., 2012). This ECE–1-dependent regulation is not confined to NK1 receptors and CLR•RAMP1, as other GPCRs are also regulated by this mechanism. Somatostatin-14 and −28 are inhibitory peptides exhibiting broad endocrine, exocrine and neuronal functions, such as the suppression of growth hormone secretion and the inhibition of pancreatic and gastrointestinal hormone release [reviewed in Olias et al. (2004)]. Somatostatin-14 and −28 exert their biological effects via activation of somatostatin receptors (sst receptors), which are expressed throughout the CNS and endocrine and immune systems. Sst receptors are also found at particularly high densities in many neuroendocrine tumours (Reubi et al., 1987a; 1987b; 1987c). This high density of sst receptors allows imaging of tumours using a radiolabelled analogue of somatostatin called octreotide, a process termed sst scintigraphy (Lamberts et al., 1990). The sst2 receptor is regulated by ECE-1 following stimulation with somatostatin-14, but not by octreotide, reflecting the ability of ECE-1 to cleave somatostatin-14, but not octreotide (Roosterman et al., 2008). A similar agonist-dependent trafficking was observed in studies with the corticotropin-releasing factor receptor 1 (CRF1). CRF1 receptors have two known agonists, corticotropin-releasing factor (CRF) and urocortin 1 (Ucn1). ECE-1 cleaves both peptides at endosonal pH, but only cleaves Unc1 at a residue critical for receptor binding (Hasdemir et al., 2012). At low agonist concentrations (30 nM), both Ucn1- and CRF-mediated intracellular calcium mobilization are dependent on ECE-1 activity; however, at high concentrations (100 nM), CRF-mediated intracellular calcium mobilization and CRF1 receptor recycling and resensitization cease to be ECE-1-dependent. This loss of ECE–1-dependent trafficking perhaps reflects a mechanism to mediate distinct CRF1 receptor trafficking and signalling, at higher concentrations of agonist (Hasdemir et al., 2012). Neurotensin is also a substrate for ECE-1 at endosomal pH (Johnson et al., 1999) and mediates intestinal inflammation and cell proliferation through activation of the neurotensin 1 receptor (NTS1) (Castagliuolo et al., 1999; Brun et al., 2005). Endosomal ECE-1 activity promotes degradation of neurotensin and recycling of NTS1 receptors (Law et al., 2012).
Figure 4.
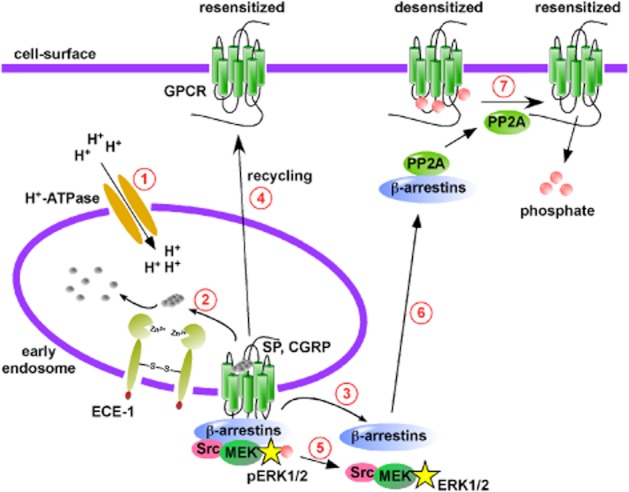
Endosomal peptidases promote GPCR recycling and resensitization. (1) Vacuolar-type H+-ATPases pump protons (H+) into vesicles, acidifying early endosomes. (2) Peptide agonists such as SP and CGRP have reduced affinity for their respective GPCRs. SP and CGRP become substrates for the endosomal peptidase, ECE-1 at low pH and are hydrolysed to inactive metabolites. (3) β-Arrestins dissociate from the GPCR, returning to the cytosol. (4) The GPCR, free from β-arrestins then recycles back to the cell-surface to mediate resensitization. (5) Certain GPCRs (e.g. neurokinin-1 receptor) signal from endosomes in a β-arrestin-dependent mechanism, phosphorylating extracellular-regulated PKs 1 and 2 (pERK1/2). ECE-1 promoted dissociation of β-arrestins terminates ERK1/2 activation. (6) β-Arrestins can recruit protein phosphatases such as protein phosphatase 2A (PP2A) to desensitized GPCRs at the cell-surface. (7) PP2A activity dephosphorylates cell-surface located GPCRs promoting resensitization.
Not all peptide-activated GPCRs are regulated by ECE-1. Studies have shown that although ECE-1 degrades bradykinin, ECE-1 does not regulate the recycling and resensitization of B2 receptors (Padilla et al., 2007). This is because of the nature of the interaction of B2 receptors with β-arrestins, B2 receptors only exhibit a transient interact with β-arrestins (Simaan et al., 2005). Thus, it is not only whether ECE-1 cleaves the agonist at endosomal pH that determines if ECE-1 regulates the GPCR, but also the duration of the association of the GPCR with β-arrestins. Moreover, agonists must be substrates for ECE-1 at endosomal pH in order for the GPCR to be regulated by ECE-1. Although angiotensin I is a substrate for ECE-1, angiotensin II is not (Johnson et al., 1999; Padilla et al., 2007) and ECE-1 does not regulate the recycling and resensitization of the AT2 receptors (Padilla et al., 2007).
An additional role for ECE-1-mediated cleavage of SP emerged following the observation that resensitization of SP-induced intracellular calcium mobilization precedes recycling of the NK1 receptor (Bennett et al., 2002; 2005; Murphy et al., 2011). It is known that agonist-unoccupied GPCRs are desensitized by phosphorylation of serine and threonine residues by the second messenger kinases, PKA and PKC (Roth et al., 1991; Pitcher et al., 1992; Dery et al., 2001). This second messenger kinase-dependent phosphorylation desensitizes the GPCR without causing internalization. In the case of the NK1 receptor, it is the dephosphorylation of cell-surface located, desensitized receptors that mediates resensitization of SP-induced signalling (Figure 4) (Murphy et al., 2011). ECE-1-dependent cleavage of SP releases β-arrestins from the endosomal GPCR complex. β-Arrestins then facilitate the recruitment of protein phosphatase 2A (PP2A), a known regulator of GPCRs (Pitcher et al., 1995) to the cell-surface, where PP2A dephosphorylates NK1 receptors to promote resensitization (Murphy et al., 2011). It is not currently known if the same or a similar mechanism controls the resensitization of other ECE-1-regulated GPCRs. Further, it is not yet known whether other endosomal peptidases regulate the trafficking of other peptide-activated GPCRs. Peptidases may represent a therapeutic target, whereby inhibitors of peptidases would prevent recycling and resensitization of GPCRs to prevent uncontrolled GPCR-mediated signalling that contributes to disease.
Proteolysis regulates endosomal signalling of GPCRs
In contrast to their role at the cell-surface in terminating G protein-dependent signalling, an additional function of β-arrestins is the recruitment of the signalling molecules to GPCRs in early endosomes. The signalling apparatus recruited to GPCRs by β-arrestins, serves to initiate a second wave of signalling that is distinct from that initiated at the cell-surface (Luttrell et al., 1999). This endosome-based signalling is a relatively new area of investigation and has been recently reviewed (von Zastrow and Sorkin, 2007; Murphy et al., 2009). For certain GPCRs, proteolysis of ligands by peptidases located in endosomes, serves as the molecular switch that determines the duration of this β–arrestin-dependent signalling. At the endosome surface, β-arrestins act as a scaffold to aid formation of mitogenic signalling complexes that include signalling proteins, such as Raf-1, MAPK kinases and ERK (Daaka et al., 1998; DeFea et al., 2000). The stability of these so-called signalosomes is dependent on the sustained interaction of β-arrestins with GPCRs in endosomes. Thus, for NK1 receptors, ECE–1-dependent hydrolysis of SP regulates the stability of the NK1 receptor•β-arrestin interaction and thus, the duration of SP-induced β–arrestin-dependent ERK activity (Cottrell et al., 2009). This endosome-derived ERK1/2 activation up-regulates and phosphorylates the nuclear death receptor, Nur77 via a mechanism that requires β-arrestins, Raf-1, MAPK kinase 2 and ERK2 (Castro-Obregon et al., 2004). Inhibition of ECE-1, which prevents the dissociation of NK1 receptors and β-arrestins (Roosterman et al., 2007), causes a sustained activation of ERK1/2, increased phosphorylation of Nur77, promoting cell death (Cottrell et al., 2009). This mechanism also operates in cultured primary myenteric neurons and in intact animals (Cottrell et al., 2009; Pelayo et al., 2011). In mice, intraplantar injection of capsaicin, an activator of transient receptor potential vanilloid ion channel 1, promotes the release of SP in the dorsal horn to cause internalization of NK1 receptors and activation of ERK1/2 (Mantyh et al., 1995; Kawasaki et al., 2004). An intrathecal injection of the highly selective ECE-1 inhibitor SM-19712 (Umekawa et al., 2000) promoted a more sustained ERK1/2 activation following injection of capsaicin (Cottrell et al., 2009). In contrast to SP-induced ERK1/2 activation, neurotensin-induced ERK1/2 activation is attenuated by ECE-1 inhibition, indicating that the recycling of NTS1 receptors promotes ERK1/2 activation (Law et al., 2012). ECE-1 inhibition similarly attenuated JNK activation, but promoted NF-κB activation and IL-8 secretion (Law et al., 2012).
Concluding remarks
It has long been known that peptidases present on the cell-surface regulate GPCR activation by generating or destroying bioactive peptides, and that many GPCRs are ultimately degraded by peptidases present in lysosomes. However, it is now apparent that proteolysis also regulates other aspects of GPCRs, including trafficking through the endocytic system and signalling from endosomes. Intracellular proteolysis of peptidic agonists by peptidases present in endosomes, not only controls the recycling and resensitization of GPCRs, but also regulates the signalling from internalized GPCRs in endosomes. Ubiquitination of GPCRs targets certain GPCRs to lysosomes, but de-ubiquitination performed by USPs is required for efficient delivery of GPCRs to lysosomes, promoting GPCR down-regulation. For other GPCRs, USPs regulate the recycling to the cell-surface and thus, resensitization of signalling. Finally, USPs act to regulate the ubiquitination of β-arrestins and downstream mitogenic signalling cascades. ACE inhibitors, such as captopril have been successfully used to treat hypertension for many years and offer hope that other peptidase inhibitors could also be used to treat disease. Inhibitors of endosomal peptidases, such as ECE-1 could be used to regulate the trafficking of CGRP and SP receptors. ECE-1 inhibitors, by preventing the recycling of CGRP receptors, could prevent sustained CGRP signalling implicated in migraine. Inhibitors of ADAM peptidases, responsible for the generation of ligands causing transactivation of ErbB receptors, may represent a new therapy to prevent proliferation of cells and thereby attenuate cancer growth. The signalling pathways arising from internalized GPCRs are distinct from pathways initiated at the cell-surface and have unique cellular consequences. Inhibitors of USPs and endosomal peptidases have been shown to regulate this signalling and thus inhibitors of these peptidases may be useful tools to regulate the signalling of internalized GPCRs. Thus, the discovery of these new peptidase-driven mechanisms of GPCR regulation opens up a new line of potential peptidase inhibitor therapies to treat GPCR-mediated diseases. A key factor in the design of these peptidase inhibitors will be targeting them to the cellular compartments where they will have the required effects.
Acknowledgments
Supported by British Heart Foundation Fellowship to G.S.C (FS/08/017/25027).
Glossary
- ADAM
a disintigrin and metallopeptidase
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin receptor-like receptor
- CRF1
receptor, corticotropin-releasing factor 1 receptor
- CXCR
C-X-C chemokine receptor
- ECE-1
endothelin-converting enzyme-1
- EGF
epidermal growth factor
- HB-EGF
membrane-anchored heparin-binding EGF-like growth factor
- NEP
neprilysin
- NK receptor
neurokinin receptor
- NTS1 receptor
neurotensin 1 receptor
- PAR
proteinase-activated receptor
- PP2A
protein phosphatase 2A
- RAMP
receptor activity-modifying protein
- sst receptor
somatostatin receptor
- SP
substance P
- USP
ubiquitin-specific peptidase
- Unc1
urocortin 1
Conflict of interest
The author declares no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- Bennett VJ, Perrine SA, Simmons MA. A novel mechanism of neurokinin-1 receptor resensitization. J Pharmacol Exp Ther. 2002;303:1155–1162. doi: 10.1124/jpet.102.040378. [DOI] [PubMed] [Google Scholar]
- Bennett VJ, Perrine SA, Simmons MA. Neurokinin-1 receptor resensitization precedes receptor recycling. J Pharmacol Exp Ther. 2005;313:1347–1354. doi: 10.1124/jpet.104.079954. [DOI] [PubMed] [Google Scholar]
- Berthouze M, Venkataramanan V, Li Y, Shenoy SK. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J. 2009;28:1684–1696. doi: 10.1038/emboj.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackhart BD, Ruslim-Litrus L, Lu CC, Alves VL, Teng W, Scarborough RM, et al. Extracellular mutations of protease-activated receptor-1 result in differential activation by thrombin and thrombin receptor agonist peptide. Mol Pharmacol. 2000;58:1178–1187. doi: 10.1124/mol.58.6.1178. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314(Pt 3):1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- Bralet J, Marie C, Mossiat C, Lecomte JM, Gros C, Schwartz JC. Effects of alatriopril, a mixed inhibitor of atriopeptidase and angiotensin I-converting enzyme, on cardiac hypertrophy and hormonal responses in rats with myocardial infarction. Comparison with captopril. J Pharmacol Exp Ther. 1994;270:8–14. [PubMed] [Google Scholar]
- Brun P, Mastrotto C, Beggiao E, Stefani A, Barzon L, Sturniolo GC, et al. Neuropeptide neurotensin stimulates intestinal wound healing following chronic intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G621–G629. doi: 10.1152/ajpgi.00140.2004. [DOI] [PubMed] [Google Scholar]
- Burger MM. Proteolytic enzymes initiating cell division and escape from contact inhibition of growth. Nature. 1970;227:170–171. doi: 10.1038/227170a0. [DOI] [PubMed] [Google Scholar]
- Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- Cain CC, Sipe DM, Murphy RF. Regulation of endocytic pH by the Na+,K+-ATPase in living cells. Proc Natl Acad Sci U S A. 1989;86:544–548. doi: 10.1073/pnas.86.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Scholten DJ, de Munnik S, Han MK, Smit MJ, Leurs R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE. 2012;7:e34192. doi: 10.1371/journal.pone.0034192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney DH, Cunningham DD. Initiation of check cell division by trypsin action at the cell surface. Nature. 1977;268:602–606. doi: 10.1038/268602a0. [DOI] [PubMed] [Google Scholar]
- Carney DH, Cunningham DD. Transmembrane action of thrombin initiates chick cell division. J Supramol Struct. 1978;9:337–350. doi: 10.1002/jss.400090305. [DOI] [PubMed] [Google Scholar]
- Castagliuolo I, Wang CC, Valenick L, Pasha A, Nikulasson S, Carraway RE, et al. Neurotensin is a proinflammatory neuropeptide in colonic inflammation. J Clin Invest. 1999;103:843–849. doi: 10.1172/JCI4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Obregon S, Rao RV, del Rio G, Chen SF, Poksay KS, Rabizadeh S, et al. Alternative, nonapoptotic programmed cell death: mediation by arrestin 2, ERK2, and Nur77. J Biol Chem. 2004;279:17543–17553. doi: 10.1074/jbc.M312363200. [DOI] [PubMed] [Google Scholar]
- Cattaruzza F, Cottrell GS, Vaksman N, Bunnett NW. Endothelin-converting enzyme 1 promotes re-sensitization of neurokinin 1 receptor-dependent neurogenic inflammation. Br J Pharmacol. 2009;156:730–739. doi: 10.1111/j.1476-5381.2008.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB, Buchanan JM. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc Natl Acad Sci U S A. 1975;72:131–135. doi: 10.1073/pnas.72.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Med Sci Monit. 2005;11:RA194–RA205. [PubMed] [Google Scholar]
- CONSENSUS. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med. 1987;316:1429–1435. doi: 10.1056/NEJM198706043162301. [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Padilla B, Pikios S, Roosterman D, Steinhoff M, Grady EF, et al. Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J Biol Chem. 2007;282:12260–12271. doi: 10.1074/jbc.M606338200. [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Padilla BE, Amadesi S, Poole DP, Murphy JE, Hardt M, et al. Endosomal endothelin-converting enzyme-1: a regulator of beta-arrestin-dependent ERK signaling. J Biol Chem. 2009;284:22411–22425. doi: 10.1074/jbc.M109.026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumashi A, Ansuini H, Celli N, De Blasi A, O'Brien PJ, Brass LF, et al. Neutrophil proteases can inactivate human PAR3 and abolish the co-receptor function of PAR3 on murine platelets. Thromb Haemost. 2001;85:533–538. [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Daull P, Benrezzak O, Arsenault D, Pheng LH, Blouin A, Cayer J, et al. Triple vasopeptidase inhibition normalizes blood pressure in conscious, unrestrained, and spontaneously hypertensive rats. Am J Hypertens. 2005;18(12 Pt 1):1606–1613. doi: 10.1016/j.amjhyper.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Daull P, Blouin A, Belleville K, Beaudoin M, Arsenault D, Leonard H, et al. Triple VPI CGS 35601 reduces high blood pressure in low-renin, high-salt Dahl salt-sensitive rats. Exp Biol Med (Maywood) 2006a;231:830–833. [PubMed] [Google Scholar]
- Daull P, Lepage R, Benrezzak O, Cayer J, Beaudoin M, Belleville K, et al. The first preclinical pharmacotoxicological safety assessment of CGS 35601, a triple vasopeptidase inhibitor, in chronically instrumented, conscious, and unrestrained spontaneously hypertensive rats. Drug Chem Toxicol. 2006b;29:183–202. doi: 10.1080/01480540600566717. [DOI] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dery O, Defea KA, Bunnett NW. Protein kinase C-mediated desensitization of the neurokinin 1 receptor. Am J Physiol Cell Physiol. 2001;280:C1097–C1106. doi: 10.1152/ajpcell.2001.280.5.C1097. [DOI] [PubMed] [Google Scholar]
- Dive V, Chang CF, Yiotakis A, Sturrock ED. Inhibition of zinc metallopeptidases in cardiovascular disease – from unity to trinity, or duality? Curr Pharm Des. 2009;15:3606–3621. doi: 10.2174/138161209789271889. [DOI] [PubMed] [Google Scholar]
- Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol. 2003;28:339–346. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, et al. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2005;32:411–419. doi: 10.1165/rcmb.2004-0274OC. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Inagami T. Signal transduction of angiotensin II type 1 receptor through receptor tyrosine kinase. Regul Pept. 2000;91:13–20. doi: 10.1016/s0167-0115(00)00126-9. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Frank GD, Mifune M, Inagami T. Metalloprotease-dependent ErbB ligand shedding in mediating EGFR transactivation and vascular remodelling. Biochem Soc Trans. 2003;31(Pt 6):1198–1202. doi: 10.1042/bst0311198. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Forgac M, Cantley L, Wiedenmann B, Altstiel L, Branton D. Clathrin-coated vesicles contain an ATP-dependent proton pump. Proc Natl Acad Sci U S A. 1983;80:1300–1303. doi: 10.1073/pnas.80.5.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh FG, Ng PY, Nilsson M, Kanke T, Plevin R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br J Pharmacol. 2009;158:1695–1704. doi: 10.1111/j.1476-5381.2009.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooz P, Dang Y, Higashiyama S, Twal WO, Haycraft CJ, Gooz M. A disintegrin and metalloenzyme (ADAM) 17 activation is regulated by alpha5beta1 integrin in kidney mesangial cells. PLoS ONE. 2012;7:e33350. doi: 10.1371/journal.pone.0033350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros C, Noel N, Souque A, Schwartz JC, Danvy D, Plaquevent JC, et al. Mixed inhibitors of angiotensin-converting enzyme (EC 3.4.15.1) and enkephalinase (EC 3.4.24.11): rational design, properties, and potential cardiovascular applications of glycopril and alatriopril. Proc Natl Acad Sci U S A. 1991;88:4210–4214. doi: 10.1073/pnas.88.10.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DM, Sweet CS, Ulm EH, Backlund EP, Morris AA, Weitz D, et al. Effect of N-[(S)-1-carboxy-3-phenylpropyl]-L-Ala-L-Pro and its ethyl ester (MK-421) on angiotensin converting enzyme in vitro and angiotensin I pressor responses in vivo. J Pharmacol Exp Ther. 1981;216:552–557. [PubMed] [Google Scholar]
- Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22:2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond-Martel I, Yu H, Affar B. Roles of ubiquitin signaling in transcription regulation. Cell Signal. 2012;24:410–421. doi: 10.1016/j.cellsig.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Hart S, Fischer OM, Prenzel N, Zwick-Wallasch E, Schneider M, Hennighausen L, et al. GPCR-induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR-independent pathways. Biol Chem. 2005;386:845–855. doi: 10.1515/BC.2005.099. [DOI] [PubMed] [Google Scholar]
- Hasdemir B, Murphy JE, Cottrell GS, Bunnett NW. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J Biol Chem. 2009;284:28453–28466. doi: 10.1074/jbc.M109.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasdemir B, Mahajan S, Bunnett NW, Liao M, Bhargava A. Endothelin-converting enzyme-1 actions determine differential trafficking and signaling of corticotropin-releasing factor receptor 1 at high agonist concentrations. Mol Endocrinol. 2012;26:681–695. doi: 10.1210/me.2011-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Hilairet S, Belanger C, Bertrand J, Laperriere A, Foord SM, Bouvier M. Agonist-promoted internalization of a ternary complex between calcitonin receptor-like receptor, receptor activity-modifying protein 1 (RAMP1), and beta-arrestin. J Biol Chem. 2001;276:42182–42190. doi: 10.1074/jbc.M107323200. [DOI] [PubMed] [Google Scholar]
- Hislop JN, von Zastrow M. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic. 2011;12:137–148. doi: 10.1111/j.1600-0854.2010.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop JN, Henry AG, Marchese A, von Zastrow M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J Biol Chem. 2009;284:19361–19370. doi: 10.1074/jbc.M109.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop JN, Henry AG, von Zastrow M. Ubiquitination in the first cytoplasmic loop of mu-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J Biol Chem. 2011;286:40193–40204. doi: 10.1074/jbc.M111.288555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM. Shedding light on ADAM metalloproteinases. Trends Biochem Sci. 2005;30:413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Ikeo T, Yabana H, Kurosawa H, Kaburaki M, Kikkawa K, Narita H, et al. Acute hemodynamic effects of the active metabolite of imidapril, (4S)-3-((2S)-2-[N-((1S)-1-carboxy-3-phenyl-propyl)amino]propionyl)-1- methyl-2-oxoimidazolidine-4-carboxylic acid, and enalaprilat in anesthetized dogs. Arzneimittelforschung. 1992;42:1109–1114. [PubMed] [Google Scholar]
- Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Zeng D, Connolly AJ, Tam C, Coughlin SR. Antibodies to protease-activated receptor 3 inhibit activation of mouse platelets by thrombin. Blood. 1998;91:4152–4157. [PubMed] [Google Scholar]
- Itoh Y, Joh T, Tanida S, Sasaki M, Kataoka H, Itoh K, et al. IL-8 promotes cell proliferation and migration through metalloproteinase-cleavage proHB-EGF in human colon carcinoma cells. Cytokine. 2005;29:275–282. doi: 10.1016/j.cyto.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076–16087. doi: 10.1074/jbc.M500109200. [DOI] [PubMed] [Google Scholar]
- Johnson GD, Stevenson T, Ahn K. Hydrolysis of peptide hormones by endothelin-converting enzyme-1. A comparison with neprilysin. J Biol Chem. 1999;274:4053–4058. doi: 10.1074/jbc.274.7.4053. [DOI] [PubMed] [Google Scholar]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Kuraishi Y, Minami M, Satoh M. Antinociceptive effect of intrathecally administered antiserum against calcitonin gene-related peptide on thermal and mechanical noxious stimuli in experimental hyperalgesic rats. Brain Res. 1989;497:199–203. doi: 10.1016/0006-8993(89)90990-6. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, et al. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimji AK, Rockey DC. Endothelin – biology and disease. Cell Signal. 2010;22:1615–1625. doi: 10.1016/j.cellsig.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Nanayama T, Ohno H, Minami M, Satoh M. Antinociception induced in rats by intrathecal administration of antiserum against calcitonin gene-related peptide. Neurosci Lett. 1988;92:325–329. doi: 10.1016/0304-3940(88)90611-8. [DOI] [PubMed] [Google Scholar]
- Kuwasako K, Shimekake Y, Masuda M, Nakahara K, Yoshida T, Kitaura M, et al. Visualization of the calcitonin receptor-like receptor and its receptor activity-modifying proteins during internalization and recycling. J Biol Chem. 2000;275:29602–29609. doi: 10.1074/jbc.M004534200. [DOI] [PubMed] [Google Scholar]
- Lamberts SW, Bakker WH, Reubi JC, Krenning EP. Somatostatin-receptor imaging in the localization of endocrine tumors. N Engl J Med. 1990;323:1246–1249. doi: 10.1056/NEJM199011013231805. [DOI] [PubMed] [Google Scholar]
- Law IK, Murphy JE, Bakirtzi K, Bunnett NW, Pothoulakis C. Neurotensin-induced proinflammatory signaling in human colonocytes is regulated by beta-arrestins and endothelin-converting enzyme-1-dependent endocytosis and resensitization of neurotensin receptor 1. J Biol Chem. 2012;287:15066–15075. doi: 10.1074/jbc.M111.327262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci U S A. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JG, Haines DS, Liu-Chen LY. Agonist-promoted Lys63-linked polyubiquitination of the human kappa-opioid receptor is involved in receptor down-regulation. Mol Pharmacol. 2008;73:1319–1330. doi: 10.1124/mol.107.042846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Benjamin CW, Jones DA. Convergence of angiotensin II and platelet-derived growth factor receptor signaling cascades in vascular smooth muscle cells. J Biol Chem. 1995;270:12563–12568. doi: 10.1074/jbc.270.21.12563. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Franco-Cereceda A, Hua X, Hokfelt T, Fischer JA. Co-existence of substance P and calcitonin gene-related peptide-like immunoreactivities in sensory nerves in relation to cardiovascular and bronchoconstrictor effects of capsaicin. Eur J Pharmacol. 1985;108:315–319. doi: 10.1016/0014-2999(85)90456-x. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- McDonald DM. Neurogenic inflammation in the rat trachea. I. Changes in venules, leucocytes and epithelial cells. J Neurocytol. 1988;17:583–603. doi: 10.1007/BF01260988. [DOI] [PubMed] [Google Scholar]
- McDonald DM, Mitchell RA, Gabella G, Haskell A. Neurogenic inflammation in the rat trachea. II. Identity and distribution of nerves mediating the increase in vascular permeability. J Neurocytol. 1988;17:605–628. doi: 10.1007/BF01260989. [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- McNeish AJ, Roux BT, Aylett S-B, Maassen Van Den Brink A, Cottrell GS. Endosomal proteolysis regulates calcitonin gene-related peptide responses in mesenteric arteries. Br J Pharmacol. 2012;167:1679–1690. doi: 10.1111/j.1476-5381.2012.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Soh UJ, Trejo J, Marchese A. Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J Biol Chem. 2012;287:9013–9027. doi: 10.1074/jbc.M111.336792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malynn BA, Ma A. Ubiquitin makes its mark on immune regulation. Immunity. 2010;33:843–852. doi: 10.1016/j.immuni.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G, Orr AG, Hamill CE, Yuan H, Pedone KH, McCoy KL, et al. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J Biol Chem. 2008;283:20600–20611. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, et al. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- Matsas R, Kenny AJ, Turner AJ. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochem J. 1984;223:433–440. doi: 10.1042/bj2230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, et al. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–9580. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, et al. G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem. 2005;280:26592–26599. doi: 10.1074/jbc.M502906200. [DOI] [PubMed] [Google Scholar]
- Milojevic T, Reiterer V, Stefan E, Korkhov VM, Dorostkar MM, Ducza E, et al. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol Pharmacol. 2006;69:1083–1094. doi: 10.1124/mol.105.015818. [DOI] [PubMed] [Google Scholar]
- Mogi M, Iwai M, Horiuchi M. New insights into the regulation of angiotensin receptors. Curr Opin Nephrol Hypertens. 2009;18:138–143. doi: 10.1097/MNH.0b013e328324f5fa. [DOI] [PubMed] [Google Scholar]
- Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Moore EL, Salvatore CA. Targeting a family B GPCR/RAMP receptor complex: CGRP receptor antagonists and migraine. Br J Pharmacol. 2012;166:66–78. doi: 10.1111/j.1476-5381.2011.01633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RH, Tuffaha A, Millman EE, Dai W, Hall HS, Dickey BF, et al. Agonist-induced sorting of human beta2-adrenergic receptors to lysosomes during downregulation. J Cell Sci. 1999;112(Pt 3):329–338. doi: 10.1242/jcs.112.3.329. [DOI] [PubMed] [Google Scholar]
- Muller S, Dennemarker J, Reinheckel T. Specific functions of lysosomal proteases in endocytic and autophagic pathways. Biochim Biophys Acta. 2012;1824:34–43. doi: 10.1016/j.bbapap.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: a legitimate platform for the signaling train. Proc Natl Acad Sci U S A. 2009;106:17615–17622. doi: 10.1073/pnas.0906541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Roosterman D, Cottrell GS, Padilla BE, Feld M, Brand E, et al. Protein phosphatase 2A mediates resensitization of the neurokinin 1 receptor. Am J Physiol Cell Physiol. 2011;301:C780–C791. doi: 10.1152/ajpcell.00096.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers TJ, Brennaman LH, Stevenson M, Higashiyama S, Russell WE, Lee DC, et al. Mitochondrial reactive oxygen species mediate GPCR-induced TACE/ADAM17-dependent transforming growth factor-alpha shedding. Mol Biol Cell. 2009;20:5236–5249. doi: 10.1091/mbc.E08-12-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal B, MacMahon S, Ohkubo T, Brnabic A, Tonkin A. Effects of the vasopeptidase inhibitor, omapatrilat, in 723 patients with coronary heart disease. J Renin Angiotensin Aldosterone Syst. 2002;3:270–276. doi: 10.3317/jraas.2002.049. [DOI] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem. 1995a;232:84–89. doi: 10.1111/j.1432-1033.1995.tb20784.x. [DOI] [PubMed] [Google Scholar]
- Nystedt S, Larsson AK, Aberg H, Sundelin J. The mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and functional expression. J Biol Chem. 1995b;270:5950–5955. doi: 10.1074/jbc.270.11.5950. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- Olias G, Viollet C, Kusserow H, Epelbaum J, Meyerhof W. Regulation and function of somatostatin receptors. J Neurochem. 2004;89:1057–1091. doi: 10.1111/j.1471-4159.2004.02402.x. [DOI] [PubMed] [Google Scholar]
- Ondetti MA, Rubin B, Cushman DW. Design of specific inhibitors of angiotensin-converting enzyme: new class of orally active antihypertensive agents. Science. 1977;196:441–444. doi: 10.1126/science.191908. [DOI] [PubMed] [Google Scholar]
- Padilla BE, Cottrell GS, Roosterman D, Pikios S, Muller L, Steinhoff M, et al. Endothelin-converting enzyme-1 regulates endosomal sorting of calcitonin receptor-like receptor and beta-arrestins. J Cell Biol. 2007;179:981–997. doi: 10.1083/jcb.200704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolillo M, Schinelli S. Therapeutic targeting of g-protein coupled receptor-mediated epidermal growth factor receptor transactivation in human glioma brain tumors. Mini Rev Med Chem. 2008;8:1418–1428. doi: 10.2174/138955708786369500. [DOI] [PubMed] [Google Scholar]
- Pedersen-Bjergaard U, Nielsen LB, Jensen K, Edvinsson L, Jansen I, Olesen J. Calcitonin gene-related peptide, neurokinin A and substance P: effects on nociception and neurogenic inflammation in human skin and temporal muscle. Peptides. 1991;12:333–337. doi: 10.1016/0196-9781(91)90022-h. [DOI] [PubMed] [Google Scholar]
- Pelayo JC, Poole DP, Steinhoff M, Cottrell GS, Bunnett NW. Endothelin-converting enzyme-1 regulates trafficking and signalling of the neurokinin 1 receptor in endosomes of myenteric neurones. J Physiol. 2011;589(Pt 21):5213–5230. doi: 10.1113/jphysiol.2011.214452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ. Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J Biol Chem. 2001;276:23155–23160. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- Pietri M, Schneider B, Mouillet-Richard S, Ermonval M, Mutel V, Launay JM, et al. Reactive oxygen species-dependent TNF-alpha converting enzyme activation through stimulation of 5-HT2B and alpha1D autoreceptors in neuronal cells. FASEB J. 2005;19:1078–1087. doi: 10.1096/fj.04-3631com. [DOI] [PubMed] [Google Scholar]
- Pitcher J, Lohse MJ, Codina J, Caron MG, Lefkowitz RJ. Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry. 1992;31:3193–3197. doi: 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Payne ES, Csortos C, DePaoli-Roach AA, Lefkowitz RJ. The G-protein-coupled receptor phosphatase: a protein phosphatase type 2A with a distinct subcellular distribution and substrate specificity. Proc Natl Acad Sci U S A. 1995;92:8343–8347. doi: 10.1073/pnas.92.18.8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Delbridge LM, Thomas WG. The angiotensin II type 2 (AT2) receptor: an enigmatic seven transmembrane receptor. Front Biosci. 2009;14:958–972. doi: 10.2741/3289. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem. 2004;279:18434–18439. doi: 10.1074/jbc.M401431200. [DOI] [PubMed] [Google Scholar]
- Raddant AC, Russo AF. Calcitonin gene-related peptide in migraine: intersection of peripheral inflammation and central modulation. Expert Rev Mol Med. 2011;13:e36. doi: 10.1017/S1462399411002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. 2009;76:791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2) J Biol Chem. 2011;286:24638–24648. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaekers CH, Wouters BG. Regulatory functions of ubiquitin in diverse DNA damage responses. Curr Mol Med. 2011;11:152–169. doi: 10.2174/156652411794859269. [DOI] [PubMed] [Google Scholar]
- Rao GN, Delafontaine P, Runge MS. Thrombin stimulates phosphorylation of insulin-like growth factor-1 receptor, insulin receptor substrate-1, and phospholipase C-gamma 1 in rat aortic smooth muscle cells. J Biol Chem. 1995;270:27871–27875. doi: 10.1074/jbc.270.46.27871. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40:D343–D350. doi: 10.1093/nar/gkr987. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regamey F, Maillard M, Nussberger J, Brunner HR, Burnier M. Renal hemodynamic and natriuretic effects of concomitant Angiotensin-converting enzyme and neutral endopeptidase inhibition in men. Hypertension. 2002;40:266–272. doi: 10.1161/01.hyp.0000030178.90322.11. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Hacki WH, Lamberts SW. Hormone-producing gastrointestinal tumors contain a high density of somatostatin receptors. J Clin Endocrinol Metab. 1987a;65:1127–1134. doi: 10.1210/jcem-65-6-1127. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Lang W, Maurer R, Koper JW, Lamberts SW. Distribution and biochemical characterization of somatostatin receptors in tumors of the human central nervous system. Cancer Res. 1987b;47:5758–5764. [PubMed] [Google Scholar]
- Reubi JC, Maurer R, von Werder K, Torhorst J, Klijn JG, Lamberts SW. Somatostatin receptors in human endocrine tumors. Cancer Res. 1987c;47:551–558. [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Roosterman D, Cottrell GS, Padilla BE, Muller L, Eckman CB, Bunnett NW, et al. Endothelin-converting enzyme 1 degrades neuropeptides in endosomes to control receptor recycling. Proc Natl Acad Sci U S A. 2007;104:11838–11843. doi: 10.1073/pnas.0701910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosterman D, Kempkes C, Cottrell GS, Padilla BE, Bunnett NW, Turck CW, et al. Endothelin-converting enzyme-1 degrades internalized somatostatin-14. Endocrinology. 2008;149:2200–2207. doi: 10.1210/en.2007-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth NS, Campbell PT, Caron MG, Lefkowitz RJ, Lohse MJ. Comparative rates of desensitization of beta-adrenergic receptors by the beta-adrenergic receptor kinase and the cyclic AMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88:6201–6204. doi: 10.1073/pnas.88.14.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133–149. doi: 10.1007/s00281-011-0289-1. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–78. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- Scase TJ, Heath MF, Evans RJ. Cloning of PAR3 cDNA from human platelets, and human erythroleukemic and human promonocytic cell lines. Blood. 1997;90:2113–2114. [PubMed] [Google Scholar]
- Schafer B, Marg B, Gschwind A, Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004;279:47929–47938. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- Sefton BM, Rubin H. Release from density dependent growth inhibition by proteolytic enzymes. Nature. 1970;227:843–845. doi: 10.1038/227843a0. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Barak LS, Xiao K, Ahn S, Berthouze M, Shukla AK, et al. Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J Biol Chem. 2007;282:29549–29562. doi: 10.1074/jbc.M700852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Modi AS, Shukla AK, Xiao K, Berthouze M, Ahn S, et al. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci U S A. 2009;106:6650–6655. doi: 10.1073/pnas.0901083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev. 2012 doi: 10.2174/157340312803217193. PMID: 22935021 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simaan M, Bedard-Goulet S, Fessart D, Gratton JP, Laporte SA. Dissociation of beta-arrestin from internalized bradykinin B2 receptor is necessary for receptor recycling and resensitization. Cell Signal. 2005;17:1074–1083. doi: 10.1016/j.cellsig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- SOLVD. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22:33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Moraes TJ, Vachon E, Ginzberg HH, Huang TT, Matthay MA, et al. Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells. Am J Respir Cell Mol Biol. 2005;33:231–247. doi: 10.1165/rcmb.2005-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. Am J Respir Cell Mol Biol. 2009;41:742–755. doi: 10.1165/rcmb.2008-0157OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275:26333–26342. doi: 10.1074/jbc.M002941200. [DOI] [PubMed] [Google Scholar]
- Tanida S, Kataoka H, Mizoshita T, Shimura T, Kamiya T, Joh T. Intranuclear translocation signaling of HB-EGF carboxy-terminal fragment and mucosal defense through cell proliferation and migration in digestive tracts. Digestion. 2010;82:145–149. doi: 10.1159/000310903. [DOI] [PubMed] [Google Scholar]
- Tanowitz M, Von Zastrow M. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J Biol Chem. 2002;277:50219–50222. doi: 10.1074/jbc.C200536200. [DOI] [PubMed] [Google Scholar]
- Thanawala V, Kadam VJ, Ghosh R. Enkephalinase inhibitors: potential agents for the management of pain. Curr Drug Targets. 2008;9:887–894. doi: 10.2174/138945008785909356. [DOI] [PubMed] [Google Scholar]
- Trapani AJ, Beil ME, Bruseo CW, Savage P, Firooznia F, Jeng AY. CGS 35601 and its orally active prodrug CGS 37808 as triple inhibitors of endothelin-converting enzyme-1, neutral endopeptidase 24.11, and angiotensin-converting enzyme. J Cardiovasc Pharmacol. 2004;44(Suppl. 1):S211–S215. doi: 10.1097/01.fjc.0000166237.57077.d6. [DOI] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao PI, von Zastrow M. Type-specific sorting of G protein-coupled receptors after endocytosis. J Biol Chem. 2000;275:11130–11140. doi: 10.1074/jbc.275.15.11130. [DOI] [PubMed] [Google Scholar]
- Umekawa K, Hasegawa H, Tsutsumi Y, Sato K, Matsumura Y, Ohashi N. Pharmacological characterization of a novel sulfonylureid-pyrazole derivative, SM-19712, a potent nonpeptidic inhibitor of endothelin converting enzyme. Jpn J Pharmacol. 2000;84:7–15. doi: 10.1254/jjp.84.7. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. 2007;177:905–916. doi: 10.1083/jcb.200610154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt D, Malik R, Vesecky AC, Marchese A. Small ubiquitin-like modifier modification of arrestin-3 regulates receptor trafficking. J Biol Chem. 2011;286:3884–3893. doi: 10.1074/jbc.M110.152116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Shirakabe K, Werb Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J Cell Biol. 2002;158:221–226. doi: 10.1083/jcb.200112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Yoshikawa T. Basic and translational research on proteinase-activated receptors: implication of proteinase/proteinase-activated receptor in gastrointestinal inflammation. J Pharmacol Sci. 2008;108:415–421. doi: 10.1254/jphs.08r31fm. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Sorkin A. Signaling on the endocytic pathway. Curr Opin Cell Biol. 2007;19:436–445. doi: 10.1016/j.ceb.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Oliver P, Lancaster JR, Jr, Schwarzenberger PO, Joshi MS, Cork J, et al. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. FASEB J. 2001;15:303–305. doi: 10.1096/fj.00-0371fje. [DOI] [PubMed] [Google Scholar]
- Zwick E, Wallasch C, Daub H, Ullrich A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and PYK2 tyrosine phosphorylation in PC12 cells. J Biol Chem. 1999;274:20989–20996. doi: 10.1074/jbc.274.30.20989. [DOI] [PubMed] [Google Scholar]