

Abstract
Background and Purpose
T16Ainh-A01 is a recently identified inhibitor of the calcium-activated chloride channel TMEM16A. The aim of this study was to test the efficacy of T16Ainh-A01 for inhibition of calcium-activated chloride channels in vascular smooth muscle and consequent effects on vascular tone.
Experimental Approach
Single channel and whole cell patch clamp was performed on single smooth muscle cells from rabbit pulmonary artery and mouse thoracic aorta. Isometric tension studies were performed on mouse thoracic aorta and mesenteric artery as well as human abdominal visceral adipose artery.
Key Results
In rabbit pulmonary artery myocytes T16Ainh-A01 (1–30 μM) inhibited single calcium (Ca2+)-activated chloride (Cl−) channels and whole cell currents activated by 500 nM free Ca2+. Similar effects were observed for single Ca2+-activated Cl− channels in mouse thoracic aorta, and in both cell types, channel activity was abolished by two antisera raised against TMEM16A but not by a bestrophin antibody. The TMEM16A potentiator, Fact (10 μM), increased single channel and whole cell Ca2+-activated Cl− currents in rabbit pulmonary arteries. In isometric tension studies, T16Ainh-A01 relaxed mouse thoracic aorta pre-contracted with methoxamine with an IC50 of 1.6 μM and suppressed the methoxamine concentration–effect curve. T16Ainh-A01 did not affect the maximal contraction produced by 60 mM KCl and the relaxant effect of 10 μM T16Ainh-A01 was not altered by incubation of mouse thoracic aorta in a cocktail of potassium (K+) channel blockers. T16Ainh-A01 (10 μM) also relaxed human visceral adipose arteries by 88 ± 3%.
Conclusions and Implications
T16Ainh-A01 blocks calcium-activated chloride channels in vascular smooth muscle cells and relaxes murine and human blood vessels.
Keywords: calcium-activated chloride channels, TMEM16A, anoctamins, Ano1, vascular smooth muscle, anti-hypertensive, patch clamp
Introduction
Calcium (Ca2+)-activated chloride (Cl−) currents (IClCa) have been recorded from a number of different cell types, including epithelial cells, secretory glands, cardiomyocytes, neurones and smooth muscle cells. Research into the physiological and pathophysiological roles of the underlying Ca2+-activated Cl− channels (CACCs) has been hampered by the lack of molecular identity and the poor selectivity of existing Cl− channel blockers such as niflumic acid (see Greenwood and Leblanc, 2007). However, the identification that expression products of the gene TMEM16A (Ano1) generate a CACC with biophysical properties identical to native CACCs (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008) has allowed the development through small-molecule screening of a new generation of reagents for investigating CACC (Namkung et al., 2010; 2011a). These efforts led to the identification of T16Ainh-A01, a compound shown to inhibit CACC activity in TMEM16A-transfected FRT cells with an IC50 of ∼1 μM (Namkung et al., 2011a). In addition, Namkung et al. (2011b) identified two agents that either activate TMEM16A independent of Ca2+ (Eact) or potentiate activated TMEM16A channels (Fact).
In vascular smooth muscle cells, Cl− ions are accumulated so that opening of CACCs leads to membrane depolarization sufficient to increase the open probability of voltage-dependent Ca2+ channels (VDCCs; Large and Wang, 1996; Kitamura and Yamazaki, 2001; Leblanc et al., 2005). Consequently, CACCs are generally considered as a mechanism for vasoconstrictors to promote membrane contraction, but the lack of reliable pharmacological tools has hampered the research on this aspect. However, TMEM16A expression was recently revealed in vascular smooth muscle that exhibits robust IClCa (Davis et al., 2010; Manoury et al., 2010; Sones et al., 2010; Thomas-Gatewood et al., 2011). Moreover, TMEM16A down-regulation of TMEM16A transcripts by interference RNA strategies inhibited IClCa in pulmonary artery (PA) smooth muscle cells in short-term culture (Manoury et al., 2010). However, there is no information on the contribution of TMEM16A to CACCs in freshly dispersed smooth muscle cells or whether TMEM16A is involved in vascular contractions. The aims of the present study were twofold. Firstly, to investigate the effect of T16Ainh-A01 and Fact on whole cell and single channel IClCa in freshly dispersed rabbit PA smooth muscle cells, which have been studied extensively by our group (Greenwood et al., 2001; 2004; Piper and Large, 2003), as well as mouse thoracic aortic (TA) smooth muscle cells that express TMEM16A. Secondly, to ascertain the effectiveness of T16Ainh-A01 on the contractility of mouse and human blood vessels in comparison to the conventional CACC blocker niflumic acid.
Methods
Initial electrophysiological experiments were performed on single smooth muscle cells isolated from the main PA from New Zealand White rabbits, killed by pentobarbitone sodium overdose in accordance with Schedule 1 of the United Kingdom Animals Act (1986). Additional electrophysiological and functional experiments were performed using TA and mesenteric arteries (MAs) from BALB/c mice (6–8 weeks old) killed by cervical dislocation (Schedule 1). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., #b1001; McGrath et al., #b1002). In addition, human visceral adipose tissue samples were taken from elective bariatric surgery at St George's Hospital, London (local ethics approval, 08/H0803/84). Arteries used in the study were dissected free from any damaged or pathologically altered tissue by the operating surgeon and micro-dissected in the laboratory within 1 h. Arteries were identified as the muscular vessel running as a pair with thin walled veins (Mulvany and Halpern, 1977).
Single cell dispersal
Initial electrophysiological experiments to characterize the effects of T16Ainh-A01 and Fact were performed in PA smooth muscle cells where Cl− currents due to the activation of CACCs (IClCa) have been studied extensively at both whole cell and single channel levels (e.g. Greenwood et al., 2001; 2004; Piper et al., 2002; Piper and Large, 2003; Angermann et al., 2006). Single channel experiments were also performed on mouse TA smooth muscle cells where expression of TMEM16A protein has been identified and whole cell IClCa have been characterized (Davis et al., 2010). Rabbit PA myocytes were isolated by incubating segments of artery in Ca2+-free physiological salt solution (PSS) containing 0.5 mg·mL−1 papain, 1 mM DTT and 2 mg·mL−1 BSA overnight at 4°C, followed by trituration through a wide bore glass pipette. TA myocytes were prepared by incubating segments of tissue in PSS containing 50 μM CaCl2, 1 mg·mL−1 collagenase type X and 1 mg·mL−1 protease type 2A for 25 min.
Whole cell currents
Similar to previous studies (e.g. Greenwood et al., 2001; 2004; Piper et al., 2002; Angermann et al., 2006), whole cell IClCa were evoked immediately upon membrane rupture when the pipette solution contained a fixed concentration of [Ca2+]. The pipette solution had the following composition (in mM): TEA (20), CsCl (106), HEPES-CsOH (10, pH 7.2), BAPTA (10), ATP.Mg (5) and GTP.diNa (0.2). To this solution, 4.2, 6.8 or 7.08 mM CaCl2 were added to achieve a free [Ca2+] of 100, 250 and 500 nM respectively. The bathing solution had the following composition (in mM): NaCl (126), HEPES-NaOH (10, pH 7.35), TEA (8.4), glucose (20), MgCl2 (1.2) and CaCl2 (1.8). Voltage protocols were the same as used previously (Greenwood et al., 2001; 2004; Angermann et al., 2006). The effect of T16Ainh-A01 (1–30 μM) was studied on IClCa evoked by 500 nM free Ca2+ only, whereas the effect of Fact was studied on IClCa evoked by a range of [Ca2+] (20–500 nM) to allow comparison with the previous work (Namkung et al., 2011b).
Single channel recordings
Single channel IClCa have been defined previously in rabbit PA myocytes by Piper and Large (2003). With 500 nM Ca2+ activating [Ca2+], channel activity is characterized by the existence of two sub-conductance states (1.8 and 1.2 pS), but a single 3.5 pS state dominates at lower [Ca2+] (Piper and Large, 2003). The effects of T16Ainh-A01, the less potent TMEM16A blocker tannic acid (Namkung et al., 2010) and Fact on single channel IClCa were studied in outside-out patches using a patch pipette solution containing (mM) NMDG-Cl [126, equimolar addition of N-methyl-D-glucamine (NMDG) and HCl], HEPES (10), MgCl2 (1.2), EGTA (0.1) and MgATP (1), pH adjusted to 7.2 with NMDG or HCl. The bathing solution for outside-out patches contained (mM) NMDG-Cl (126), CaCl2 (1.5), MgCl2 1.2 and HEPES (10), pH adjusted to 7.2 with HCl. The effect of TMEM16A-specific antibodies on IClCa was studied using the inside-out configuration with a patch pipette solution containing (mM) NMDG-Cl, (126), MgCl2 (1.2) CaCl2 (10) and HEPES (10), pH set at 7.2 with NMDG or HCl. The bathing (intracellular) solution had the same composition as the patch pipette solution used in outside-out patches. Single Cl− channel currents were recorded and analysed according to protocols detailed in Piper and Large (2003).
Immunopharmacological studies
Previously, Manoury et al. (2010) showed that siRNA targeted against TMEM16A decreased whole cell IClCa in rat PA cells in short-term culture. We used an antibody-based approach to ascertain if TMEM16A contributes to native IClCa in freshly dispersed cells. Consequently, we used an antibody-based approach that has been used previously to characterize TRPC channels in our laboratory (e.g. Albert et al., 2009) to ascertain if TMEM16A contributes to native IClCa in freshly dispersed cells. In an attempt to circumvent off target effects, we used two different antibodies raised in different hosts (rabbit and goat) against different epitopes and we controlled for non-specific binding by comparing the effect of serum from non-immunized rabbits or goats (see Johnstone and Thorpe, 1996). In addition, an antibody against bestrophin-3, which underlies the cGMP-dependent CACC (Matchkov et al., 2008), was also applied to excised patches as a control. Single channels were recorded in the inside-out configuration with bath [Ca2+] set to 500 nM in both PA and mouse TA cells. In single channel recordings using rabbit PA cells either sc-69343, a goat polyclonal antibody targeted to an internal epitope of human TMEM16A (Santa Cruz Biotechnology, Insight Biotech, Wembley, UK) or the bestrophin-3 antibody (ab94904, Abcam Ltd., Cambridge, UK) was applied. In the case of single channel electrophysiology using mouse cells, both sc-69343 and ab72984, a rabbit polyclonal raised against a peptide derived from the N-terminal region of human TMEM16A (Abcam) was applied. We have characterized ab72984 by Western blot in our previous work (Davis et al., 2010).
Isometric tension recordings
The functional effects of T16Ainh-A01, tannic acid and niflumic acid were investigated in segments of mouse TA and MA as well as human visceral adipose arteries. All segments were transferred to Krebs' solution and then mounted on a wire myograph (DMT, Aarhus, Denmark). All resistance vessels were normalized to 90% of the diameter of the vessel at 100 mmHg using a passive stretch/tension relationship (Mulvany and Halpern, 1977), whereas the mouse TA segments were set to an initial tension of 12 mN (Yeung et al., 2007). After a 30 min period of equilibration, segments were challenged twice with 60 mM KCl for 5 min. After washout and stabilization of tension, the vessels were then pre-contracted with either 3 μM methoxamine (TA) or 100 nM U46619 (MA/human arteries; Tocris, Bristol, UK) to obtain robust, stable contractions. After the response had stabilized either dimethyl sulfoxide (DMSO) or the following Cl− channel blockers – niflumic acid, T16Ainh-A01 or tannic acid – were applied at increasing concentrations to generate dose–response relationships. A second protocol was also employed in the mouse TA where increasing concentrations of methoxamine (100 nM–10 μM) were applied cumulatively. Tissues were then washed with Krebs' solution and incubated in either DMSO, T16Ainh-A01 (5 μM), Fact (10 μM), tannic acid (30 μM) or niflumic acid (10 and 100 μM) for 30 min before a second methoxamine concentration–effect curve was constructed in the presence of these agents. The composition of the Kreb's solution (mM) is as follows: NaCl (125), KCl (4.6), CaCl2 (2.5), NaHCO3 (25.4), Na2HPO4 (1), MgSO4 (0.6) and glucose (10) bubbled with 95%O2/5%CO2 at 37°C. PSS for dispersal (mM) is as follows: NaCl (126), KCl (6), glucose (10), HEPES (11), MgCl2 (1.2) and CaCl2 (1.5), with pH adjusted to 7.2 with NaOH.
Endpoint PCR
Total RNA was extracted from arteries, as well as pools of single smooth muscle cells from either rabbit PA or mouse TA, and reverse transcribed to cDNA as described previously (Davis et al., 2010). Primer sequences and expected amplicon size are found in the Online Supplement (Table S1).
Immunohistochemistry
Protein detection in slices of artery was performed as described previously (Davis et al., 2010; Ng et al., #b1003) and further details are included in the Online Supplement.
Immunocytochemistry
TMEM16A protein was detected in single smooth muscle cells from mouse TA using methodology described previously (Davis et al., 2010). Cells were imaged using a Zeiss confocal microscope (Zeiss, Welwyn, UK) and membrane localization of TMEM16A protein in any Z-section was determined by overlap of fluorescence from an α1 sodium (Na+)–potassium (K+) ATPase antibody (Abcam; ab7671). As no control blocking peptide was supplied with the TMEM16A antibody, we used a boiled version of this sample in accordance with the previous work in cerebral arteries (Thomas-Gatewood et al., 2011).
Materials
All chemicals were purchased from Sigma (Poole, Dorset, UK), unless otherwise stated.
Statistical analysis
All data are shown as the mean of n animals/patients (where appropriate) ± SEM. Statistical significance was determined by unpaired Student's t-test using GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA). All nomenclature is consistent with Alexander et al. (2011).
Results
In agreement with previous studies (Davis et al., 2010; Sones et al., 2010), expression of TMEM16A was identified in whole mouse TA and MA by endpoint PCR (Figure 1A). Amplicons for TMEM16A were also identified using RNA extracted from a pool of smooth muscle cells from both rabbit PA and mouse TA, as well as in all samples of human visceral adipose artery studied (confirmed by sequencing of product). Translation of TMEM16A gene to protein was confirmed by immunohistochemistry (Figure 1B) using an antibody that has been characterized thoroughly in overexpression systems and native protein by Davis et al. (2010). Figure 1B shows that TMEM16A localized to the smooth muscle region in human blood vessels as defined by the smooth muscle α-actin staining. In immunocytochemical experiments of enzymatically isolated smooth muscle cells, TMEM16A expression products were observed in the cytoplasm but predominantly co-localized with α1 sodium potassium ATPase, suggesting membrane localization (Figure 1C).
Figure 1.

Identification of TMEM16A expression in mouse and human arteries. (A) Endpoint PCR analysis of TMEM16A amplicons from whole mouse TA (mTA) and mesenteric artery (mMA) tissue (Ai), enzymatically isolated smooth muscle cells from rabbit pulmonary artery (rbPA) and mTA (Aii) and human visceral adipose arteries (hVAA; Aiii) confirms expression of TMEM16A gene in all tissues tested. The # denotes a different RNA extraction from new material. NTC = no template control and M = 100 base pair (bp) ladder. (B) Immunohistochemical analysis of transverse sections of mTA, mMA and hVAA show specific TMEM16A expression (red staining/Alexa Fluor 546) in the smooth muscle α-actin layer (green staining/Alexa Fluor 488) of all tissues tested. All images presented were exposure time-matched and are representative of four separate experiments, with four different animal/patient samples. Staining surrounding the vessels represents extraneous fat and (in the case of mMA) the paired vein can also be seen and were used to aid in the identification of the arteries. (C) Immunocytochemical analysis of enzymatically isolated mTA smooth muscle cells shows distinct overlap of TMEM16A expression (red staining/Alexa Fluor 546) with α1 sodium potassium ATPase (green staining/Alexa Fluor 488; a plasma membrane marker).
Electrophysiology
Consistent with previous studies (Greenwood et al., 2001; 2004; Angermann et al., 2006), distinctive whole cell IClCa with characteristic voltage-dependent kinetics were evoked immediately upon membrane rupture with a pipette solution containing 500 nM free Ca2+. These currents exhibited considerable rundown within the first 2 min of recording (see Angermann et al., 2006) but then remained stable (data not shown). Application of 10 μM T16Ainh-A01 consistently inhibited a component of the evoked current after an initial lag period of a few minutes (Figure 2A,B) and this was readily reversed upon washout (Figure 2A). Inhibition of the evoked current was also observed with 1 μM T16Ainh-A01 (Figure 2D), while 30 μM T16Ainh-A01 had no greater effect than 10 μM T16Ainh-A01 (Figure 2D). Previously, we found that niflumic acid had inhibitory and stimulatory effects on whole cell IClCa in rabbit PA smooth muscle cells that were manifested as an increase in currents at negative test potentials and enhancement of the current to approximately 200% of pre-drug levels upon washout (Piper et al., 2002; see also Supporting Information Figure S1). However, no stimulatory effect was observed with T16Ainh-A01 either in the presence of the agent or upon washout (Figure 2, current after 5 min washout of 10 μM T16Ainh-A01 was 105 ± 5% of pre-drug levels, n = 4). In contrast to T16Ainh-A01, the TMEM16A potentiator Fact (10 μM) had no significant effect on IClCa evoked by 500 nM free Ca2+ (Figure 3A) with the current at +90 mV being 15. ± 7 and 14.5 ± 6 pA·pF−1 in the absence and presence of Fact respectively. However, when IClCa was evoked by pipette solutions containing 100 or 250 nM free Ca2+, the currents were enhanced considerably (Figure 3B,C).
Figure 2.

Effect of T16Ainh-A01 on whole cell IClCa in rabbit PA smooth muscle cells. (A) shows a representative time course for the effect of 10 μM T16Ainh-A01 and subsequent washout on whole cell IClCa recorded at +70 mV. The gap represents the application of a current-voltage protocol. (B) shows representative currents recorded at voltages between −100 and +100 mV in the absence (Bi) and presence (Bii) of 10 μM T16Ainh-A01. (C) shows the T16Ainh-A01-sensitive current determined as (Bi) and (Bii). The mean T16Ainh-A01-sensitive current at different concentrations is shown in (D). Each point is the mean of 4–6 cells ± SEM. *P < 0.05; **P < 0.01.
Figure 3.
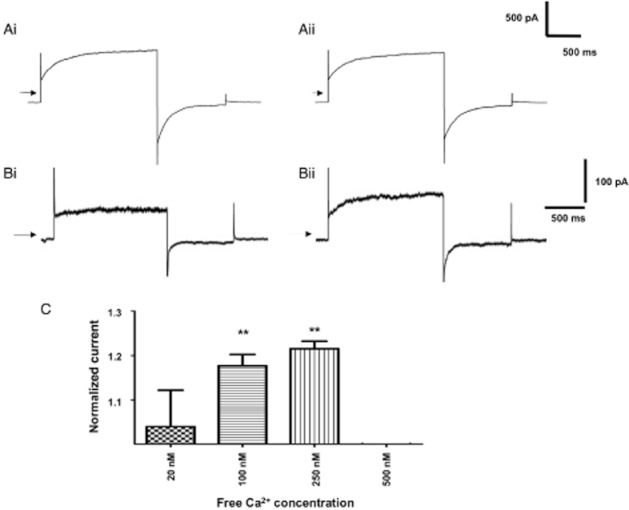
Effect of Fact on whole cell IClCa in rabbit PA smooth muscle cells. (A) shows currents evoked by 500 nM Ca2+ before (Ai) and after application of 10 μM Fact for 5 min (Aii). (B) shows currents evoked by 100 nM Ca2+ before (Bi) and after application of 10 μM Fact for 5 min (Bii). (C) shows the mean increase in current produced by 10 μM Fact at different [Ca2+]. Current in the presence of the drug was normalized to the pre-drug amplitude. Each bar represents the mean of 4–7 cells ± SEM; **P < 0.01, paired Student's t-test.
Single channel studies
In outside-out patches from PA smooth muscle cells, no channel activity was present when the pipette solution was Ca2+ free (n = 5, Figure 4A). However, inclusion of 500 nM free Ca2+ in the patch pipette solution activated single IClCa channel currents (Figure 4B), which had a mean open probability (NPo) of 0.68 ± 0.07 (n = 12) at −100 mV. IClCa channel currents evoked by 500 nM free Ca2+i had two amplitudes of −0.16 ± 0.02 pA (n = 12) and −0.29 ± 0.02 pA (n = 12) at −100 mV. These two channel amplitudes are consistent with the two sub-conductance levels of 1.8 and 3.5 pS reported for IClCa in these cells previously (Piper and Large, 2003). Similar channel activity was evoked in inside-out patches by a bath solution containing 500 nM Ca2+. Figure 4C shows that co-application of an anti-TMEM16A antibody (1:100 sc-69343), but not an anti-Bestrophin-3 antibody (1:200; ab94904), significantly reduced IClCa channel activity evoked by 500 nM Ca2+I, with NPo decreasing from 0.61 ± 0.07 to 0.03 ± 0.01 (n = 7). In control experiments on smooth muscle cells from rabbit MA, bath application of the anti-bestrophin-3 antibody blocked IClcGMP channel activity by 95 ± 2% (n = 5, Figure 4D).
Figure 4.
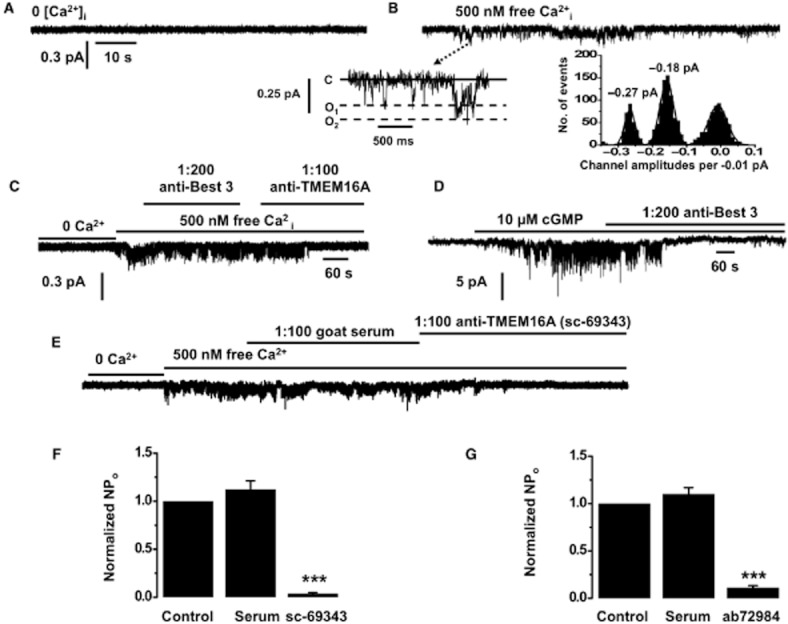
Single channel recordings in rabbit PA and mouse TA smooth muscle cells. IClCa Activity in outside-out patches rabbit PA smooth muscle cells is evoked following inclusion of 500 nM free Ca2+i in the patch pipette solution (A,B). In the insert, (C) denotes closed state, O1 and O2 are different open states. Subpanels in (B) show the different channel amplitudes of individual opening on a faster time scale and their amplitude histogram. (C) shows a representative effect of antisera raised against TMEM16A (sc-69343). Antisera against bestrophin-3 had no effect on this channel activity (D) but inhibited cGMP-stimulated IClCa channel activity in rabbit mesenteric artery cells. (E) shows a representative trace for the effect of a TMEM16A antibody (sc-69343) on single Cl− channel activity in mouse TA smooth muscle cells. (F,G) show the mean effect of both TMEM16A antibodies, sc-69343 (1:100) and ab72984 (1:500) on IClCa channel activity in inside-out patches from mouse TA cells evoked by applying 500 nM free Ca2+ (n = 5). Note that the corresponding rabbit and goat non-immune serum had no effect on IClCa channel activity. *P < 0.05; ***P < 0.001.
Identical single channel activity was recorded in smooth muscle cells from mouse TA upon application of 500 nM Ca2+i (Figure 4E). Application of the TMEM16A antibody sc-69343 (raised in goat) inhibited the activity of IClCa channels by 90 ± 3% (n = 5), similar to the data in rabbit PA (Figure 4C). A second TMEM16A antibody (ab72984, raised in rabbit) also inhibited channel activity markedly (97 ± 1%; Figure 4G). In contrast, serum from non-immunized goat or rabbit had no effect on IClCa channel activity (Figure 4F,G). Consequently, IClCa channel activity in rabbit PA and mouse TA was decreased considerably by two TMEM16A antibodies raised in different hosts and against different epitopes.
Effects of pharmacological agents on single channel IClCa
Experiments were performed to determine the effect of the TMEM16A modulators on single channel IClCa in rabbit PA and mouse TA smooth muscles. In outside-out recordings from rabbit PA smooth muscle cells application of 10 μM T16Ainh-01 inhibited IClCa channel activity evoked by 500 nM free Ca2+i by 93 ± 2% (n = 6) at −100 mV, which was partially reversed upon washout (Figure 5A,B). IClCa channel activity was also blocked by bath application of 30 μM tannic acid, which is a less potent blocker of TMEM16A channels (Namkung et al., 2010; 2011a). Figure 5C shows that T16Ainh-01 also inhibited the single channel activity in mouse TA smooth muscle cells that was readily reversed upon washout.
Figure 5.
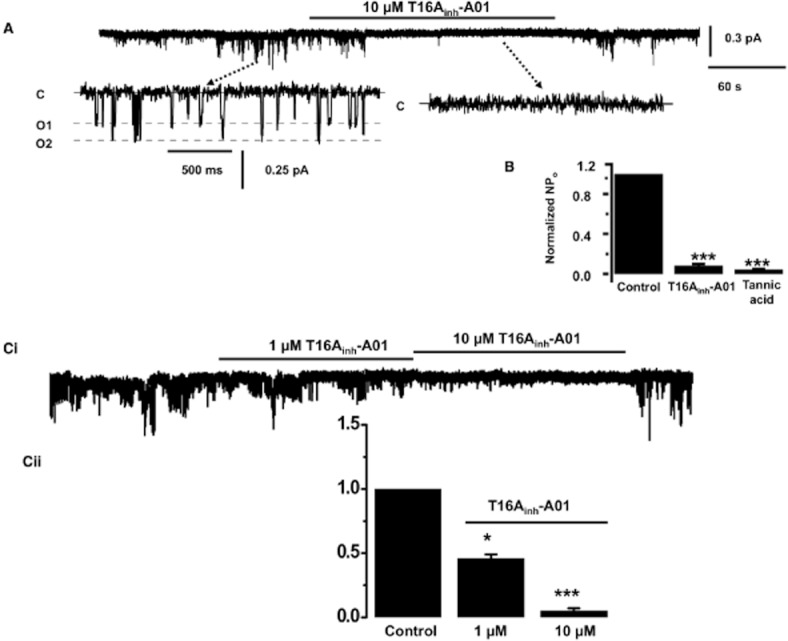
Effect of T16Ainh-A01 on single channel recordings in rabbit PA and mouse TA smooth muscle cells. (A) shows an example of the inhibitory effect of 10 μM T16Ainh-A01 on IClCa activity in outside-out patches from rabbit PA cells evoked following inclusion of 500 nM free Ca2+i in the patch pipette solution. In the insert, (C) denotes closed state, O1 and O2 are different open states. Subpanels show the different channel amplitudes of individual opening on a faster time scale and their amplitude histogram. (B) shows the mean change in NPo produced by 10 μM T16Ainh-A01 and 30 μM tannic acid. (C) shows the concentration-dependent inhibition produced by 1 and 10 μM T16Ainh-A01 (n = 6) of channel activity evoked by 500 nM Ca2+ in mouse TA smooth muscle cells. *P < 0.05; ***P < 0.001.
A final series of experiments investigated the effect of the TMEM16A potentiator Fact on single channel IClCa from rabbit PA smooth muscle cells activated with either 100 or 500 nM free Ca2+. With the lower [Ca2+], single channel activity was considerably less (NPo was 0.65 ± 0.1 vs. 0.14 ± 0.02, n = 5) and was dominated by transitions to the larger sub-conductance state (cf. Figures 5A and 6A). In agreement with the whole cell data, Fact had no significant effect upon single channel activity evoked by 500 nM Ca2+ (NPo was 0.65 ± 0.12 and 0.76 ± 0.08 before and after Fact, n = 3) but produced a marked increase in channel activity evoked by 100 nM Ca2+ (Figure 6A,Cii).
Figure 6.

Effect of Fact on single channel recordings in rabbit PA smooth muscle cells. (A) shows an example of the effect of 10 μM Fact on single channel evoked with 100 nM Ca2+ in the pipette solution. (B) shows amplitude histograms in the absence (i) and presence of 10 μM Fact (ii). Mean channel activity ± Fact for 500 nM and 100 nM Ca2+ is shown in (Ci) and (Cii) respectively (n = 3–5).
Whole tissue functional studies
The simple working hypothesis is that TMEM16A functions as a CACC that when opened produces membrane depolarization sufficient to increase Ca2+ influx through VDCC. Consequently, blockers of TMEM16A should relax pre-contracted vessels and suppress vascular contractions. Pre-contraction of mouse TA by application of the α1-adrenoceptor agonist methoxamine produced robust contractions that were inhibited in a concentration-dependent manner by T16Ainh-A01 with an apparent IC50 of 1.7 μM derived from the averaged data (Figure 7A,D), whereas the equivalent volume of DMSO had negligible effect (Figure 7C; mean relaxation was 2 ± 1%, n = 5). The effect of T16Ainh-A01 was relatively slow to develop, with a maximum effect occurring after 18 ± 4 min for 10 μM (n = 8) but was well maintained for at least 60 min (data not shown). The prototypic CACC blocker niflumic acid (Figure 7B,D), currently considered the most effective blocker of IClCa (see Greenwood and Leblanc, 2007), inhibited contractions in a concentration-dependent manner, although at higher concentrations compared with T16Ainh-A01 (Figure 7A,B). Tannic acid was a less effective relaxant of mouse TA segments (Figure 7D), although subsequent application of niflumic acid or T16Ainh-A01 produced marked relaxation of the tissues in the continued presence of tannic acid (n = 4, data not shown).
Figure 7.

Effect of T16Ainh-A01 in isometric tension recordings from murine TA. Representative effects of 1 μM and 10 μM T16Ainh-A01 (A), niflumic acid (10–100 μM, B) and equivalent DMSO on TA pre-contracted by 3 μM methoxamine (applied at •). (D) shows relaxant concentration–effect curves for niflumic acid, T16Ainh-01 and tannic acid on pre-contracted mouse TA. All data are the mean of 5–8 experiments ± SEM. (E) shows the mean effect of cumulative application of methoxamine in the presence of DMSO, 5 μM T16Ainh-A01, 30 μM tannic acid and 10 μM niflumic acid. All points are the mean of 5 experiments ± SEM.
Incubation of TA segments with a submaximal concentration of T16Ainh-A01 (5 μM) suppressed the subsequent methoxamine concentration–effect curve to a greater extent than 30 μM tannic acid or 10 μM niflumic acid (Figure 7E). In contrast, Fact had no significant effect on the methoxamine-induced contractions (n = 5, data not shown). Furthermore, 100 μM niflumic acid abrogated the methoxamine concentration-dependent contractions (data not shown) but also abolished contractions induced by 60 mM KCl, suggesting a direct effect on VDCCs. T16Ainh-A01 had no significant effect on contractions produced by application of 60 mM KCl (1.93 ± 0.6 vs. 1.7 ± 0.5 mN, n = 5) and the inhibitory effect of 10 μM T16Ainh-A01 was not affected by incubation of the TA with a cocktail of K+ channel blockers. Thus, the relaxation to 10 μM T16Ainh-A01 in 10 μM glibenclamide (ATP-sensitive K+ channel blocker), 10 μM paxilline (BKCa blocker) and 1 μM linopirdine (Kv7 channel blocker, Yeung et al., 2007) was 82 ± 6% (n = 4) compared with 74 ± 5% in the absence of these agents (n = 8). These studies suggest that T16Ainh-A01 does not relax mouse TA through direct block of VDCCs or activation of K+ channels.
Effect of T16Ainh-A01 in mouse and human mesenteric arteries
Having established that T16Ainh-A01 was an effective vasorelaxant of mouse TA, experiments were performed to ascertain the impact of this agent in arteries that have a crucial role in blood pressure regulation. Figure 8A shows that 10 μM T16Ainh-A01 was a highly effective relaxant of mouse MA pre-contracted with the stable thromboxane mimetic, U46619, compared with the equivalent vehicle (P < 0.0001, n = 4). Figure 8B shows that 10 μM T16Ainh-A01 also relaxed U46619-induced contractions of human visceral adipose arteries. The mean data for the experiments on mouse MA and human visceral adipose arteries are shown in Figure 8C. These data provide evidence for a functional role of TMEM16A in the vasoconstrictor responses of murine TA and MA as well as human resistance vessels.
Figure 8.
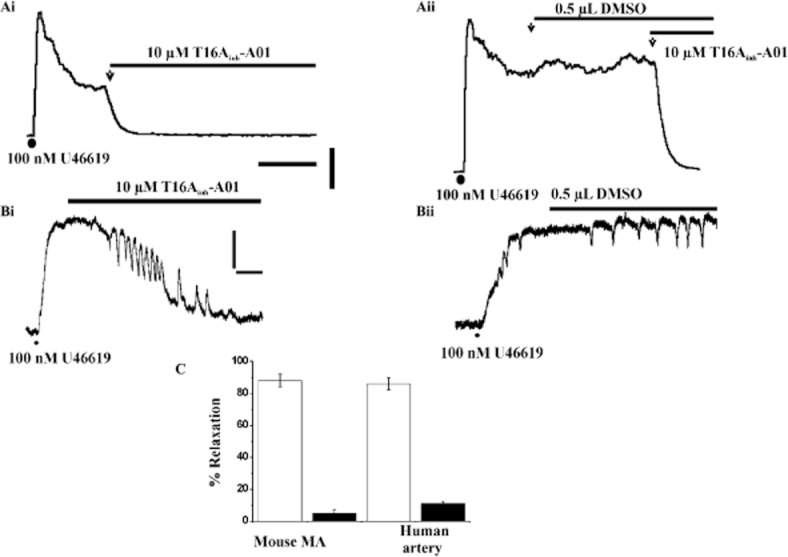
Effect of T16Ainh-A01 in isometric tension recordings from mouse and human resistance arteries. Ai shows the rapid effect of 10 μM T16Ainh-A01 on mouse MA pre-contracted with U46619. Aii shows the lack of effect of DMSO equivalent. Scale bars denote 8 min and 2 mN. Representative traces for the effect of 10 μM T16Ainh-A01 (Bi) and the equivalent DMSO volume (Bii) on segments of human visceral adipose arteries pre-contracted with U46619. Scale bars denote 2 mN and 8 min for both (Bi and Bii). (C) shows the mean relaxant effect for 10 μM T16Ainh-A01 in human visceral adipose arteries and mouse MA. Each column is the mean of 4-6 animals ± SEM. Open bars show the DMSO equivalent. Application of vasoconstrictor in all panels is shown by (•).
Discussion
In the present study, we have shown for the first time that a small molecule blocker of TMEM16A (T16Ainh-A01; Namkung et al., 2011a) and the recently identified TMEM16A activator Fact (Namkung et al., 2011b) have marked effects on native IClCa recorded in vascular smooth muscle cells. These data add weight to the view that TMEM16A constitutes a major component of native IClCa in freshly dispersed smooth muscle cells and agree with Manoury et al. (2010) who showed that TMEM16A targeted siRNA reduced IClCa in rat PA smooth muscle cells maintained in short-term culture. Moreover, single channel IClCa in smooth muscle cells from rabbit PA and mouse TA were abolished by two TMEM16A-specific antibodies raised in different hosts and against different epitopes. These inhibitory effects were not seen with non-immune serum from the host animals or antisera against bestrophin-3, which comprises the cGMP-sensitive IClCa in rat MA (Matchkov et al., 2008) and is expressed in rabbit PA (Leblanc et al., 2005). While there are caveats to the use of antibodies as pharmacological tools and the precise mechanism by which these polyclonal antibodies produced an inhibitory effect is not known, antibodies have been used as blocking molecules in the characterization of a number of different ion channels (e.g. Albert et al., 2009), including CACC studies (Thomas-Gatewood et al., 2011). Consequently, the fact that two structurally different antibodies targeted against TMEM16A proteins, and a TMEM16A blocker, exert an inhibitory effect on IClCa provides evidence that TMEM16A contributes to native CACCs. However, it is likely that the native CACC in vascular smooth muscle is not composed solely of TMEM16A. Firstly, the single channel conductance of native CACCs (1–3 pS) determined in the rabbit PA (present study, Piper and Large, 2003) as well as other blood vessels (Klöckner, 1993; Van Renterghem and Lazdunski, 1993; Hirakawa et al., 1999) is smaller than that determined for TMEM16A channels in overexpression studies (∼8 pS; Yang et al., 2008), although it is similar to channels generated by heterologously expressed TMEM16B (Pifferi et al., 2009). Secondly, in our whole cell recordings, only a component of the evoked current was inhibited by T16Ainh-A01 over the same concentration range where full blockade of TMEM16A-generated currents is seen (Namkung et al., 2011a). These differences may represent variable contribution from the different TMEM16A splice variants (Ferrera et al., 2009) that are expressed in vascular smooth muscle (Davis et al., 2010; Manoury et al., 2010) or the native CACC comprises TMEM16A proteins in association with other proteins. These aspects will be the focus of future studies.
The present study shows that T16Ainh-A01 relaxed pre-contracted mouse TA with an IC50 of 1.7 μM, which is similar to the derived value for inhibition of epithelial short circuit current (Namkung et al., 2011a) and for reduction of IClCa in the present study. T16Ainh-A01 at 10 μM was also a remarkably effective relaxant of pre-contracted resistance arteries and human visceral adipose arteries that we showed express TMEM16A and has recently been shown to relax 5HT-precontracted rat pulmonary arteries (Sun et al., 2012). T16Ainh-A01 was considerably more potent vasorelaxant than conventional Cl channel blockers such as niflumic acid (see Criddle et al., 1996 and present study). These findings are consistent with a simplified working model, whereby activation of a membrane receptor leads to a release of calcium sufficient to activate CACCs, which leads to Cl− efflux because smooth muscle cells accumulate Cl− ions (Chipperfield and Harper, 2000), and the membrane depolarization increases Ca2+ influx through voltage-dependent Ca2+ channels (VDCCs, Kitamura and Yamazaki, 2001; Leblanc et al., 2005). A blocker of CACCs would therefore limit the agonist-induced membrane depolarization and impair voltage-dependent calcium influx. The ability of a TMEM16A-specific blocker to impair vasoconstriction therefore suggests that TMEM16A expression products contribute to the native CACCs or some undefined part of the working model. However, using pharmacological agents to define a functional role requires a degree of caution. For instance, a similar degree of vasorelaxation would be produced by direct block of calcium channels or activation of potassium channels that would indirectly close the same channels. With respect to this point, T16Ainh-A01 had no significant effect on contractions produced by high KCl and the inhibitory effect of this agent was unaffected by paxilline, glibenclamide or linopirdine, effectively repudiating a role for ATP-sensitive K+ channels, KCa3.1 or KCNQ-encoded K+ channels that are present in this tissue. In addition, T16Ainh-A01 was equally as effective against methoxamine-induced or U46619-induced contractions, suggesting that this agent is not functioning as a receptor blocker. Having shown that the TMEM16A potentiator, Fact, increased single channel and whole cell IClCa, we hypothesized that this agent would augment the methoxamine-induced concentration–effect curve in line with the model described earlier. However, this was not the case and Fact did not produce a contraction by itself. It is possible that the activator was more effective at stimulating the current at positive than at negative potentials, which could have resulted in minimal effects of the drug in the physiological range of membrane potentials influenced by methoxamine and future studies will investigate the vascular effects of this agent and Fact (Namkung et al., 2011b) further.
In summary, the identification of TMEM16A expression products as constituents of native CACCs has energized the CACC research field and has allowed for novel reagents to be developed, which can provide insight on the physiological role of these proteins in various tissues. We now provide further evidence that TMEM16A proteins comprise native CACCs in vascular smooth muscle cells and inhibiting TMEM16A has marked anti-contractile effects in various mouse and human arteries.
Acknowledgments
We thank all the patients and associated staff at St George's Hospital for providing human samples. We also thank Mr Thomas Jepps and the Image Resource Facility at SGUL for their support with the immunological experiments. This work was supported by the British Heart Foundation grants awarded to I. A. G. (PG/09/104 and PG\07\127\24235), APA (PG/08/042/2506, BB/J007226/1) and by grants to N. L. from the National Institutes of Health (Grants 5 R01 HL 075477 and 3 R01 HL075477-04S1).
Glossary
- CACCs
calcium-activated chloride currents
- IClCa
calcium-activated chloride currents
- MA
mesenteric artery
- PA
pulmonary artery
- TA
thoracic aorta
- VDCCs
voltage-dependent calcium channels
Conflict of interests
None of the authors have any conflict of interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of niflumic acid on whole cell IClCa evoked in rabbit PA smooth muscle cells.
Table S1 PCR primer pair sequences used to probe for TMEM16A in various tissues.
References
- Albert AP, Saleh SN, Large WA. Identification of canonical transient receptor potential (TRPC) channel proteins in native vascular smooth muscle cells. Curr Med Chem. 2009;16:1158–1165. doi: 10.2174/092986709787581815. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angermann JE, Sanguinetti AR, Kenyon JL, Leblanc N, Greenwood IA. Mechanism of the inhibition of Ca2+-activated Cl− currents by phosphorylation in pulmonary arterial smooth muscle cells. J Gen Physiol. 2006;128:73–87. doi: 10.1085/jgp.200609507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Chipperfield AR, Harper AA. Chloride in smooth muscle. Prog Biophys Mol Biol. 2000;74:175–221. doi: 10.1016/s0079-6107(00)00024-9. [DOI] [PubMed] [Google Scholar]
- Criddle DN, de Moura RS, Greenwood IA, Large WA. Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. Br J Pharmacol. 1996;118:1065–1071. doi: 10.1111/j.1476-5381.1996.tb15507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, et al. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol. 2010;299:C948–C959. doi: 10.1152/ajpcell.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, et al. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl− and K+ channels. Trends Pharmacol Sci. 2007;28:1–5. doi: 10.1016/j.tips.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Ledoux J, Leblanc N. Differential regulation of Ca2+-activated Cl− currents in rabbit arterial and portal vein smooth muscle cells by Ca2+-calmodulin-dependent kinase. J Physiol. 2001;534:395–408. doi: 10.1111/j.1469-7793.2001.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Ledoux J, Sanguinetti A, Perrino BA, Leblanc N. Calcineurin Aα but not Aβ augments ICl(Ca) in rabbit pulmonary artery smooth muscle cells. J Biol Chem. 2004;279:38830–38837. doi: 10.1074/jbc.M406234200. [DOI] [PubMed] [Google Scholar]
- Hirakawa Y, Gericke M, Cohen RA, Bolotina VM. Ca2+-dependent Cl− channels in mouse and rabbit aortic smooth muscle cells: regulation by intracellular Ca2+ and NO. Am J Physiol. 1999;277:H1732–H1744. doi: 10.1152/ajpheart.1999.277.5.H1732. [DOI] [PubMed] [Google Scholar]
- Johnstone AP, Thorpe RC. Immunochemistry in Practice. 3rd edn. Oxford: Wiley-Blackwell; 1996. [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Yamazaki J. Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol. 2001;85:351–357. doi: 10.1254/jjp.85.351. [DOI] [PubMed] [Google Scholar]
- Klöckner U. Intracellular calcium ions activate a low-conductance chloride channel in smooth muscle cells isolated from human mesenteric artery. Pflugers Arch. 1993;424:231–237. doi: 10.1007/BF00384347. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am J Physiol. 1996;271:C435–C454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O'Driscoll K, et al. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–556. doi: 10.1139/y05-040. [DOI] [PubMed] [Google Scholar]
- Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–2314. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, et al. Bestrophin-3 (vitelliform macular dystrophy2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res. 2008;103:864–872. doi: 10.1161/CIRCRESAHA.108.178517. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24:4178–4186. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011a;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011b;25:4048–4062. doi: 10.1096/fj.11-191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A, et al. Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifferi S, Dibattista M, Menini A. TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch. 2009;458:1023–1038. doi: 10.1007/s00424-009-0684-9. [DOI] [PubMed] [Google Scholar]
- Piper AS, Large WA. Multiple conductance states of single Ca2+-activated Cl− channels in rabbit pulmonary artery smooth muscle cells. J Physiol. 2003;547:181–196. doi: 10.1113/jphysiol.2002.033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper AS, Greenwood IA, Large WA. Dual effect of blocking agents on Ca2+-activated Cl− currents in rabbit pulmonary artery smooth muscle cells. J Physiol. 2002;539:119–131. doi: 10.1113/jphysiol.2001.013270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sones WR, Davis AJ, Leblanc N, Greenwood IA. Cholesterol depletion alters amplitude and pharmacology of vascular calcium-activated chloride channels. Cardiovasc Res. 2010;87:476–484. doi: 10.1093/cvr/cvq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Xia Y, Paudel O, Yang XR, Sham JS. Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol. 2012;590:3507–3521. doi: 10.1113/jphysiol.2012.232520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, et al. TMEM16A channels generate Ca2+-activated Cl− currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2011;301:H1819–H1827. doi: 10.1152/ajpheart.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Renterghem C, Lazdunski M. Endothelin and vasopressin activate low conductance chloride channels in aortic smooth muscle cells. Pflugers Arch. 1993;425:156–163. doi: 10.1007/BF00374516. [DOI] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Pucovský V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for Kv7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.