

Abstract
Despite considerable amount of research, the poor prognosis of patients diagnosed with glioblastoma multiforme (GBM) critically needs new drug development to improve clinical outcomes. The development of an inflammatory microenvironment has long been considered important in the initiation and progression of glioblastoma; however, the success of developing therapeutic approaches to target inflammation for GBM therapy has yet been limited. Here, we summarize the accumulating evidence supporting a role for inflammation in the pathogenesis of glioblastoma, discuss anti-inflammatory targets that could be relevant for GBM treatment and provide a perspective on the challenges faced in the development of drugs that target GBM inflammation. In particular, we will review the function of IL-1β, IL-6 and IL-8 as well as the potential of kinase inhibitors targeting key players in inflammatory cell signalling cascades such as JAK, JNK and p38 MAPK.
Keywords: glioblastoma, tumour microenvironment, ILs, inflammation, kinase inhibitors
Introduction
Glioblastoma multiforme (GBM) is the most common and lethal primary malignant cancer of the CNS. The GBM tumour is characterized by extensive heterogeneity, as implicated in its full name (multiforme: having or occurring in many forms). From the cellular point of view, GBM harbour multiple cell types, some with increased tumourigenicity and stem-like capacity, which are thought to be responsible for tumour relapse. However, the tumour-associated parenchymal cells – such as vascular cells, microglia, peripheral immune cells and neural precursor cells – also play a vital role in controlling the course of pathology. From a molecular standpoint, GBM heterogeneity is evidenced by the presence of various site- or tumour-specific mutations. For example, O6-methylguanine-DNA methyltransferase (MGMT) promoter hypermethylation, which is indicative of GBM response to the chemotherapy with the DNA-alkylating agent temozolomide (TMZ), is detected in some but not all GBM tumours, or only in distinct areas of hypermethylation-positive cells surrounded by cells lacking any sign of increased MGMT activity within the same tumour (Von Deimling et al., 2011). Other oncogenic markers not uniformly distributed in GBMs include overexpression of the EGFR mutant EGFRvIII (also known as ΔEGFR) or loss of phosphatase and tensin homolog (PTEN), resulting in differential activation/silencing of multiple signalling pathways (The Cancer Genome Atlas Research Network, 2008). The complex network of these cellular and molecular differences is believed to confer GBM with unpredictable and often very poor response to therapeutic interventions. The median survival of GBM patients is 15 months, and the overall 5 year survival remains extremely low at approximately 5% (Stupp et al., 2005). Little improvement in overall or progression-free survival has been achieved in the past five decades, reflecting an unmet medical need in the treatment of this cancer.
The deadly nature of GBMs resides in their capacity to diffusely infiltrate throughout the brain tissue, their extreme growth characteristics and their intrinsic resistance to current therapies. The ability of GBM cells to invade healthy brain tissue is critical for the tumour recurrence that accounts for the fatal outcome of the disease. Invading cells, besides seeding micro-tumours, are refractory to most lines of treatment, probably because they transiently arrest from mitosis (Lefranc et al., 2005; Kislin et al., 2009). The identification of glioblastoma stem cells (GSCs) within the tumour (Galli et al., 2004; Singh et al., 2004) had further implications for understanding its resistance to treatment. GSCs are highly invasive and proliferative cells (Cheng et al., 2011), and the standard therapy targets predominantly non-stem cells, while sparing the small population of GSCs (Huang et al., 2010; Beier et al., 2011). Recently, a quiescent subset of endogenous glioblastoma cells has been identified. These cells have properties similar for cancer stem cells and sustain long-term tumour growth through the production of highly proliferative cells (Chen et al., 2012). Finally, the inflammatory microenvironment, which is known to be the driving force for the progression of incipient neoplasias into highly malignant tumours (Mantovani et al., 2008; Solinas et al., 2010), is also found at the GBM lesions. Consistent with its role in other malignancies, inflammatory cytokines greatly enhance the proliferation, invasiveness and/or stemness of GBM cells and thus actively contribute to the global phenotype of GBM.
Glioblastomas are surrounded by a pool of pro-inflammatory cytokines, chemokines and growth factors. Here, we particularly review the tumourigenic effects of inflammatory IL-1β, IL-6 and IL-8 in GBM pathophysiology. Significant up-regulation of these cytokines is found in GBM cell lines as well as in patient samples (Table 1); and some of these interleukins (ILs) have prognostic potential. For example, IL-6 gene amplification in GBM tissue correlates with GBM aggressiveness and decreased patient survival (Rolhion et al., 2001; Tchirkov et al., 2007). A number of cell culture and xenograft studies support the hypothesis that targeting these inflammatory mediators could be beneficial in controlling the insidious behaviour of GBM. Thus, anti-inflammatory agents in conjunction with cytotoxic agents could offer a new and possibly improved approach to GBM therapy. We will therefore also discuss molecular therapeutics that could be used to target the production and activity of these ILs, focusing on inhibitors of signalling cascades activated by IL-1β, IL-6 and IL-8, namely the JAK, p38 MAPK and JNK pathways.
Table 1.
Expression and function of interleukins in glioblastoma
| Cytokine | Expression | Function |
|---|---|---|
| IL-1β | GBM clinical samples (Sharma et al., 2011b; Yeung et al., 2012) GBM cell lines (Yamanaka et al., 1994) | ↑ ERK activity ↑ proliferation (Meini et al., 2008) ↑ p38 MAPK/JNK activity ↑ VEGF (Yoshino et al., 2006) ↑ JNK and sphingosine kinase-1 activity ↑ proliferation and invasiveness (Paugh et al., 2009) ↑ p38 MAPK activity ↑ IL-6 (Tanabe et al., 2011; Yeung et al., 2012) ↑ stemness factor genes ↑ invasiveness ↑ drug resistance (Wang et al., 2012) |
| IL-6 | GBM clinical samples (Tchirkov et al., 2001; 2007; Kudo et al., 2009; Inda et al., 2010; Yeung et al., 2012) GBM cell lines (Yamanaka et al., 1994) Plasma of GBM patients (Reynes et al., 2011) | IL-6-deficient mice failed to develop GBM (Weissenberger et al., 2004) ↑ JAK2–STAT3 activation ↑ migration and invasiveness (Liu et al., 2010; Senft et al., 2011; Zhang et al., 2012) ↑ JAK2–STAT3 activation ↑ proliferation ↓ apoptosis (Rahaman et al., 2002) ↑ JAK2–STAT3 and Jagged–Notch activation ↑ stemness ↑ tumour heterogeneity and formation (Jin et al., 2012) ↑ tumour heterogeneity (Inda et al., 2010) |
| IL-8 | GBM clinical samples (de la Iglesia et al., 2008b; Hong et al., 2009; Bonavia et al., 2011) GBM cell lines (Yamanaka et al., 1994) | ↑ proliferation and invasiveness (de la Iglesia et al., 2008a) ↑ angiogenesis and tumour growth (Bonavia et al., 2011) ↑ tumour growth in an autocrine manner (Sun et al., 2011) ↑ invasiveness (Wakabayashi et al., 2004) |
Role of ILs in GBM pathophysiology
IL-1β
IL-1β is a master pro-inflammatory cytokine that triggers a number of malignant processes by activating various cells to up-regulate key molecules that drive oncogenic events. Elevated levels of IL-1β were observed in a panel of GBM cell lines, including CCF3 and U87MG (Lu et al., 2007), and in human GBM tumour specimens (Sharma et al., 2011b; Yeung et al., 2012). IL-1β receptor (IL-1R) is found in GBM cells and tissues (Sasaki et al., 1998), and we observed increased IL-1R levels in U87MG cells stably overexpressing the EGFRvIII variant (unpubl. data). IL-1β binding to the IL-1R resulted in the activation of NF-κB and MAPK signalling pathways, which cooperatively induced expression of various target genes (Griffin and Moynagh, 2006; McCulloch et al., 2006). IL-1β-induced ERK activation had mitogenic effects on human U373MG cells and significantly increased GBM cell proliferation (Meini et al., 2008). IL-1β-dependent activation of NF-κB, p38 MAPK and JNKs pathways in GBM cells (Figure 1) also led to the up-regulation of VEGF and sphingosine kinase 1, which promoted angiogenesis, migration and invasion respectively (Yoshino et al., 2006; Paugh et al., 2009). Furthermore, IL-1β-mediated up-regulation of hypoxia-inducible factor-1 (Sharma et al., 2011b) mediated molecular responses to hypoxia, which is a crucial component in GBM progression. In addition, GBM cells responded to IL-1β with an exacerbation of the inflammatory environment by secreting high levels of IL-6 and IL-8 (Spooren et al., 2011; Tanabe et al., 2011; Yeung et al., 2012) or up-regulation of COX-2 (Sharma et al., 2011a), an inflammatory enzyme detected in brain tumours and associated with poor prognosis (Xu and Shu, 2007; Myung et al., 2010). The recent finding that GBM cells without self-renewal capacity can gain this ability after exposure to IL-1β and TGF-β further adds to the detrimental role of IL-1β in the tumour microenvironment. In the GBM cell line LN-229, which did not form neurospheres and generated only small tumours in vivo, IL-1β in combination with TGF-β induced up-regulation of stemness factor genes, increased invasiveness and drug resistance that resulted in amplified tumour growth in vivo (Wang et al., 2012). Hence, selectively blocking IL-1β production and/or activity could contribute to control GBM progression.
Figure 1.

Simplified overview of inflammation-orchestrated signal transduction that induces pro-tumourigenic responses in glioblastoma. GBM tissue and microenvironment contains high levels of IL-1β, IL-6 and IL-8. Binding of IL-6 and IL-8 to IL-6/gp130 complex and GPCRs (CXCR1/2), respectively, leads to the activation (phosphorylation) of JAK2 that stimulates rapid, transient activation of STAT3 transcription factor. PI3K–Akt pathway is also a downstream target of CXCR1/2. IL-1β via its receptor IL-1R is a well-known p38 MAPK and JNK activating cytokine. Upon activation, p38 MAPK and JNK induce the activation of key oncogenic transcription factors NFκB and AP-1 or post-transcriptional gene regulation. This includes p38 MAPK-dependent HuR-shuttling and Mnk1/2 activation, leading to the control of mRNA stability and protein translation. Via these mechanisms, inflammation-activated JAK2, p38 MAPK and JNK signalling pathways are linked to proliferation, invasiveness, angiogenesis, stemness and tumour growth in GBM pathophysiology. Furthermore, JAK2 and JNK2 are also activated by EGFR and constitutively active EGFRvIII. Persistent activation JAK2 and JNK2 pathways via these growth factor receptors possibly contributes to the inflammation-driven GBM progression.
IL-6
IL-6 is produced by malignant cells as a response to external stimuli or intrinsic factors, such as oncogenic mutations. For example, the master pro-inflammatory cytokines IL-1β or TNF-α activate various signalling pathways resulting in stabilization of IL-6 mRNA and increased IL-6 biosynthesis (Tanabe et al., 2010; Spooren et al., 2011; Yeung et al., 2012). Furthermore, as discussed in more detail below, the EGFRvIII variant can induce IL-6 biosynthesis and secretion into the tumour microenvironment (Inda et al., 2010; Jin et al., 2011). The crucial role of IL-6 in glioblastoma development was convincingly demonstrated by Weissenberger et al. (2004), who showed that transgenic mice expressing the src oncogene in astrocytes failed to develop GBM on an IL-6-deficient background. Recent studies investigating IL-6 function in GBM progression further improve our understanding on how proliferation, differentiation and invasiveness require IL-6 signalling. Canonical IL-6 signal transduction is initiated by IL-6 binding to heteromeric plasma membrane receptor complexes formed by the IL-6 receptor (IL-6R) and the common signal transducing receptor glycoprotein 130 (gp130). Importantly, IL-6R and gp130 were expressed in GBM tissues and GBM-derived primary cells, including stem cells (Krona et al., 2005; Wang et al., 2009), underscoring that crucial components of this IL-6 circuitry are associated with the tumour cells. Alternatively, IL-6 trans-signalling is triggered by interaction of IL-6 with the soluble form of IL-6Rα and subsequent binding of this complex to the membrane-bound gp-130. In both pathways, upon receptor activation, intracellular signalling is propagated by JAK family members (JAK1-3), leading to activation of STAT transcription factors, particularly STAT3 (Figure 1). Indeed, activated STAT3 was found in GBM patients when compared with lower-grade brain tumours (Rahaman et al., 2002; Weissenberger et al., 2004; Abou-Ghazal et al., 2008; Lo et al., 2008; de Groot et al., 2010).
IL-6-induced activation of STAT3 promoted invasion and migration in U251, T98G and U87MG glioblastoma cells (Liu et al., 2010) and correlated with increased expression and secretion of MMP-2, a protease implicated in GBM motility (Li et al., 2010). Furthermore, IL-6 derived from neighbouring cells, in particular microglia, strongly stimulated GBM cell invasion (Zhang et al., 2012). This is in line with data showing that STAT3 inhibition attenuated the invasive and migratory potential of GBM cells (Senft et al., 2011; Michaud-Levesque et al., 2012). Interestingly, IL-6 depleted U87MG cells grafted on the chick chorioallantoic membrane formed a reduced primary tumour (volume <2% compared with negative controls), although a significant number of infiltrated malignant cell clusters was still detected, indicating that sole targeting of IL-6 production might be insufficient to completely block GBM invasiveness. In fact, invasion of GBM cells in this model was reversed upon combination of IL-6 depletion with the administration of VEGF monoclonal antibody bevacizumab (Avastin; Saidi et al., 2009). Despite its potent anti-angiogenic activities, in the model developed by de Groot et al. (2010), bevacizumab promoted GBM invasiveness, suggesting that combined inhibition of IL-6 and VEGF might be more efficient in reducing invasiveness and tumour growth.
IL-6-mediated STAT3 activation fostered proliferation and inhibits apoptosis of GBM cells (Rahaman et al., 2002), including GBM stem cells where STAT3 was identified as a downstream mediator of pro-survival IL-6 signalling. When comparing non-stem and stem cells in human GBM tissues, GSCs expressed higher IL-6R levels, whereas IL-6 ligand levels were higher in non-stem cells. Knock-down of IL-6R or IL-6 significantly reduced growth and neurosphere formation capacity while increasing apoptosis. Targeting IL-6R or its ligand in GSCs thus increased the survival of mice bearing the intracranial human GBM xenografts (Wang et al., 2009). In support of IL-6 promoting stemness of GBM cells, Jin et al. (2012) showed that IFN regulatory factor 7-derived IL-6 played a pivotal role in maintaining GBM stem cell properties via JAK/STAT mediated activation of the Jagged–Notch signalling pathway. The same group reported that IL-6–Jagged–Notch cascades conferred GSCs-like features and that IL-6 accelerated formation of GBMs in vivo. Furthermore, tumours derived from U87MG cells overexpressing IL-6 were positive for markers of neural stem cells, oligodendrocytes, neurons and astrocytes, as well as endothelial cells, thus showing tumour heterogeneity. In sharp contrast, tumours derived from IL-6-depleted cells were homogenous (Jin et al., 2011). This study confirms and expands the findings from Inda et al. (2010) revealing a new tumourigenic function for IL-6. Their study identified IL-6 derived from EGFRvIII-positive GBM cells as a growth factor for GBM cells carrying wild-type EGFR and described IL-6 as a messenger that enhances GBM cellular heterogeneity and tumour growth.
IL-8
IL-8 is highly expressed and secreted from GBM cell lines, stem cells and human specimens (Table 1). Similar to the regulation of IL-6, IL-8 expression was enhanced by TNF-α, IL-1β or macrophage infiltration (Hong et al., 2009; Yeung et al., 2012). In particular, TNF-α dramatically increased IL-8 synthesis in GBM cells through IL-8 mRNA stabilization (Nabors et al., 2003). In GBM pathophysiology, IL-8 is known as a potent angiogenic factor (Brat et al., 2005). Furthermore, the oncogenic EGFRvIII mutant and loss of tumour suppressor PTEN were linked to significantly higher IL-8 expression levels, which correlated with increased proliferation, neovascularization, invasiveness and in vivo tumour growth (de la Iglesia et al., 2008a; Bonavia et al., 2011). IL-8 also acted as an inflammatory chemoattractant for GBM cells promoting their invasiveness (Raychaudhuri and Vogelbaum, 2011; Wakabayashi et al., 2004). Moreover, it is secreted by tumour cells to promote growth in an autocrine manner (Wakabayashi et al., 2004; Sun et al., 2011). It is believed that biological effects of IL-8 are mediated by two highly related G-protein coupled chemokine receptors CXCR1 and CXCR2. However, only CXCR1 has yet been found to be expressed in GBM cell lines (Raychaudhuri and Vogelbaum, 2011). PI3K is one of the main downstream targets of CXCR1/2; with Raf–MAPK/ERK kinase (MEK)–ERK, p38 MAPK and JAK2–STAT3 (Figure 1) also being activated (Waugh and Wilson, 2008).
Underlying molecular mechanism of inflammation in GBM
The pro-tumourigenic activities of inflammatory mediators, in particular the IL-1β, IL-6 and IL-8, are well established, pointing at the need to better understand the signalling mechanisms that underlie their production and activities so that new therapies can be designed.
At present, several models of inflammatory events contributing to the development of cancer exist. First, repetitive injury or infections resulting in a chronic inflammatory response can eventually lead to cancer. Well-known examples for this pathway are Helicobacter pylori infection and the development of gastric cancer or hepatitis virus causing hepatocellular carcinoma (Mantovani et al., 2008). Second, cancers that evolve without underlying chronic inflammation can also exhibit tumour-associated inflammation. This inflammatory microenvironment is typically generated by tumour-associated macrophages, which significantly contribute to the tumour mass. Third, malignant cells and oncogenes, such as Ras and Myc, are also potent producers of inflammatory mediators (Sparmann and Bar-Sagi, 2004; Ancrile et al., 2007). Fourth, cell senescence induced by DNA damage enhances the transcription of pro-inflammatory cytokines and the role of senescence-associated secretory phenotype (SASP) in cancer is increasingly being investigated (Rodier et al., 2009). Fifth, chemotherapy or radiotherapy-induced cell death is often necrotic in nature and accompanied by release of inflammatory cytokines. In the healthy individual, the cytokine milieu ensures that primarily humoral immune responses are generated in order to prevent damage due to inflammation (Becker, 2006). However, in cancer patients, these normal humoral responses are skewed and immunosuppressive cytokines (e.g. TGF-β; Barcellos-Hoff et al., 2009) or pro-inflammatory cytokines IL-6 (Pasi et al., 2010) are predominantly expressed. These cytokines then activate oncogenic transcription factors in the remaining cancer cells and stimulate their survival and proliferation.
In GBM pathophysiology, the link between predisposing inflammation and GBM development has not been established; thus, glioblastoma is probably not an inflammation-induced cancer. The inflammation in GBM appears to be driven by oncogenic mutations as well as changes in the microenvironment and is best described as cancer-induced inflammation. Importantly, the evolving inflammation is a response not only to the disease but also to the therapy.
Oncogene- and tumour suppressor-induced inflammation
This intrinsic pathway of cancer-related inflammation is initiated and orchestrated by the tumour cells themselves without stimulation by prior inflammation. For example, oncogenic H-RasG12V-mediated production of pro-inflammatory cytokines IL-6, IL-8 and growth-regulated alpha protein (GRO1, also known as CXCL1) had emerged as an important contributor to the induction of an inflammatory microenvironment in cancer (Sparmann and Bar-Sagi, 2004; Ancrile et al., 2007). The Ras-dependent up-regulation of IL-6 or IL-8 is predominantly mediated by the activation of signalling pathways that impinge on the transcriptional machinery controlling their expression. Oncogenic Ras is not common in GBM pathophysiology; however, the overexpression of EGFR and/or its mutation EGFRvIII, leading to deregulation of EGFR effector pathways, including Ras and PI3K/Akt, is a hallmark of approximately 88% GBMs (The Cancer Genome Atlas Research Network, 2008). Indeed, the EGFRvIII mutation in GBM tissues, cell lines and glioblastoma stem cells was associated with strong induction and secretion of IL-6 and related leukaemia inhibitory factor (LIF; Inda et al., 2010). The underlying mechanism appeared to involve EGFR- or EGFRvIII-mediated activation of the Akt–Smad5 pathway, which in turn promoted up-regulation of inhibitor of the differentiation 3 (ID3) protein. EGFRvIII- and EGFR-driven ID3 induction in GBM cells then up-regulated expression of IL-6 and significantly enhanced angiogenesis and neurosphere formation. Pharmacological inhibition utilizing EGFR inhibitors or sh-RNA targeting ID3 confirmed ID3 as the major regulator of EGFR-driven IL-6 expression (Jin et al., 2011).
Several pathways have been identified that facilitate IL-8 up-regulation via EGFRvIII in GBM. For example, EGFRvIII enhanced IL-8 expression through activation of NF-κB, activating protein-1 (AP-1) and CCAAT-enhancer-binding protein (cEBP) transcription factors. Selective pharmacological or genetic targeting of NF-κB or AP-1 pathways blocked IL-8 promoter activity and cytokine secretion (Bonavia et al., 2011). EGFRvIII-dependent GBM cell transformation was also associated with simultaneous overexpression of tissue factor (TF) and its receptors PAR1/2. Consequently, EGFRvIII-positive GBM cells became hypersensitive to TF and subsequent PAR1/2 activation produced abundant amounts of IL-8 (Magnus et al., 2010). Finally, high IL-8 levels correlated with loss of the tumour suppressor PTEN and an inability to activate STAT3. In PTEN-deficient GBM cells, activated STAT3 functioned as a tumour suppressor by occupying endogenous IL-8 promoter and directly repressing IL-8 transcription. Via this mechanism, activated STAT3 inhibited GBM cell proliferation, invasiveness and spreading (de la Iglesia et al., 2008a,b). Together these observations suggest that several pathways activated by oncogenic mutations or loss of tumour suppressors cooperate to increase IL-8 production in GBM cells. Thus, blockade of one pathway will probably not be sufficient to fully inhibit production of IL-8.
Therapy-induced inflammation
After surgical removal of GBM, conventional treatment consisting of radiation and chemotherapy with the DNA-alkylating agent temozolomide aims to control tumour progression by causing death of remaining cancer cells (Nagasawa et al., 2012). However, almost all GBM tumours recur within 12 months after initial diagnosis. Intriguingly, most recurrent tumours are located at the site of origin and within the irradiated microenvironment.
In the last decade, it has become evident that the long-term consequence of radiation therapy is not only an amalgam of genetic damage and cell loss but also a change in the irradiated microenvironment. Exposure of cells to ionizing radiation (IR) induces the release of various proteins, which can trigger counteractive effects in the irradiated and neighbouring non-irradiated cells (i.e. bystander effect). This function of the radiation-induced microenvironment is exemplified by the IR-induced TGF-β activation in normal and cancer cells (reviewed in (Barcellos-Hoff et al., 2009). TGF-β released from irradiated T98G glioblastoma cells is a key signalling factor in the radiation-induced bystander effect by further inducing free radicals and DNA damage in the non-targeted bystander cells (Shao et al., 2008). Just recently, Hardee et al. (2012) showed that targeting TGF-β in the microenvironment significantly improved response of GBM cells and stem cell to radiation and increased the effectiveness of the therapy. Exposure of GBM cells to radiation also caused upregulation of IL-6 and, with different kinetics, IL-8 (Pasi et al., 2010), suggesting these cytokines are differentially induced by IR. Importantly, IL-8 expression levels were elevated in tumours irradiated in vivo (Kim et al., 2010).
GBM tissues often contain non-malignant cells, in particular tumour-associated microglia. Microglia are macrophages of the brain that constitute up to 30% of the tumour mass. Activated microglia are the primary source of inflammatory cytokines in non-malignant neuronal diseases (e.g. Alzheimer's disease) (Bachstetter and Van Eldik, 2010). Interestingly, although the morphological appearance of microglia found in or around GBM tumours suggested microglia activation, these cells did not release the prototypical inflammatory cytokines such as TNF-α or IL-6 (Sliwa et al., 2007; Gabrusiewicz et al., 2011). This is in agreement with in vitro data showing that GBM cells down-regulated the production of TNF-α from activated microglia (Kostianovsky et al., 2008). However, irradiation of primary and transformed microglial cells led to an increase of IL-1β and TNF-α mRNA levels (Kyrkanides et al., 1999; Ramanan et al., 2008; Schnegg et al., 2012). This correlates with in vivo studies showing that irradiation was associated with microglial activation in rodents (Monje et al., 2003). It is thus conceivable that microglia in recurrent GBM tumours exposed to IR therapy could contribute to local inflammation.
A couple of studies suggest that treatment with temozolomide increases production of inflammatory mediators. For instance, long-term TMZ-treated astroglia cells developed resistance to TMZ, which correlated with up-regulated chemokines CXCL2, CXCL3 and IL-8 (Bruyere et al., 2011). Another study, although limited and not directly addressing chemotherapy-induced inflammation, revealed that TMZ at clinically relevant concentration was not cytotoxic or anti-proliferative for microglia but significantly increased cellular protein synthesis (Vairano et al., 2004). Individual proteins had not been investigated, but given that another alkylating agent cyclophosphamide induced microglial activation and neuroinflammation in vivo (Christie et al., 2012), it is tempting to speculate that TMZ could also activate microglia to produce microglia-specific cytokines such as IL-1β and IL-6. This hypothesis is further supported by the discovery that DNA-damaging therapy such as IR can induce cell senescence accompanied with a striking release of pro-inflammatory cytokines (Rodier et al., 2009).
Senescence-induced inflammation in cancer
Cellular senescence, which is normally associated with ageing, is a process by which cells enter a state of permanent cell cycle arrest. In malignancy, this process constitutes a potent tumour suppressive mechanism. However, cellular senescence can also exert harmful effects in ageing or cancer. This is related to the acquisition of the SASP phenotype that is manifested by a remarkable secretion of inflammatory cytokines, especially IL-6 and IL-8. As SASP develops after DNA damage with IR, chemotherapy or potent mitogenic stimulation by oncogenic Ras, it is now well documented that cellular senescence is a source of inflammatory factors and thus has tumour-promoting rather than tumour-suppressing properties (Krtolica et al., 2001; Parrinello et al., 2005; Rodier et al., 2009).
Whether cellular senescence contributes to the development of the inflammatory microenvironment in GBM is yet not established. However, literature summarized here suggests that SASP could be one of the sources of inflammatory cytokines found in GBMs. As such, the PTEN status of GBM cells appeared to determine the cell fate after irradiation. PTEN-deficient U87, U251 and U373 cells underwent senescence after IR, whereas PTEN-proficient LN18 and LN428 cells entered apoptosis (Lee et al., 2011). Thus, PTEN-deficient GBM cells might stop to proliferate after IR but are likely to produce inflammatory cytokines that in turn could activate the radioresistant GBM stem cells. Cell surface-bound IL-1α was identified as an essential regulator of the senescence associated IL-6/IL-8 network (Orjalo et al., 2009), and GBM cell lines as well GBM specimens were found to express IL-1α mRNA and protein (Tada et al., 1994; Kam et al., 2007; Marcus et al., 2010). Furthermore, glucocorticoids by interfering with IL-1α signalling suppressed the production of IL-6 and IL-8 from senescent cells (Laberge et al., 2012). Despite their severe side effects, glucocorticoids are used in GBM therapy to control tumour-associated oedema, and their anti-inflammatory efficacy (Piette et al., 2009) could also be attributed to the control of senescence-associated inflammation.
Finally, the contribution of age-associated senescence to the inflammation in GBM pathophysiology should be considered. Age-associated senescent microglia do not function normally, as they fail to respond to certain stimuli and show inflammatory hyper-responsiveness (Lukiw, 2004). As such, ageing microglia are characterized by increased mRNA expression of inflammatory cytokines (TNF-α, IL-1β, IL-6; Sierra et al., 2007). Given that (a) every third cell in GBM tumour mass is a microglia (Graeber et al., 2002), (b) age is the most significant risk factor for GBM (average disease onset is >60 years) (Behin et al., 2003; Ricard et al., 2012) and (c) that expression of pro-inflammatory genes is increased during aging (Sparkman and Johnson, 2008), one can postulate that not only therapy-induced cell senescence but also age-associated microglial senescence contributes to the development of a permissive inflammatory microenvironment allowing cancer cells to thrive.
Therapeutic interventions
Antibodies neutralizing cytokine levels
Cytokine function in an inflammatory disease state can be successfully targeted with antibodies that will neutralize cytokines or their receptors. For example, anti-TNF antibodies infliximab or adalimumab are approved for the treatment of rheumatoid arthritis and investigated for an array of other inflammatory disorders, including cancers (Larkin et al., 2010; Diaz et al., 2011). Similarly, monoclonal antibody against IL-6, siltuximab, has been trialled for ovarian, prostate and renal cell cancer (Rossi et al., 2010; Coward et al., 2011; Fizazi et al., 2012). In GBM, IL-6R antibody tocilizumab has only been tested in vitro and exerted anti-proliferative effects in U87MG cells (Kudo et al., 2009). The only antibody against a protein in the tumour microenvironment investigated in GBM clinical trials is bevacizumab (Avastin). Avastin targets VEGF and is the only molecularly targeted drug that is FDA-approved for use in recurrent GBM. Unfortunately, although Avastin has potent anti-angiogenic efficacy in GBM patients, it strongly promotes tumour infiltration (Bai et al., 2011), which does not support its use in GBM therapy.
Large molecules like antibodies have limited brain penetration due to their limited ability to cross the blood–brain barrier (BBB). The anti-angiogenic efficacy of bevacizumab suggests that systematically administered antibodies may be useful as anti-glioblastoma therapies. However, bevacizumab targets VEGF, which has crossed from the brain into the vasculature, and the primary target for signal transduction inhibition appear to be endothelial cells lining the capillary walls and not the tumours cells themselves (Patel et al., 2012). Hence, one must be careful when interpreting the therapeutic potential of monoclonal antibodies, and promising results seen with IL-6 antibody tested against GBM in the absence of brain-specific delivery restrictions (Wang et al., 2009) are difficult to translate into the clinic. Along these lines, limited penetration of the EGFR inhibitor gefitinib into the CNS has been documented in GBM patients (Lassman et al., 2005). Hence, although BBB might be disrupted and leaky at the site of surgical resection and radiotherapy, the delivery of drugs to GBM tumours appears to be facing the challenge of all CNS-targeted drugs. Therefore, clinical development of antibodies focuses on novel drug delivery techniques to bypass the BBB by using viral vectors, liposomal carriers, nanoparticles or regional drug delivery to the brain (Allhenn et al., 2012). Alternatively, small molecules with BBB permeability might offer a more promising way to control inflammation in GBM. These agents could inhibit the interconnected circuits of inflammatory signal transduction pathways such as JAK–STAT, p38 MAPK, JNK and/or NF-κB. In this context, one of the most investigated pathways linking cancer with inflammation is the NF-κB pathway, which has been reviewed in great detail elsewhere (Grivennikov and Karin, 2010; Nogueira et al., 2011). In the following, we will summarize the potential of JAK–STAT3, p38 MAPK and JNK inhibitors for GBM therapy.
JAK–STAT inhibitors
The thorough investigation of IL-6 induced JAK–STAT signalling (Figure 1) in GBM pathophysiology and the availability of efficacious JAK–STAT inhibitors resulted in considerable pharmacological investigation for GBM therapy. JAK inhibitor JSI-124 (cucurbitacin I, Table 2) potently inhibits growth of U251 and A172 GBM cells where decreased levels of activated STAT3 led to down-regulation of cyclin B1 and cdc2 expression and induced apoptosis and cell cycle arrest (Su et al., 2008). The same inhibitor proved efficacious in inhibiting STAT3 activation in GBM stem cells and inhibited GSCs proliferation and survival (Wang et al., 2009). In vivo, inhibition of JAK–STAT3 activity by curcumin (administered as curcumin-fortified diet) reduced the proliferation of tumour cells as well as growth and midline crossing of intracranially implanted tumours (Weissenberger et al., 2010). The inhibitor AZD1480 (Table 2) also effectively blocked JAK1, JAK2 and STAT3 phosphorylation in GBM cells, leading to a decrease in cell proliferation (Ioannidis et al., 2011). In vivo, AZD1480 inhibits the growth of subcutaneous tumours and increases survival of mice bearing human intracranial GBM tumours (McFarland et al., 2011). WP1066 (Table 2) inhibits the STAT3 pathway by targeting JAK2 (Verstovsek et al., 2008). In vitro, this compound induced apoptosis and potently inhibited viability of U87MG and U373MG cell lines. Systematic i.p. administration of WP1066 in mice attenuated the growth of s.c. GBM xenografts. Immunohistochemical analysis of excised tumours confirmed that the anti-growth efficacy resulted from inhibiting the STAT3 activity as levels of activated STAT3 (p-STAT3) in the treatment group remained inhibited 3 weeks after initial WP1066 injection, whereas tumours from the control group continuously expressed high levels of p-STAT3 (Iwamaru et al., 2007).
Table 2.
Overview of kinase inhibitors profiled in the review
| Inhibitor | Structure | Primary Targets | Activity in GBM-related models |
|---|---|---|---|
| JSI-124 | 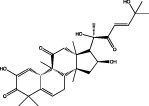 |
JAK2–STAT3 pathway (Blaskovich et al., 2003) | Inhibits growth of U251 and A172 GBM cells (Su et al., 2008) Inhibits GSCs proliferation and survival (Wang et al., 2009) Sensitizes GBM cells to gefitinib and temozolomide (Lo et al., 2008) |
| AZD1480 | 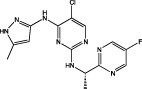 |
JAK2 IC50 = 0.058 μM JAK3 IC50 = 1.36 μM (Ioannidis et al., 2011) | Inhibits GBM cell proliferation (Ioannidis et al., 2011) Inhibits growth of subcutaneous GBM tumours (McFarland et al., 2011) |
| WP1066 | 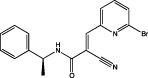 |
STAT3 in vitro IC50 not available IC50 (U87MG) = 5.6 μM IC50 (U373MG) = 3.7 μM (Iwamaru et al., 2007) | Induces apoptosis, inhibits viability of U87MG and U373MG cells, attenuates the growth of s.c. GBM xenografts (Iwamaru et al., 2007) Potentiates TMZ efficacy in TMZ-resistant GBM cells (Kohsaka et al., 2012) |
| Ruxolitinib | 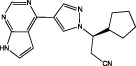 |
JAK1 IC50 = 3.3 nM JAK2 IC50 = 2.8 nM JAK3 IC50 = 428 nM (Quintás-Cardama et al., 2010) | FDA approved for treatment of myeloproliferative neoplasms |
| SB202190 | 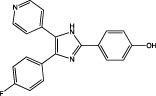 |
p38α MAPK IC50 = 50 nM (binding assay) IC50 = 67 nM (kinase activity assay) (Gallagher et al., 1997) | Abolishes the GBM-conditioned media-triggered increase in microglial expression of membrane type 1 MMP (MT1-MMP) (Markovic et al., 2009) Abrogates the GBM conditioned media-induced MT1-MMP activity (Markovic et al., 2009) |
| SB203580 | 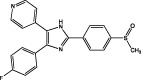 |
p38α MAPK IC50 = 42 nM (binding assay) IC50 = 74 nM (kinase activity assay) (Gallagher et al., 1997) | Sensitizes mismatch repair-proficient tumour cells to temozolomide (Hirose et al., 2003) Inhibits synthesis of IL-6, IL-8, VEGF in GBM cells (Yoshino et al., 2006; Yeung et al., 2012) Inhibits LPA-induced GBM cell migration (Malchinkhuu et al., 2005) |
| LY479754 | 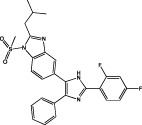 |
p38α MAPK IC50 = 4.9 nM p38β MAPK IC50 = 11.9 nM (de Dios et al., 2005) | Sensitizes non-migrating GBM cells to cytotoxic therapy with temozolomide (Demuth et al., 2007) Reduces tumour growth in vivo (Campbell et al., 2005) |
| 069A | 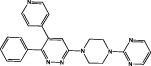 |
p38α MAPK IC50 = 0.8 μM (Munoz et al., 2007) | Inhibits inflammatory response, migration and invasiveness of U251 GBM cells (Yeung et al., 2012) |
| BIRB796 | 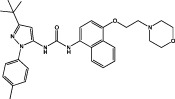 |
p38α MAPK KD = 0.097 nM (Regan et al., 2002) JNK2 KD = 4.6 nM JNK3 KD = 62 nM (Gruenbaum et al., 2009) | |
| SP600125 | 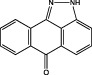 |
JNK1 IC50 = 40 nM JNK2 IC50 = 40 nM JNK3 IC50 = 90 nM (Bennett et al., 2001) | Inhibits LPA-induced migration and invasiveness of GBM cells (Malchinkhuu et al., 2005) Inhibits adenosine receptor stimulated increase of MMP-9 (Gessi et al., 2010) Amplifies senescence in TMZ-treated U87MG cells (Ohba et al., 2009) Inhibits IL-1β-induced IL-6, VEGF and sphingosine kinase 1 upregulation (Yoshino et al., 2006; Paugh et al., 2009; Tanabe et al., 2011) Blocks IL-8 promoter activity and expression in EGFRvIII-bearing cells (Bonavia et al., 2011) |
In the context of GBM development, it is important to note that STAT3 is activated not only by JAKs but also by EGFR and EGFRvIII (Figure 1), which are overexpressed in 40–50% of GBMs (see above). STAT3 activation and de-regulated EGFR correlated in 27.2% of investigated tumours. Thus, targeting JAKs alone might not be sufficient to halt growth of tumours with increased EGFR activity. Sorafenib, an oral inhibitor targeting several receptor tyrosine kinases including EGFR, inhibited proliferation in primary and established GBM cell lines. Effects of sorafenib were associated with inhibiting STAT phosphorylation via inhibiting JAK1 and JAK2 (Yang et al., 2010). Combination of EGFR inhibitor gefitinib (Iressa) and JAK2 inhibitor JSI-124 synergistically suppressed STAT3 activation and potently killed GBM cells that expressed EGFR or EGFRvIII. JSI-124 also sensitized GBM cells to TMZ, an alkylating agent used in GBM therapy (Lo et al., 2008). Kohsaka et al. (2012) observed increased sensitivity towards TMZ and reported significant positive correlation between expression levels of MGMT and p-STAT3 in 44 GBM specimens. In vitro, up-regulation of IL-6, STAT3 and MGMT was accompanied with acquisition of TMZ resistance. Importantly, STAT3 inhibitor WP1066 potentiated TMZ efficacy in TMZ-resistant GBM cell lines by post-transcriptionally suppressing MGMT protein levels. However, the study failed to show in vivo efficacy of a WP1066–TMZ combination, suggesting that fast metabolism of STAT3 inhibitor and short half-life of TMZ hindered in vivo evaluation.
Together, the encouraging results from these studies indicate that pharmacological inhibition of the JAK2-STAT3 pathway could be considered for the treatment of GBM patients. Availability of ruxolitinib (Table 2), safe and efficacious JAK2 inhibitor recently approved by FDA for the treatment of myelofibrosis (Verstovsek et al., 2010; 2012; Mesa et al., 2012) and currently in clinical trials for the treatment of other malignancies (invasive metastatic breast cancer, multiple myeloma), is a promising starting point for anti-inflammatory GBM therapy.
p38 MAPK inhibitors
Limited studies have yet addressed the therapeutic potential of p38 MAPK inhibition in GBM, but rather focussed on improving the understanding of the role of p38 MAPK isoforms (α, β, γ, δ) in various aspects of GBM pathophysiology.
p38 MAPK activity contributes to the highly invasive capacity of GBM cell lines and regulates the expression of MMPs, which are responsible for the proteolytic degradation of extracellular matrix, which is the major obstacle for cell motility. MMP secretion from GBM cells stimulates the migratory response in a p38 MAPK-dependent manner. Accordingly, inhibition of p38 MAPK blocked MMP secretion and invasion of GBM cells (Park et al., 2002; Malchinkhuu et al., 2005). IR-induced EGFR activation, which triggers p38 MAPK activation, along with Akt and PI3K signalling, also increased MMP2 expression and heightened invasiveness of PTEN deficient GBM cells (Park et al., 2006). Furthermore, GBM-released factors manipulated the tumour-associated microglia via toll-like receptor induced p38 MAPK signalling, which up-regulates membrane type 1-MMP to activate pro-MMP2 and thereby support GBM expansion (Markovic et al., 2009).
In addition, p38 MAPK may affect GBM malignancy via modulating cytokines and growth factors in the tumour microenvironment. p38 MAPK has a key role in the production of TNF-α, IL-1β and IL-6 from activated microglia and also regulates signalling of these cytokines in GBM cells (Figure 1 ), further exacerbating local inflammation. These cytokines exert their pro-tumourigenic effects in GBM pathology by increased expression of other inflammatory (e.g. COX-2; Xu and Shu, 2007), invasiveness-promoting (e.g. MMPs; Markovic et al., 2009; Sarkar and Yong, 2009) or angiogenic (e.g. IL-8 and VEGF; Yoshino et al., 2006) mediators. GBM cells could potentially up-regulate these mediators via p38 MAPK-dependent phosphorylation of transcription factors such as NF-κB (Figure 1). Post-transcriptional gene regulation has also been implicated as one mechanism of the tumour response to the inflammatory microenvironment, and in this regard, p38 MAPK-mediated shuttling of the mRNA stabilizing human antigen R (HuR, Figure 1) protein is linked to increased stability of VEGF, TGF-β, IL-6, IL-8 and TNF-α mRNA in GBM cells (Nabors et al., 2003). HuR depletion led to transcript destabilization, reduced protein expression, a significant decrease in tumour volume and increased sensitivity to chemotherapeutic drugs (Filippova et al., 2011). Further supporting p38 MAPK activity in regulating mRNA stability, p38 MAPK-mediated hyperphosphorylation deactivated the RNA destabilizer tristetraprolin (TTP), resulting in stabilization of IL-8 and VEGF mRNA, and promoted GBM cell proliferation and viability (Suswam et al., 2008). Moreover, MAPK-interacting kinase 1 and 2 (Mnk1/2) are translation-controlling serine/threonine kinases that are activated by p38 MAPK (Figure 1) and Mnk1 knockdown in U87MG reduced tumourigenic activity in nude mice (Ueda et al., 2010). Finally, activity of p38 MAPK influences the response of GBM cells to DNA-alkylating agents and pharmacological inhibition of p38 MAPK sensitized GBM cells to TMZ-induced toxicity (Hirose et al., 2003; 2004).
Despite increasing evidence for p38 MAPK as an anti-GBM target, currently in vivo data are limited in order to validate the effectiveness of p38 MAPK inhibition in GBM. Indirectly supporting the use of p38 MAPK inhibitors for GBM therapy comes from research on minocycline, a semi-synthetic broad-spectrum and lipophilic tetracycline antibiotic approved by the FDA that is able to cross the BBB and inhibit microglial activation and inflammation in CNS disease models. One of the main targets of minocycline is p38 MAPK (Nikodemova et al., 2006), and the anti-inflammatory effect of minocycline correlates with inhibition of microglial p38 MAPK phosphorylation and decreased GBM invasiveness and expansion in vitro and in vivo (Liu et al., 2011; Markovic et al., 2011).
Commonly, the prototypical p38 MAPK inhibitors SB203580 (Table 2) and SB202190 (Table 2) have been utilized to examine p38 MAPK in GBM malignancy (Hirose et al., 2003; Malchinkhuu et al., 2005; Markovic et al., 2009; Bonavia et al., 2011). A better clinical candidate, the p38 MAPK inhibitor LY479754 (Table 2), was shown to inhibit GBM invasiveness in vitro. In addition, LY479754 also sensitized arrested, non-migrating cells to cytotoxic therapy with TMZ (Demuth et al., 2007). We recently showed that a BBB-permeable p38 MAPK inhibitor, 069A (Table 2, Munoz et al., 2007), can potentially target both malignant and non-malignant microglial cells in GBM tumours and attenuate the development of an inflammatory microenvironment and GBM invasiveness (Yeung et al., 2012). Possibly in combination with other treatments, these promising results mandate further development and testing of suitable p38 MAPK inhibitors against invasive glioblastoma in vivo. In clinic, however, BBB permeability might be problematic as p38 MAPK inhibitors have been developed for rheumatoid arthritis therapy and were intentionally made highly polar in order to avoid BBB permeability. This restricts the use of current p38 MAPK inhibitors for CNS-related diseases. Certainly, a p38 MAPK inhibitor with satisfying preclinical safety might be able to control the local inflammation at the tumour site, but a new p38 MAPK inhibitor with improved BBB permeability could have far more potential in GBM therapy.
JNK inhibitors
JNKs are an evolutionarily conserved sub-group of MAPKs activated by MAPK kinases 4 and 7 (MMK4 and MKK7), which integrate a wide array of stimuli to phosphorylate and stimulate JNKs. The major JNK target is the transcription factor AP-1, which is composed of Fos and c-Jun family members. Thereby, the JNK/c-Jun pathway modulates expression of a plethora of AP-1 target genes that control inflammatory response, cell proliferation, apoptosis as well as invasion. Besides regulating proto-oncogenes Jun, Fos and Myc; JNKs have also been linked to p53-dependent senescence and apoptosis. Overall, pro- and anti-apoptotic effects of JNK signalling on tumour development appear to be determined by stimuli and tissue specificity, signal intensity and crosstalk between JNK isoforms (Wagner and Nebreda, 2009).
In GBM, JNKs and c-Jun phosphorylation correlate with the grade of malignancy and patients' age (Antonyak et al., 2002). JNK2 is the major activated JNK isoform in GBM (Tsuiki et al., 2003). This isoform is unique among all MAPKs as it possesses autophosphorylation activity and constitutive substrate kinase activity in vitro and in vivo (Nitta et al., 2008). JNK2 is also activated by oncogenic EGFRvIII (Figure 1) (Antonyak et al., 1998) and supports tumourigenesis in vivo through increased proliferation and tumour formation (Cui et al., 2006). In addition to its role in apoptosis, JNKs have been linked to glial-derived neurotrophic factor (Lu et al., 2010) and lysophosphatidic acid-induced migration and invasiveness of GBM (Malchinkhuu et al., 2005). SP600125-mediated JNK inhibition antagonized adenosine receptor stimulated increase of MMP-9 levels (Gessi et al., 2010). Furthermore, SP600125 (Table 2) amplified senescence in TMZ-treated U87MG cells, suggesting that SP600125 potentiates TMZ-dependent cell death pathways (Ohba et al., 2009).
Within the tumour microenvironment, JNKs likely potentiate carcinogenesis by promoting local inflammatory responses. For example, IL-1β-induced IL-6 production, which contributes to increased tumour development and invasiveness, is inhibited by SP600125 in an inflammatory model of GBM (Tanabe et al., 2011). Inhibition of JNK signalling also suppressed angiogenesis and invasiveness of GBM cell via inhibiting IL-1β-induced VEGF production and sphingosine kinase 1 upregulation, respectively (Yoshino et al., 2006; Paugh et al., 2009). Together, these studies indicate that therapeutic manipulation of oncogenic IL-1β activities can be managed with JNK inhibitors; however, the JNK isoforms responsible for these effects have yet to be identified. Interference with the JNK pathway also decreased EGFR-dependent tissue factor expression (Rong et al., 2009). TF is the main cellular initiator of coagulation that significantly contributes to two fundamental features of GBM, namely hypoxia and necrosis. EGFR-mediated TF expression depends on AP-1 transcriptional activity and is associated with JNK and JunD activation. These mechanisms are likely to work in vivo as elevated/mutated EGFR highly correlated with TF expression in GBM specimen (Rong et al., 2009). Recently, Bonavia et al. (2011) showed that the pharmacological targeting of the JNK–NF-κB pathway with SP600125 efficiently blocked IL-8 promoter activity and expression in EGFRvIII-bearing cells. However, interpretation of results derived from the pan-JNK inhibitor SP600125 should have taken into consideration the vast number of off-target kinases that this compound inhibits (Bain et al., 2007).
A significant number of potent and selective JNK inhibitors have recently been developed, some with BBB permeability (He et al., 2011; Plantevin Krenitsky et al., 2012). These inhibitors either target peripheral JNK1 in obesity and diabetes, or the JNK3 isoform expressed in CNS to reduce neurodegeneration. Yet JNK2 appears to be the main JNK isoform involved in GBM pathophysiology and JNK2 selective inhibitors are yet to be developed. Interestingly, BIRB796 (Table 2), a potent type II inhibitor of p38 MAPK that also binds with high affinity to JNK2 (Gruenbaum et al., 2009), could provide an opportunity as dual-kinase inhibitor.
Conclusion
ILs play a major role in various inflammatory diseases including cancer. For example, IL-13 is a cytokine that modulates inflammation, apoptosis and/or tumour immunosurveillance in many different cancers (Wynn, 2003). However, its beneficial activity in glioblastoma is limited by increased expression of IL-13 receptor α2 (IL-13Rα2) on GBM cells. IL-13Rα2 binds and internalizes IL-13 with high affinity, but it does not mediate signal transduction. Accordingly, the decoy receptor IL-13Rα2 is a promising drug target (Hsi et al., 2011; Balyasnikova et al., 2012). As we review here, IL-1β, IL-6 and IL-8 in the close vicinity of malignant cells promote carcinogenesis through activation of proliferative mechanisms and/or activation of signalling cascades involved in angiogenesis, migration and invasiveness. Furthermore, these ILs are potent activators of signalling pathways, which regulate cell survival and promote resistance to chemotherapy. Hence, targeting these inflammatory cytokines in the GBM microenvironment with small molecule kinase inhibitors in combination with cytotoxic agents could achieve therapeutic benefits for patients with recurrent GBM. As demonstrated recently, resistance of GBM and stem cells to radiation can be attenuated by inhibiting TGF-β in the tumour microenvironment (Hardee et al., 2012). The magnitude of radiosensitization seen in this study is comparable with the radiosensitization achieved by adding the cytotoxic agent TMZ to the radiation therapy. Given that concurrent combination of TMZ and radiation is the standard and best available approach to treat GBM, it is reasonable to expect increasing basic and clinical research in targeting the GBM microenvironment. Among the diverse signalling networks involved in inflammation, JAK, p38 MAPK and JNK pathways have emerged as critical convergent points that warrant further pursuit as drug targets. Availability of ruxolitinib, assuming it will reach therapeutic concentrations in the brain, and significant in vivo data supporting beneficial JAK inhibition in GBM pathophysiology, could launch trials assessing anti-IL-6 targeted approaches for GBM therapy in the near future. Testing of p38 MAPK or JNK inhibitors in GBM therapy could be next, but in vivo data and FDA-approved inhibitors of these kinases are not yet available.
It is important to note that success of anti-inflammatory therapies in GBM does not entirely depend on the availability of suitable drugs. Treatment of glioblastoma is challenged by the extreme heterogeneity of these tumours requiring a highly personalized approach. EGFR, PTEN, MGMT and p53 status, isocitrate dehydrogenase (IDH) mutation and/or deletion of chromosome arms 1p and 19q (Riemenschneider et al., 2010; Ricard et al., 2012) are known to determine the response of GBM cells to radiation and chemotherapy, as well as efficacies of newly developed kinase inhibitors. Hence, the future success of kinase inhibitors, including those targeting inflammatory pathways, lies in the ability to identify vulnerable tumours based on their specific genetic alterations.
Research summarized in this review suggests that the future direction for GBM therapy could benefit from the design of synergistic multi-target drug combinations that would target both oncogenic cellular signalling in cancer cells as well as the inflammatory microenvironment. Although the significance of inflammation for GBM pathology and in vivo evaluation of therapies directly targeting inflammation is still missing, accumulating in vitro evidence indicates that inflammation-based therapies could provide useful tools in combating GBMs, most likely in the combination with standard therapeutic regimens. The extreme heterogeneity of GBMs and the lack of kinase inhibitors with sufficient BBB permeability remain challenging aspects for future research. In addition, the plethora of possible drug combinations might exhibit unknown and unacceptable toxicity profiles. Importantly, however, future research in the inflammatory microenvironment will not only improve our understanding of GBM development, progression and therapy resistance but also provide new opportunities for therapeutic strategies.
Glossary
- AP
activating protein
- BBB
blood–brain barrier
- cEBP
CCAAT-enhancer binding protein
- GSC
glioblastoma stem cell
- GBM
glioblastoma multiforme
- HIF
hypoxia inducible factor
- HuR
human antigen R
- ID
inhibitor of differentiation
- IDH
isocitrate dehydrogenase
- IR
irradiation
- LIF
leukaemia inducible factor
- MGMT
O6-methylguanine-DNA methyltransferase
- MEK
MAPK kinase
- MKK
MAPK kinase
- Mnk
MAPK-interacting kinase
- PTEN
phosphatase and tensin homologue
- SASP
senescence associated secretory phenotype
- TMZ
temozolomide
- TF
tissue factor
- TTP
tristetraprolin
Conflict of interest
All authors declare no conflict of interest.
References
- Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, Kong L-Y, et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res. 2008;14:8228–8235. doi: 10.1158/1078-0432.CCR-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allhenn D, Boushehri MAS, Lamprecht A. Drug delivery strategies for the treatment of malignant gliomas. Int J Pharm. 2012;436:299–310. doi: 10.1016/j.ijpharm.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Ancrile B, Lim K-H, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonyak MA, Moscatello DK, Wong AJ. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem. 1998;273:2817–2822. doi: 10.1074/jbc.273.5.2817. [DOI] [PubMed] [Google Scholar]
- Antonyak MA, Kenyon LC, Godwin AK, James DC, Emlet DR, Okamoto I, et al. Elevated JNK activation contributes to the pathogenesis of human brain tumors. Oncogene. 2002;21:5038–5046. doi: 10.1038/sj.onc.1205593. [DOI] [PubMed] [Google Scholar]
- Bachstetter A, Van Eldik L. The p38 MAP kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010;1:199–211. [PMC free article] [PubMed] [Google Scholar]
- Bai R-Y, Staedtke V, Riggins GJ. Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med. 2011;17:301–312. doi: 10.1016/j.molmed.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balyasnikova IV, Wainwright DA, Solomaha E, Lee G, Han Y, Thaci B, et al. Characterization and immunotherapeutic implications for a novel antibody targeting interleukin (IL)-13 Receptor α2. J Biol Chem. 2012;287:30215–30227. doi: 10.1074/jbc.M112.370015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Newcomb EW, Zagzag D, Narayana A. Therapeutic targets in malignant glioblastoma microenvironment. Semin Radiat Oncol. 2009;19:163–170. doi: 10.1016/j.semradonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker Y. Molecular immunological approaches to biotherapy of human cancers – a review, hypothesis and implications. Anticancer Res. 2006;26:1113–1134. [PubMed] [Google Scholar]
- Behin A, Hoang-Xuan K, Carpentier AF, Delattre J-Y. Primary brain tumours in adults. Lancet. 2003;361:323–331. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- Beier D, Schulz J, Beier C. Chemoresistance of glioblastoma cancer stem cells – much more complex than expected. Mol Cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- Bonavia R, Inda MM, Vandenberg S, Cheng S-Y, Nagane M, Hadwiger P, et al. EGFRvIII promotes glioma angiogenesis and growth through the NFkB, interleukin-8 pathway. Oncogene. 2011;31:4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005;7:122–133. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruyere C, Mijatovic T, Lonez C, Spiegl-Kreinecker S, Berger W, Kast R, et al. Temozolomide-induced modification of the CXC chemokine network in experimental gliomas. Int J Oncol. 2011;38:1453–1464. doi: 10.3892/ijo.2011.964. [DOI] [PubMed] [Google Scholar]
- Campbell RM, Xin X, Xia X, Ye X, Lee P, Schultz RM, et al. Effect of a selective p38-MAPK inhibitor (LY479754) on the tumor microenvironment: implication of macrophage-derived p38α-MAPK in the growth of murine P815 mastocytoma tumors. 2005. AACR Meeting Abstracts 2005: 613-c-614.
- Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–527. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN, et al. Elevated invasive potential of glioblastoma stem cells. Biochem Biophys Res Commun. 2011;406:643–648. doi: 10.1016/j.bbrc.2011.02.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie L-A, Acharya MM, Parihar VK, Nguyen A, Martirosian V, Limoli CL. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res. 2012;18:1954–1965. doi: 10.1158/1078-0432.CCR-11-2000. [DOI] [PubMed] [Google Scholar]
- Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17:6083–6096. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Han S-Y, Wang C, Su W, Harshyne L, Holgado-Madruga M, et al. c-Jun NH2-terminal kinase 2α2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 2006;66:10024–10031. doi: 10.1158/0008-5472.CAN-06-0136. [DOI] [PubMed] [Google Scholar]
- Demuth T, Reavie L, Rennert J, Nakada M, Nakada S, Hoelzinger D, et al. MAP-ing glioma invasion: mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol Cancer Ther. 2007;6:1212–1222. doi: 10.1158/1535-7163.MCT-06-0711. [DOI] [PubMed] [Google Scholar]
- Diaz L, Messersmith W, Sokoll L, Sinibaldi V, Moore S, Carducci M, et al. TNF-blockade in patients with advanced hormone refractory prostate cancer. Invest New Drugs. 2011;29:192–194. doi: 10.1007/s10637-009-9346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Dios A, Shih C, López de Uralde B, Sánchez C, del Prado M, Martín Cabrejas LM, et al. Design of potent and selective 2-aminobenzimidazole-based p38α MAP kinase inhibitors with excellent in vivo efficacy. J Med Chem. 2005;48:2270–2273. doi: 10.1021/jm048978k. [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotech. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Filippova N, Yang X, Wang Y, Gillespie GY, Langford C, King PH, et al. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res. 2011;9:648–659. doi: 10.1158/1541-7786.MCR-10-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48:85–93. doi: 10.1016/j.ejca.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. Plos ONE. 2011;6:e23902. doi: 10.1371/journal.pone.0023902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher TF, Seibel GL, Kassis S, Laydon JT, Blumenthal MJ, Lee JC, et al. Regulation of stress-induced cytokine production by pyridinylimidazoles; inhibition of CSBP kinase. Bioorg Med Chem. 1997;5:49–64. doi: 10.1016/s0968-0896(96)00212-x. [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Gessi S, Sacchetto V, Fogli E, Merighi S, Varani K, Baraldi PG, et al. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem Pharmacol. 2010;79:1483–1495. doi: 10.1016/j.bcp.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–259. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- Griffin BD, Moynagh PN. Persistent interleukin-1β signaling causes long term activation of NFκB in a promoter-specific manner in human glial cells. J Biol Chem. 2006;281:10316–10326. doi: 10.1074/jbc.M509973200. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot JF, Fuller GN, Kumar A, Piao Y, Eterovic K, Ji Y, et al. Tumour invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum LM, Schwartz R, Woska JJR, DeLeon RP, Peet GW, Warren TC, et al. Inhibition of pro-inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem Pharmacol. 2009;77:422–432. doi: 10.1016/j.bcp.2008.10.032. [DOI] [PubMed] [Google Scholar]
- Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning S, et al. Resistance of glioblastoma initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β (TGFβ) Cancer Res. 2012;72:4119–4129. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Kamenecka TM, Shin Y, Song X, Jiang R, Noel R, et al. Synthesis and SAR of novel quinazolines as potent and brain-penetrant c-jun N-terminal kinase (JNK) Inhibitors. Bioorg Med Chem Lett. 2011;21:1719–1723. doi: 10.1016/j.bmcl.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23:8306–8315. doi: 10.1128/MCB.23.22.8306-8315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose Y, Katayama M, Berger MS, Pieper RO. Cooperative function of Chk1 and p38 pathways in activating G2 arrest following exposure to temozolomide. J Neurosurg. 2004;100:1060–1065. doi: 10.3171/jns.2004.100.6.1060. [DOI] [PubMed] [Google Scholar]
- Hong T-M, Teng L-J, Shun C-T, Peng M-C, Tsai J-C. Induced interleukin-8 expression in gliomas by tumor-associated macrohages. J Neurooncol. 2009;93:289–301. doi: 10.1007/s11060-008-9786-z. [DOI] [PubMed] [Google Scholar]
- Hsi LC, Kundu S, Palomo J, Xu B, Ficco R, Vogelbaum MA, et al. Silencing IL-13Rα2 promotes glioblastoma cell death via endogenous signaling. Mol Cancer Ther. 2011;10:1149–1160. doi: 10.1158/1535-7163.MCT-10-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Cheng L, Guryanova O, Wu Q, Bao S. Cancer stem cells in glioblastoma -molecular signaling and therapeutic targeting. Protein Cell. 2010;1:638–655. doi: 10.1007/s13238-010-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia N, Konopka G, Lim K-L, Nutt CL, Bromberg JF, Frank DA, et al. Deregulation of a STAT3-Interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci. 2008a;28:5870–5878. doi: 10.1523/JNEUROSCI.5385-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia Nr, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008b;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda M, Bonavia R, Mukasa A, Narita Y, Sah D, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, Davies AM, et al. Discovery of 5-Chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J Med Chem. 2011;54:262–276. doi: 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–2444. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- Jin X, Yin J, Kim S-H, Sohn Y-W, Beck S, Lim YC, et al. EGFR-AKT-Smad signaling promotes formation of glioma stem-like cells and tumor angiogenesis by ID3-driven cytokine induction. Cancer Res. 2011;71:7125–7134. doi: 10.1158/0008-5472.CAN-11-1330. [DOI] [PubMed] [Google Scholar]
- Jin X, Kim S-H, Jeon H-M, Beck S, Sohn Y-W, Yin J, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling. Brain. 2012;135:1055–1069. doi: 10.1093/brain/aws028. [DOI] [PubMed] [Google Scholar]
- Kam AYF, Tse TTM, Kwan DHT, Wong YH. Formyl peptide receptor like 1 differentially requires mitogen-activated protein kinases for the induction of glial fibrillary acidic protein and interleukin-1α in human U87 astrocytoma cells. Cell Signal. 2007;19:2106–2117. doi: 10.1016/j.cellsig.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Kim SM, Oh JH, Park SA, Ryu CH, Lim JY, Kim D-S, et al. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells. 2010;28:2217–2228. doi: 10.1002/stem.543. [DOI] [PubMed] [Google Scholar]
- Kislin K, McDonough W, Eschbacher J, Armstrong B, Berens M. NHERF-1: modulator of glioblastoma cell migration and invasion. Neoplasia. 2009;11:377–387. doi: 10.1593/neo.81572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, et al. STAT3 Inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11:1289–1299. doi: 10.1158/1535-7163.MCT-11-0801. [DOI] [PubMed] [Google Scholar]
- Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE. Astrocytic regulation of human monocytic/microglial activation. J Immunol. 2008;181:5425–5432. doi: 10.4049/jimmunol.181.8.5425. [DOI] [PubMed] [Google Scholar]
- Krona A, Jarnum S, Salford L, Widegren B, Aman P. Oncostatin M signaling in human glioma cell lines. Oncol Rep. 2005;13:807–811. [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez P-Y, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo M, Jono H, Shinriki S, Yano S, Nakamura H, Makino K, et al. Antitumor effect of humanized anti-interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation. J Neurosurg. 2009;111:219–225. doi: 10.3171/2008.12.JNS081284. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, Olschowka J, Williams J, Hansen J, O'Banion M. TNFalpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J Neuroimmunol. 1999;95:95–106. doi: 10.1016/s0165-5728(98)00270-7. [DOI] [PubMed] [Google Scholar]
- Laberge R-M, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PLJ, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. 2012;11:569–578. doi: 10.1111/j.1474-9726.2012.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ferguson T, Pickering L, Edmonds K, James M, Thomas K, et al. A phase I/II trial of sorafenib and infliximab in advanced renal cell carcinoma. Br J Cancer. 2010;103:1149–1153. doi: 10.1038/sj.bjc.6605889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassman AB, Rossi MR, Razier JR, Abrey LE, Lieberman FS, Grefe CN, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11:7841–7850. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- Lee J-J, Kim B, Park M-J, Lee Y-S, Kim Y-N, Lee B, et al. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011;18:666–677. doi: 10.1038/cdd.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- Li R, Li G, Deng L, Liu Q, Dai J, Shen J, et al. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep. 2010;23:1553–1559. doi: 10.3892/or_00000795. [DOI] [PubMed] [Google Scholar]
- Liu Q, Li G, Li R, Shen J, He Q, Deng L, et al. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J Neurooncol. 2010;100:165–176. doi: 10.1007/s11060-010-0158-0. [DOI] [PubMed] [Google Scholar]
- Liu W-T, Lin C-H, Hsiao M, Gean P-W. Minocycline inhibits the growth of glioma by induced autophagy. Autophagy. 2011;7:165–175. doi: 10.4161/auto.7.2.14043. [DOI] [PubMed] [Google Scholar]
- Lo H-W, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14:6042–6054. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D-Y, Leung Y-M, Cheung C-W, Chen Y-R, Wong K-L. Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem Pharmacol. 2010;80:1201–1209. doi: 10.1016/j.bcp.2010.06.046. [DOI] [PubMed] [Google Scholar]
- Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104:4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw W. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem Res. 2004;29:1287–1297. doi: 10.1023/b:nere.0000023615.89699.63. [DOI] [PubMed] [Google Scholar]
- McCulloch CA, Downey GP, El-Gabalawy H. Signalling platforms that modulate the inflammatory response: new targets for drug development. Nat Rev Drug Discov. 2006;5:864–876. doi: 10.1038/nrd2109. [DOI] [PubMed] [Google Scholar]
- McFarland BC, Ma J-Y, Langford CP, Gillespie GY, Yu H, Zheng Y, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10:2384–2393. doi: 10.1158/1535-7163.MCT-11-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood. 2010;116:815–818. doi: 10.1182/blood-2009-10-250639. [DOI] [PubMed] [Google Scholar]
- Malchinkhuu E, Sato K, Horiuchi Y, Chihir M, Ohwada S, Ishiuchi S, et al. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene. 2005;24:6676–6688. doi: 10.1038/sj.onc.1208805. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Marcus H, Carpenter K, Price S, Hutchinson P. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol. 2010;97:11–23. doi: 10.1007/s11060-009-9990-5. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106:12530–12535. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, Glass R, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun. 2011;25:624–628. doi: 10.1016/j.bbi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Meini A, Sticozzi C, Massai L, Palmi M. A nitric oxide/Ca2+/calmodulin/ERK1/2 mitogen-activated protein kinase pathway is involved in the mitogenic effect of IL-1β in human astrocytoma cells. Br J Pharmacol. 2008;153:1706–1717. doi: 10.1038/bjp.2008.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa RA, Yasothan U, Kirkpatrick P. Ruxolitinib. Nat Rev Drug Discov. 2012;11:103–104. doi: 10.1038/nrd3652. [DOI] [PubMed] [Google Scholar]
- Michaud-Levesque J, Bousquet-Gagnon N, Béliveau R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp Cell Res. 2012;318:925–935. doi: 10.1016/j.yexcr.2012.02.017. [DOI] [PubMed] [Google Scholar]
- Monje M, Toda H, Palmer T. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, et al. A novel p38alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J, Cho B-K, Kim Y-S, Park S-H. Snail and Cox-2 expressions are associated with WHO tumor grade and survival rate of patients with gliomas. Neuropathology. 2010;30:224–231. doi: 10.1111/j.1440-1789.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Tumor necrosis factor α induces angiogenic factor up-regulation in malignant glioma cells. Cancer Res. 2003;63:4181–4187. [PubMed] [Google Scholar]
- Nagasawa DT, Chow F, Yew A, Kim W, Cremer N, Yang I. Temozolomide and other potential agents for the treatment of glioblastoma multiforme. Neurosurg Clin N Am. 2012;23:307–322. doi: 10.1016/j.nec.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Nitta RT, Chu AH, Wong AJ. Constitutive activity of JNK2α2 is dependent on a unique mechanism of MAPK activation. J Biol Chem. 2008;283:34935–34945. doi: 10.1074/jbc.M804970200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira L, Ruiz-Ontañon P, Vazquez-Barquero A, Moris F, Fernandez-Luna JL. The NFκB pathway: a therapeutic target in glioblastoma. Oncotarget. 2011;2:646–653. doi: 10.18632/oncotarget.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba S, Hirose Y, Kawase T, Sano H. Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced cytotoxicity in human glioma cells. J Neurooncol. 2009;95:307–316. doi: 10.1007/s11060-009-9929-x. [DOI] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C-M, Park M-J, Kwak H-J, Lee H-C, Kim M-S, Lee S-H, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66:8511–8519. doi: 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- Park M, Park I, Hur H, Kim M, Lee H, Woo S, et al. Modulation of phorbol ester -induced regulation of matrix metalloproteinases and tissue inhibitors of metalloproteinases by SB203580, a specific inhibitor of p38 mitogen-activated protein kinase. J Neurosurg. 2002;97:112–118. doi: 10.3171/jns.2002.97.1.0112. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Coppe J-P, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasi F, Facoetti A, Nano R. IL-8 and IL-6 bystander signaling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 2010;30:2769–2772. [PubMed] [Google Scholar]
- Patel M, Vogelbaum MA, Barnett GH, Jalali R, Ahluwalia MS. Molecular targeted therapy in recurrent glioblastoma: current challenges and future directions. Expert Opin Investig Drugs. 2012;21:1247–1266. doi: 10.1517/13543784.2012.703177. [DOI] [PubMed] [Google Scholar]
- Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM, Singh SK, et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem. 2009;284:3408–3417. doi: 10.1074/jbc.M807170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piette C, Deprez M, Roger T, Noël A, Foidart J-M, Munaut C. The dexamethasone-induced inhibition of proliferation, migration, and invasion in glioma cell lines is antagonized by macrophage migration inhibitory actor (MIF) and can be enhanced by specific MIF inhibitors. J Biol Chem. 2009;284:32483–32492. doi: 10.1074/jbc.M109.014589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantevin Krenitsky V, Nadolny L, Delgado M, Ayala L, Clareen SS, Hilgraf R, et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–1438. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum M, Haque S. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- Ramanan S, Kooshki M, Zhao W, Hsu F-C, Robbins ME. PPARα ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-κB and AP-1 pathways. Free Radic Biol Med. 2008;45:1695–1704. doi: 10.1016/j.freeradbiomed.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri B, Vogelbaum M. IL-8 is a mediators of NF-kB induced invasion by gliomas. J Neurooncol. 2011;101:227–235. doi: 10.1007/s11060-010-0261-2. [DOI] [PubMed] [Google Scholar]
- Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, et al. Pyrazole urea-based inhibitors of p38 MAP kinase:? from lead compound to clinical candidate. J Med Chem. 2002;45:2994–3008. doi: 10.1021/jm020057r. [DOI] [PubMed] [Google Scholar]
- Reynes G, Vila V, Martin M, Parada A, Fleitas T, Reganon E, et al. Circulating markers of angiogenesis, inflammation, and coagulation in patients with glioblastoma. J Neurooncol. 2011;102:35–41. doi: 10.1007/s11060-010-0290-x. [DOI] [PubMed] [Google Scholar]
- Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre J-Y. Primary brain tumours in adults. Lancet. 2012;379:1984–1996. doi: 10.1016/S0140-6736(11)61346-9. [DOI] [PubMed] [Google Scholar]
- Riemenschneider M, Jeuken J, Wesseling P, Reifenberger G. Molecular diagnostics of gliomas: state of the art. Acta Neuropathol. 2010;120:567–584. doi: 10.1007/s00401-010-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe J, Patil C, Hoeijmakers W, Munoz D, Raza S, et al. Persistant DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion C, Penault-Llorca Fdr KJ-L, Lemaire J-J, Jullien C, Labit-Bouvier C, et al. Interleukin-6 overexpression as a marker of malignancy in human gliomas. J Neurosurg. 2001;94:97–101. doi: 10.3171/jns.2001.94.1.0097. [DOI] [PubMed] [Google Scholar]
- Rong Y, Belozerov VE, Tucker-Burden C, Chen G, Durden DL, Olson JJ, et al. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/Activator protein-1 transcriptional activity. Cancer Res. 2009;69:2540–2549. doi: 10.1158/0008-5472.CAN-08-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J-M, Negrier S, James N, Kocak I, Hawkins R, Davis H, et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br J Cancer. 2010;103:1154–1162. doi: 10.1038/sj.bjc.6605872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidi A, Hagedorn M, Allain N, Verpelli C, Sala C, Bello L, et al. Combined targeting of interleukin-6 and vascular endothelial growth factor potently inhibits glioma growth and invasiveness. Int J Cancer. 2009;125:1054–1064. doi: 10.1002/ijc.24380. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Yong V. Inflammatory cytokine modulation of matrix metalloproteinase expression and invasiveness of glioma cells in a 3-dimensional collagen matrix. J Neurooncol. 2009;91:157–164. doi: 10.1007/s11060-008-9695-1. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Tamura M, Hasegawa M, Ishiuchi S, Hirato J, Nakazato Y. Expression of interleukin-1beta mRNA and protein in human gliomas assessed by RT-PCR and immunohistochemistry. J Neuropathol Exp Neurol. 1998;57:653–663. doi: 10.1097/00005072-199807000-00002. [DOI] [PubMed] [Google Scholar]
- Schnegg CI, Kooshki M, Hsu F-C, Sui G, Robbins ME. PPARδ prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-κB and inhibition of the PKCα/MEK1/2/ERK1/2/AP-1 pathway. Free Radic Biol Med. 2012;52:1734–1743. doi: 10.1016/j.freeradbiomed.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft C, Priester M, Polacin M, Schroder K, Seifert V, Kogel D, et al. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J Neurooncol. 2011;101:393–403. doi: 10.1007/s11060-010-0273-y. [DOI] [PubMed] [Google Scholar]
- Shao C, Folkard M, Prise K. Role of TGF-beta1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene. 2008;27:434–440. doi: 10.1038/sj.onc.1210653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Dixit D, Ghosh S, Sen E. COX-2 regulates the proliferation of glioma stem like cells. Neurochem Int. 2011a;59:567–571. doi: 10.1016/j.neuint.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Sharma V, Dixit D, Koul N, Mehta VS, Sen E. Ras regulates interleukin-1b-induced HIF-1alpha transcriptional activity in glioblastoma. J Mol Med. 2011b;89:123–136. doi: 10.1007/s00109-010-0683-5. [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- Singh S, Hawkins C, Clarke I, Squire J, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Sliwa M, Markovic D, Gabrusiewicz K, Synowitz M, Glass R, Zawadzka M, et al. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain. 2007;130:476–489. doi: 10.1093/brain/awl263. [DOI] [PubMed] [Google Scholar]
- Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 2010;29:243–248. doi: 10.1007/s10555-010-9227-2. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–330. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Spooren A, Mestdagh P, Rondou P, Kolmus K, Haegeman G, Gerlo S. IL-1β potently stabilizes IL-6 mRNA in human astrocytes. Biochem Pharmacol. 2011;81:1004–1015. doi: 10.1016/j.bcp.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z, et al. JSI-124 inhibits glioblastoma multiforme cell proliferation through G2/M cell cycle arrest and apoptosis augmentation. Cancer Biol Ther. 2008;7:1243–1249. doi: 10.4161/cbt.7.8.6263. [DOI] [PubMed] [Google Scholar]
- Sun S, Wang Q, Giang A, Cheng C, Soo C, Wang C-Y, et al. Knockdown of CypA inhibits interleukin-8 (IL-8) and IL-8-mediated proliferation and tumor growth of glioblastoma cells through down-regulated NF-κB. J Neurooncol. 2011;101:1–14. doi: 10.1007/s11060-010-0220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, et al. Tristetraprolin down-regulates interleukin-8 and vascular endothelial growth factor in malignant glioma cells. Cancer Res. 2008;68:674–682. doi: 10.1158/0008-5472.CAN-07-2751. [DOI] [PubMed] [Google Scholar]
- Tada M, Diserens A, Desbaillets I, Jaufeerally R, Hamou M, de Tribolet N. Production of interleukin-1 antagonist by human glioblastoma cells in vitro and in vivo. J Neuroimmunol. 1994;50:187–194. doi: 10.1016/0165-5728(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Matsushima-Nishiwaki R, Yamaguchi S, Iida H, Dohi S, Kozawa O. Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J Neuroinflammation. 2010;7:16. doi: 10.1186/1742-2094-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Kozawa O, Iida H. Midazolam suppresses interleukin-1beta-induced interleukin-6 release from rat glial cells. J Neuroinflammation. 2011;8:68. doi: 10.1186/1742-2094-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchirkov A, Rolhion C, Bertrand S, Dore JF, Dubost JJ, Verrelle P. IL-6 gene amplification and expression in human glioblastomas. Br J Cancer. 2001;85:518–522. doi: 10.1054/bjoc.2001.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchirkov A, Khalil T, Chautard E, Mokhtari K, Veronese L, Irthum B, et al. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer. 2007;96:474–476. doi: 10.1038/sj.bjc.6603586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuiki H, Tnani M, Okamoto I, Kenyon LC, Emlet DR, Holgado-Madruga M, et al. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63:250–255. [PubMed] [Google Scholar]
- Ueda T, Sasaki M, Elia AJ, Chio IIC, Hamada K, Fukunaga R, et al. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A. 2010;107:13984–13990. doi: 10.1073/pnas.1008136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairano M, Graziani G, Tentori L, Tringali G, Navarra P, Russo CD. Primary cultures of microglial cells for testing toxicity of anticancer drugs. Toxicol Lett. 2004;148:91–94. doi: 10.1016/j.toxlet.2003.12.058. [DOI] [PubMed] [Google Scholar]
- Verstovsek S, Manshouri T, Quintás-Cardama A, Harris D, Cortes J, Giles FJ, et al. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin Cancer Res. 2008;14:788–796. doi: 10.1158/1078-0432.CCR-07-0524. [DOI] [PubMed] [Google Scholar]
- Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Deimling A, Korshunov A, Hartmann C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol. 2011;21:74–87. doi: 10.1111/j.1750-3639.2010.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Wakabayashi T, Kambe F, Cao X, Murakami R, Mitsuyama H, Nagaya T, et al. Inhibitory effects of cyclosporin A on calcium mobilization-dependent interleukin-8 expression and invasive potential of human glioblastoma U251MG cells. Oncogene. 2004;23:6924–6932. doi: 10.1038/sj.onc.1207778. [DOI] [PubMed] [Google Scholar]
- Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Balivada S, Shrestha T, Bossmann S, Pyle M, et al. Interleukin-1beta and transforming growth factor-beta cooperate to induce neurosphere formation and increase tumorigenicity of adherent LN-229 glioma cells. Stem Cell Res Ther. 2012;3:5. doi: 10.1186/scrt96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh DJJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A, Afanasieva TA, et al. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–3316. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- Weissenberger J, Priester M, Bernreuther C, Rakel S, Glatzel M, Seifert V, et al. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin Cancer Res. 2010;16:5781–5795. doi: 10.1158/1078-0432.CCR-10-0446. [DOI] [PubMed] [Google Scholar]
- Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- Xu K, Shu H-KG. EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 2007;67:6121–6129. doi: 10.1158/0008-5472.CAN-07-0141. [DOI] [PubMed] [Google Scholar]
- Yamanaka R, Tanaka R, Saitoh T, Okoshi S. Cytokine gene expression on glioma cell lines and specimens. J Neurooncol. 1994;21:243–247. doi: 10.1007/BF01063773. [DOI] [PubMed] [Google Scholar]
- Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells via dephosphorylation of STAT3. Mol Cancer Ther. 2010;9:953–962. doi: 10.1158/1535-7163.MCT-09-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung Y, Bryce N, Adams S, Braidy N, Konayagi M, McDonald K, et al. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and invasiveness of human U251 glioblastoma cells. J Neurooncol. 2012;109:35–44. doi: 10.1007/s11060-012-0875-7. [DOI] [PubMed] [Google Scholar]
- Yoshino Y, Aoyagi M, Tamaki M, Duan L, Morimoto T, Ohno K. Activation of p38 MAPK and/or JNK contributes to increased levels of VEGF secretion in humanmalignant glioma cells. Int J Oncol. 2006;29:981–987. [PubMed] [Google Scholar]
- Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33:312–319. doi: 10.1093/carcin/bgr289. [DOI] [PubMed] [Google Scholar]