

Abstract
The asymmetric bioreduction of a library of β-cyanoacrylate esters using ene-reductases was studied with the aim to provide a biocatalytic route to precursors for GABA analogues, such as pregabalin. The stereochemical outcome could be controlled by substrate-engineering through size-variation of the ester moiety and by employing stereochemically pure (E)- or (Z)-isomers, which allowed to access both enantiomers of each product in up to quantitative conversion in enantiomerically pure form. In addition, stereoselectivities and conversions could be improved by mutant variants of OPR1, and the utility of the system was demonstrated by preparative-scale applications.
Introduction
In recent years, ene-reductases became prominent for providing a green alternative for the highly selective production of chiral molecules.1 The enzymes are members of the flavin-dependent old yellow enzyme (OYE) family and catalyze the asymmetric reduction of a broad variety of activated alkenes at the expense of a nicotinamide cofactor.2,3 Ene-reductases have been utilized for the synthesis of diverse enantiomerically pure products, including asymmetric building blocks such as chiral acyloins4 and the “Roche-ester”.5 Further examples encompass enantiopure α- and β-amino acid derivatives,6,7 molecules for pharma applications,8 and compounds of commercial importance, such as the fragrances Lilial or Lysmeral.9
Pregabalin [(S)-3-aminomethyl-5-methylhexanoic acid], the active ingredient in Lyrica, is a β-substituted γ-amino acid belonging to the class of drugs known as GABA analogues. The manufacture of pregabalin begins with the enzymatic resolution of a β-cyanodiester10 followed by decarboxylation to give the penultimate intermediate, (S)-ethyl 3-cyano-5-methylhexanoate (S)-4b. Reduction of the nitrile moiety gives pregabalin.11 While this chemo-enzymatic process achieved a large increase in efficiency over the initial manufacturing protocol,12 we were interested in evaluating stereoselective approaches independent of kinetic resolution protocols with the objective of further increasing process efficiency. We postulated that asymmetric bioreduction of a β-cyanoacrylate ester by an ene-reductase could provide access to (S)-4b, thus enabling a novel route to pregabalin, and set out to evaluate the feasibility of this potential strategy (SCHEME 1). This approach could also be useful for the stereoselective synthesis of related β-substituted γ-amino acids (Baclofen) or derivatives (Brivaracetam).
Scheme 1. Asymmetric Bioreduction of β-Cyanoacrylic Esters.

Results and Discussion
β-Cyanoacrylate esters (E)-1a–5a and (Z)-1a–5a were prepared from the appropriate β-keto ester via enol triflate precursors using the procedure described by Frantz et al.13 (Scheme 2). The selectivity for (Z)-enol triflates was excellent with no trace of (E)-isomers present at the end of the reaction, and (Z)-enol triflates were isolated in 80–92% yield after chromatography. The conditions to generate (E)-analogues gave poorer selectivity (>50:1 to 5:1), as the steric bulk of the R1 group increased from Me to i-Bu. Furthermore, lower isolated yields (54–71%) were observed for the (E)-enol triflates, which was due to less effective extraction of the product from the Me4N+OH–/water mixture going in hand with competing hydrolysis of triflic anhydride. Treatment of (E)- or (Z)-enol triflates with Zn(CN)2 in the presence of a palladium(0) catalyst using the procedure of Tebben et al.14 generated the desired β-cyanoacrylate esters in 51–86% yield. Substrates (Z)-6a and (E)-7a were obtained by transesterification from (Z)-3a and (E)-5a, respectively.
Scheme 2. Synthesis of Substrates (E)- and (Z)-1a–5a.

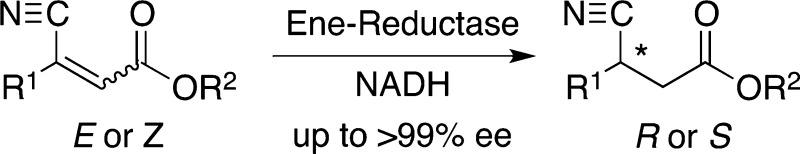
Since the (E/Z)-configuration of alkenes has been shown to have a dramatic impact on the stereochemical outcome of ene-reductions,15−18 stereochemically pure substrates 1a–4a were used. In addition, the size of the alcohol moiety of the (activating) ester group was varied in order to test its influence on the substrate-docking mode of substrates 5a–7a within the active site.4,6,18 All substrates were tested using a set of native ene-reductases, which have shown to possess a broad substrate spectrum on acrylic ester derivatives5,6,15,18,19 (Scheme 1, Table 1).
Table 1. Asymmetric Bioreduction of β-Cyanoacrylic Esters Using Native OYEsa.
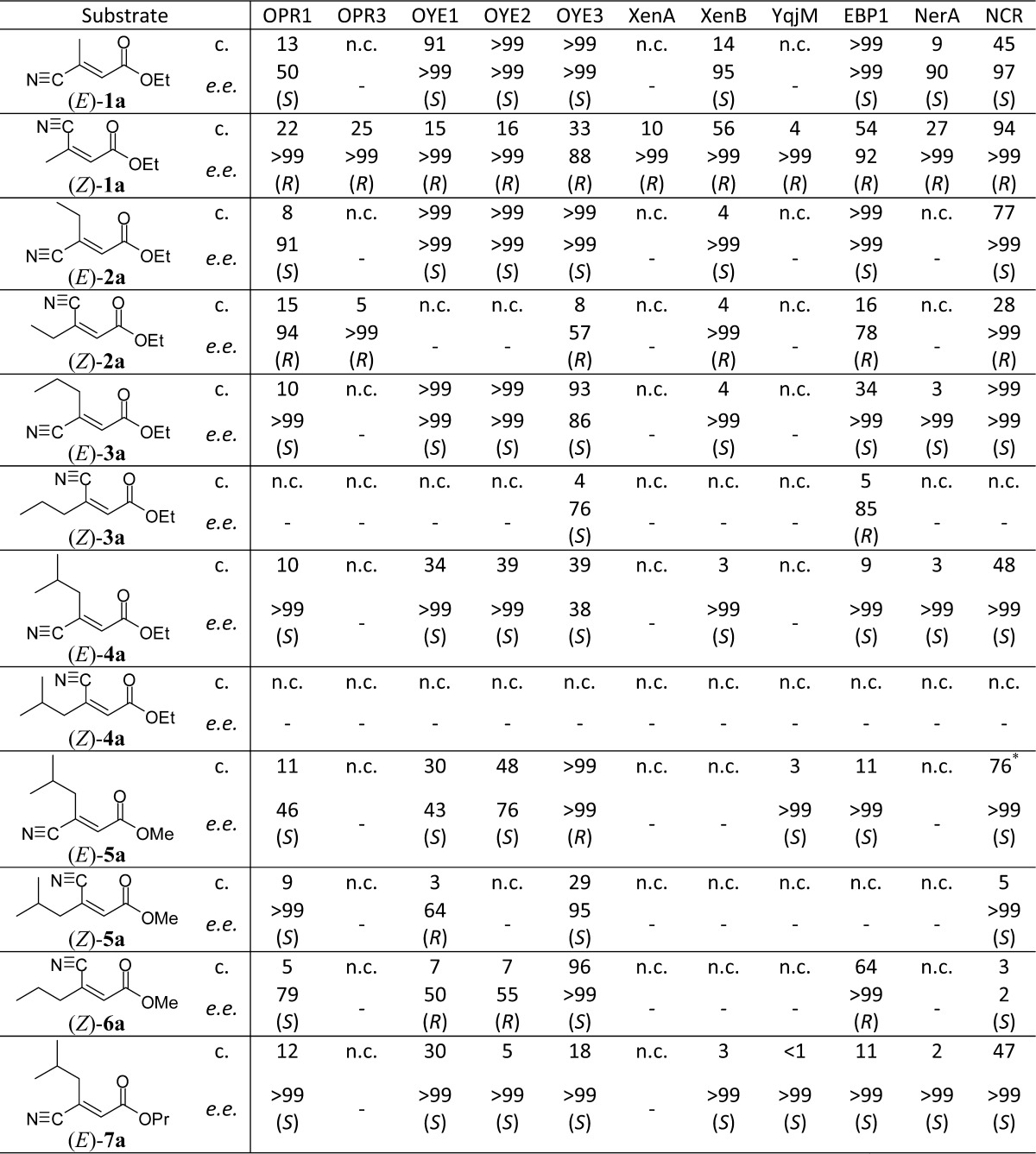
Key: c. = conversion [%]; e.e. = enantiomeric excess of products 1b–7b [%]; n.c. = no conversion.
double enzyme and half substrate amount: >99% conversion, >99 e.e. (S).
We envisaged that the appropriate combination of substrate- and enzyme-engineering would enable us to access both stereoisomers of GABA-precursors with high conversion and perfect stereoselectivity. Interestingly, in line with previous reports,15−18 (E)-β-cyanoacrylate esters generally were converted to the (S)-products while the bioreduction of (Z)-analogues gave the enantiomeric (R)-β-cyano esters. Accordingly, (S)-1b was obtained from (E)-1a in perfect conversion and stereoselectivity using OYE2, OYE3, and EBP1, while (R)-1b was formed from (Z)-1a using NCR (ee > 99, c 94%). The same enzymes showed similar activities toward (E)-2a and (Z)-2a: OYEs 1–3 and NCR produced (S)-2b from (E)-2a (ee > 99, c >99%) and (R)-2b could be obtained with perfect ee from (Z)-2a using OPR3, XenB, and NCR. In analogy, (S)-3b could be produced from (E)-3a (ee > 99, c >99%) and the reduction of (E)-4a afforded (S)-4b with perfect stereoselectivity in up to 48% conversion using OYE1, OYE2, and NCR. However, with increasing steric demand of the substrates, the conversion of (Z)-β-cyanoacrylate esters dropped, resulting in low reactivity toward (Z)-3a and no activity toward (Z)-4a at all. In contrast, decreasing the size of the ester moiety from ethyl to methyl had a strongly positive impact on reaction rates: By applying methyl (E)-β-cyanoacrylate (Z)-6a instead of the ethyl ester (Z)-3a, the activity of EBP1 forming the (R)-enantiomer could be improved from 5% conversion (85% ee) to 64% conversion and perfect stereoselectivity. In the same way, ethyl ester (Z)-4a was not accepted by any of the enzymes, but the corresponding methyl ester (Z)-5a was converted with modest rates (OYE3, c 29%). For the bulkier substrates 4a, 5a, and 7a bearing the isobutyl pharmacophor side chain of Pregabalin, the stereorecognition was not as homogeneously conserved as for the smaller analogues. Whereas the less bulky E-configured substrates strictly yielded S-products with the majority of enzymes in up to quantitative conversion, the stereopreference of OYE3 switched to R (c >99, ee > 99) for (E)-5a. This exceptional behavior of OYE3 has been observed before6 and depends on the steric demand of the ester group, going from absolute R-selectivity for the methyl ester (E)-5a to moderate S-preference for the ethyl ester (E)-4a (ee 38%). In order to push the incomplete S-selectivity, the size of the ester group was increased to n-propyl [(E)-7a], which successfully resulted in (S)-7b as the sole product. Overall, OPR3, XenA, and YqjM showed poor activities toward all tested substrates and NerA and XenB accepted only the smaller substrates 1a–3a. Using OYE2 in combination with a recently developed ADH-based NADH-cofactor recycling system,20 (S)-1b–3b could be produced on preparative scale (>1 g) with perfect ee and good isolated yields (up to 83%), which nicely demonstrates the applicability of this bioreduction protocol to asymmetric syntheses.
In order to improve stereoselectivities of native enzymes, mutant variants were constructed.21,22 A basic sequence alignment of OPR1 identified many potential sites for mutation of which positions Cys20, Ile287, and His245 were selected for investigation. His245 (neutral at pH 7.5) was mutated to a negatively charged Asp to enhance the polarity around the active site entrance for improved substrate binding. The sequence alignment showed a high variability of 14 amino acids at position Ile287, which is located in a flexible loop close to the active site entrance extending from Arg279 to Thr290. In order to make this region more rigid, lipophilic Ile287 was exchanged by a more polar His.43 Cys20 located at the surface opposite to the active site was exchanged by a Tyr to probe whether such a distant mutation had any effects on the chiral recognition process.44
Results from bioreduction screens on OPR1 variants (Table 2) showed improved conversions for variant Cys20Tyr with (E)-4a and (E)-5a compared to wild-type OPR1, while Ile287His did not give improved conversions for any of the substrates. However, Cys20Tyr and Ile287His both gave greatly enhanced stereoselectivities in the bioreduction of (E)-5a and slight improvements for (E)-2a and (Z)-2a. Variant His245Asp gave the highest increase in selectivity for the bioreduction of (E)-5a and also showed the highest improvements in conversion for several of the substrates.
Table 2. Asymmetric Bioreduction of β-Cyanoacrylic Esters Using Wild-Type and Mutant Variants of OPR1a.
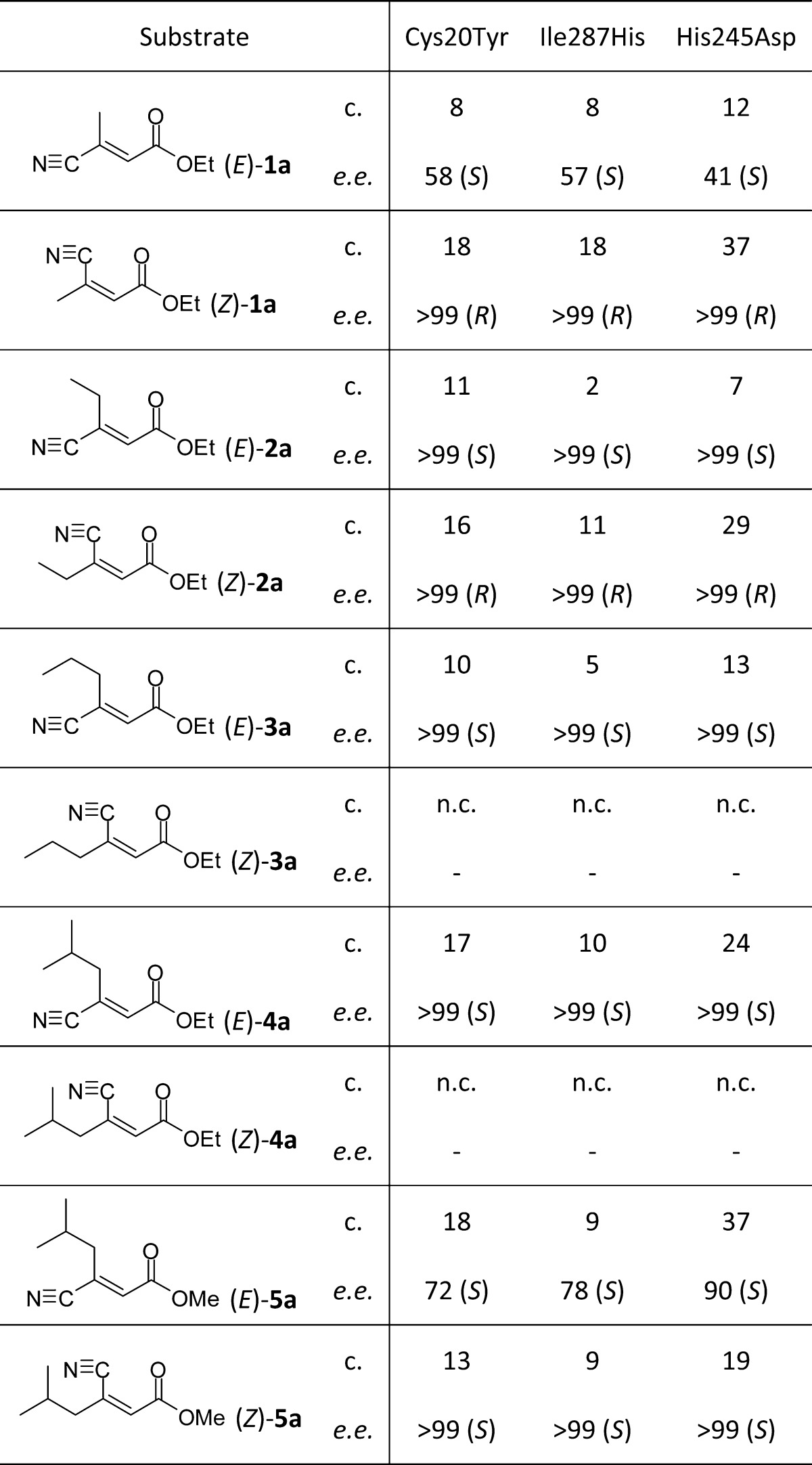
Key: c. = conversion [%]; e.e. = enantiomeric excess of products 1b–5b [%]; n.c. = no conversion.
Conclusion
The asymmetric bioreduction of β-cyanoacrylate esters using ene-reductases provides an elegant alternative to metal-dependent hydrogenation protocols for the asymmetric synthesis of precursors for GABA analogues, such as pregabalin. This concept was successfully applied to the production of a library of chiral β-cyano esters. The stereochemical outcome could be switched via substrate-engineering through size-variation of the alkoxy moiety and by employing stereochemically pure (E)- or (Z)-isomers, which allowed to access both enantiomers of each product with up to quantitative conversion in enantiomerically pure form. In addition, stereoselectivities and conversions could be improved by mutant variants of OPR1. Finally, the utility of the system for preparative-scale applications was demonstrated by the isolation of gram-amounts of β-cyano esters with perfect ee. Upscaling to industrial production should be feasible as the procedure utilized inexpensive cosubstrate to recycle the nicotinamide cofactor, improvement of the volumetric efficiency would be feasible by development of more active enzymes via enzyme engineering and/or screening natural diversity. Further process efficiency could be achieved by the implementation of a hydrolase-reductase cascade process:45 In the case of pregabalin synthesis, the addition of a hydrolase to the bioreduction of (E)-4a could result in a one-pot process to (S)-3-cyano-5-methylhexanoic acid, the penultimate precursor to pregabalin,11 thus eliminating an additional step for hydrolysis of (S)-4b. In conclusion, this study demonstrates the applicability of ene-reductases as a general synthetic tool by meeting the demand for green and highly stereoselective synthetic methods.31
Experimental Section
General Methods
GC analyses were carried out using a Chiraldex G-TA column (30 m × 0.25 mm id, 0.12 μm film) or a Hydrodex-β-TBDAc capillary column (250 m × 0.25 mm id). NMR spectra were obtained on a 400 MHz or a 300 MHz NMR spectrometer. Chemical shifts are reported relative to TMS (δ 0.00), and coupling constants (J) are given in Hz. High resolution mass spectra were obtained either on a oa-TOF mass spectrometer with direct insertion and electron-impact ionization (70 eV) or on a ion trap FT-MS using electrospray ionization in positive mode or APCI ionization in negative mode. TLC plates were run on silica gel, and compounds were visualized by spraying with phosphomolybdic acid stain or by UV (254 nm). Lactobacillus brevis alcohol dehydrogenase was obtained from X-Zyme.
Source of Enzymes
12-Oxophytodienoate reductase isoenzymes OPR1 and OPR3 from Lycopersicon esculentum and the OYE homologue YqjM from Bacillus subtilis were overexpressed and purified as reported.23−25 The cloning, purification, and characterization of OYE isoenzymes from yeast (OYE1 from Saccharomyces pastorianus, OYE2 and OYE3 from Saccharomyces cerevisiae) and nicotinamide-dependent cyclohexenone reductase (NCR) from Zymomonas mobilis were performed according to literature methods.26,27 Xenobiotic reductases XenA and XenB from Pseudomonas putida and P. fluorescens, respectively, and glycerol trinitrate reductase NerA from Agrobacterium radiobacter were obtained as recently published.28−30Lycopersicon esculentum OPR1 variants Cys20Tyr, Ile287His, and His245Asp were constructed in pET26b using QuikChange Lightning Site-Directed Mutagenesis Kits (Stratagene, La Jolla, CA) and were expressed as HIS-tagged proteins in E. coli BL21 cells. The cloning and purification of EPB1 from Candida albicans and of Lycopersicon esculentum OPR1 variants Cys20Tyr, Ile287His, and His245Asp can be found in the Supporting Information.
General Procedure for Enzymatic Bioreduction
An aliquot of enzyme (OPR1-wt, OPR3, OYE1-3, XenA, XenB, YqjM, EBP1, NerA, or NCR; protein concentration in biotransformations: 75–125 μg/mL) was added into a 1.5 mL microcentrifuge tube containing a Tris-HCl buffer solution (0.8 mL, 50 mM, pH 7.5), the substrate (10 mM), and the cofactor (NADH, 15 mM). The mixture was shaken at 30 °C and 120 rpm. After 24 h, the products were extracted with ethyl acetate (0.6 mL). The organic phase was dried over Na2SO4 and analyzed on GC to determine enantiomeric excess and conversion. For every screening a control experiment without the addition of enzyme was included.
Determination of Absolute Configuration of Products 1b–3b
Absolute configurations of 1b, 2b, and 3b were determined by preparation of (S)-1b, (S)-2b, and (S)-3b via bioreductions of (E)-1a, (E)-2a, and (E)-3a at preparative scale and subsequent conversions to suitable derivatives and comparison of specific rotations of the latter with literature values. For determination of the absolute configuration of 4b, both reference materials (R)- and (S)-4b were prepared. For the assignment of the absolute configurations of 5b, 6b, and 7b, enantiomerically pure (S)-isomers were prepared by transesterification of the corresponding ethyl esters (S)-4a and (S)-3a.
Synthesis and Characterization of Substrates (E)- and (Z)-1a–5a
General Procedure A: Synthesis of Keto Esters
The ketone (1.0 equiv) was added slowly, over 15 min, to a stirred suspension of NaH (60% in mineral oil, 1.2 equiv) in dry THF (500 mL) at 0 °C. After the addition was complete, the cooling bath was removed, and the resulting mixture was stirred at rt for 40 min. The reaction was cooled to 0 °C, and the carbonate ester (1.5 equiv) was added slowly over 1 h. After the addition was complete, the reaction was allowed to slowly warm to rt with stirring (18 h). The reaction was quenched and acidified to pH ∼6 by careful addition of acetic acid (60 mL). Water (400 mL) was added, and the resulting mixture was extracted with Et2O. The organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure to give the crude β-ketoester.
Ethyl 3-Oxopentanoate32
Using general procedure A, 2-butanone (51.6 mL, 41.5 g, 576 mmol), NaH (60% in mineral oil, 27.6 g, 691 mmol) in dry THF (500 mL), and diethyl carbonate (105 mL, 102 g, 864 mmol) gave the crude product as a clear yellow oil, which was distilled twice under reduced pressure (60 °C, 4 mbar) resulting in a 2:1 mixture of the desired product and the regioisomer ethyl 2-methyl-3-oxobutanoate as a clear colorless oil (31.9 g, 38%): 1H NMR (300 MHz, CDCl3) δ 1.09 (t, J = 7.5 Hz, 3H), 1.28 (t, J = 7.0 Hz, 3H), 2.56 (q, J = 7.5 Hz, 2H), 3.43 (s, 1H), 4.19 (q, J = 7.0 Hz, 2H). Ethyl 2-methyl-3-oxobutanoate: 1H NMR (300 MHz, CDCl3) δ 1.28 (t, J = 7.0 Hz, 3H), 1.34 (d, J = 7.0 Hz, 3H), 2.24 (s, 3H), 3.49 (q, J = 7.0 Hz, 1H), 4.19 (q, J = 7.0 Hz, 2H).
Ethyl 3-Oxohexanoate33
Using general procedure A, 2-pentanone (61.5 mL, 49.6 g, 576 mmol), NaH (60% in mineral oil, 27.6 g, 691 mmol) in dry THF (500 mL), and diethyl carbonate (104.9 mL, 102.1 g, 864 mmol) gave the crude product as a clear yellow oil, which was distilled under reduced pressure (60 °C, 4 mbar) yielding the desired product as a clear colorless oil (39.9 g, 44%): 1H NMR (300 MHz, CDCl3) δ 0.93 (t, J = 7.5 Hz, 3H), 1.28 (t, J = 7.0 Hz, 3H), 1.56–1.70 (m, 2H), 2.51 (t, J = 7.0 Hz, 2H), 3.42 (s, 2H), 4.19 (q, J = 7.0 Hz, 2H).
Ethyl 5-Methyl-3-oxohexanoate34
Using general procedure A, 4-methyl-2-pentanone (608.0 mL, 487.0 g, 4.86 mol), NaH (60% in mineral oil, 256.7 g, 1.32 equiv) in dry THF (4.87 L), and diethyl carbonate (884.0 mL, 1.5 equiv) gave the crude product as an orange oil. The crude product was distilled under reduced pressure (64–82 °C, 7–10 mbar) the desired product as a pale yellow oil (661.2 g, 79% isolated yield): 1H NMR (300 MHz, CDCl3) δ 0.86 (d, J = 6.4 Hz, 6H), 1.21 (t, J = 7.2 Hz, 3H), 2.09 (J = 6.7 Hz, 1H), 2.35 (d, J = 6.9 Hz, 2H), 3.34 (s, 2H), 4.12 (q, J = 7.2 Hz, 2H).
Methyl 5-Methyl-3-oxohexanoate35
Using general procedure A, 4-methyl-2-pentanone (72.1 mL, 57.7 g, 576 mmol), NaH (60% in mineral oil, 27.6 g, 691 mmol) in dry THF (500 mL), and dimethyl carbonate (72.8 mL, 77.8 g, 864 mmol) gave the crude product as a cloudy yellow oil. The crude product was distilled under reduced pressure (60 °C, 4 mbar) to give the desired product as a clear colorless oil (61.9 g, 68%): 1H NMR (300 MHz, CDCl3) δ 0.93 (d, J = 6.5 Hz, 6H), 2.09–2.22 (m, 1H), 2.41 (d, J = 7.0 Hz, 2H), 3.42 (s, 2H), 3.73 (s, 3H).
General Procedure B: Synthesis of (Z)-Triflates
Lithium triflate (2.0 equiv) was added to a stirred solution of the keto ester (1.0 equiv) in dry CH2Cl2 (150 mL). The resulting mixture was cooled to 0 °C, and diisopropylethylamine (1.1 equiv) was added dropwise over 10 min. After the addition was complete the mixture was stirred at 0 °C for 20 min before triflic anhydride (1.1 equiv) was added slowly over 30 min. The resulting mixture was stirred at 0 °C for 1 h before carefully being quenched by addition of saturated aqueous NH4Cl (60 mL). The reaction was allowed to warm to rt and an additional portion of CH2Cl2 (30 mL) was added. The layers were separated, and the organic layer was washed with aqueous HCl (1 M, 2 × 30 mL), water (30 mL), and brine (30 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. Et2O (60 mL) was added to the resulting semisolid, and the mixture was filtered. The solids were extracted with more Et2O (2 × 20 mL), and the combined ether extracts were concentrated under reduced pressure to give the crude product.
(Z)-Ethyl 3-(Trifluoromethylsulfonyloxy)but-2-enoate13
Using general procedure B, lithium triflate (24.0 g, 154 mmol), ethyl acetoacetate (9.7 mL, 10.0 g, 76.8 mmol) in dry CH2Cl2 (150 mL), diisopropylethylamine (14.7 mL, 10.9 g, 84.5 mmol) and triflic anhydride (14.22 mL, 23.9 g, 84.5 mmol) gave the crude product as a clear yellow oil (18.6 g, 92%) which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 1.30 (t, J = 7.0 Hz), 2.16 (s, 3H), 4.24 (q, J = 7.0 Hz, 2H), 5.75 (q, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.2, 21.1, 61.4, 113.0, 118.5 (q, J = 319.8 Hz, CF3), 155.2, 162.5.
(Z)-Ethyl 3-(Trifluoromethylsulfonyloxy)pent-2-enoate13
Using general procedure B, lithium triflate (21.6 g, 139 mmol), ethyl 3-oxopentanoate (10.0 g, 69.4 mmol) in dry CH2Cl2 (150 mL), diisopropylethylamine (13.3 mL, 9.9 g, 76.3 mmol), and triflic anhydride (12.8 mL, 21.5 g, 76.3 mmol) gave the crude product as a clear dark orange oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc, 9:1) afforded the desired product as a clear pale yellow oil (17.7 g, 92% yield): 1H NMR (300 MHz, CDCl3) δ 1.18 (t, J = 7.5 Hz, 3H), 1.31 (t, J = 7.0 Hz, 3H), 2.44 (qd, J = 7.5, 1.0 Hz, 2H), 4.25 (q, J = 7.0 Hz, 2H), 5.75 (t, J = 1.0 Hz, 1H).
(Z)-Ethyl 3-(Trifluoromethylsulfonyloxy)hex-2-enoate
Using general procedure B, lithium triflate (19.7 g, 126 mmol), ethyl 3-oxohexanoate (10.0g, 63.2 mmol) in dry CH2Cl2 (150 mL), diisopropylethylamine (12.1 mL, 9.0 g, 69.5 mmol), and triflic anhydride (11.7 mL, 19.6 g, 69.5 mmol) gave the crude product as a clear dark red oil (17.0 g, 93% yield) which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 0.99 (t, J = 7.5 Hz, 3H), 1.30 (t, J = 7.0 Hz, 3H), 1.55–1.69 (m, 2H), 2.37 (t, J = 7.5 Hz, 2H), 4.25 (q, J = 7.0 Hz, 2H), 5.74 (t, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 14.2, 19.5, 36.5, 61.5, 112.1, 118.6 (q, J = 319.8 Hz, CF3), 158.9, 162.7; HRMS (EI+) m/z calcd for C9H13F3O5S [M]+ 290.0439, found 290.0436.
(Z)-Ethyl 5-Methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate34
A 20 L jacketed reactor was charged with CH2Cl2 (2.45 L), ethyl 3-oxo-5-methylhexanoate (163.3 g; 1 equiv), and lithium triflate (295.86 g; 2 equiv) under an inert atmosphere. The resulting white suspension was stirred and cooled to 0 °C. N,N-Diisopropylethylamine (134.8 g; 182 mL; 1.1 equiv) was added over 10 min at <10 °C. The suspension was stirred for 20 min at 0 °C, and then triflic anhydride (294.28 g; 175 mL; 1.1 equiv) was added slowly to the mixture over 30 min at <10 °C. The suspension was stirred at 0 °C for 1 h. The mixture then was allowed to warm from 0 to 25 °C over 1 h while quenching with satd NH4Cl solution (980 mL). Dichloromethane (490 mL) was added, and the mixture was stirred for 5 min and then allowed to settle, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (490 mL). The combined organic phases were then washed with 1 M HCl (2 × 490 mL), water (490 mL), and 20% brine (490 mL) and then stirred for 30 min with MgSO4 (∼165 g) for 15–30 min. The solids were removed by filtration and washed with CH2Cl2 (165 mL). The filtrate was concentrated under vacuum at ≤40 °C to give an orange/brown oil containing crystalline solids. The crude product was triturated with MTBE (980 mL). The mixture was filtered and the solids were washed with MTBE (2 × 165 mL). The filtrate was concentrated under vacuum at ≤40 °C to give the title product as an orange/brown oil: overall yield 271 g (94%; corrected for 2.1% w/w MTBE content); 87.7% area purity by GC; 1H NMR (400 MHz, CDCl3): δ 0.94 (6H, d, J = 6.7 Hz), 1.24 (3H, t, J = 7.2 Hz), 1.89 (1H, hept, J = 6.8 Hz), 2.17 (2H, d, J = 7.2 Hz), 4.18 (2H, q, J = 7.2 Hz), 5.67 (1H, s).
(Z)-Methyl 5-Methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate
Using general procedure B, lithium triflate (19.7 g, 126 mmol), methyl 5-methyl-3-oxohexanoate (10.0 g, 63.2 mmol) in dry CH2Cl2 (150 mL), diisopropylethylamine (12.1 mL, 9.0 g, 69.5 mmol), and triflic anhydride (11.7 mL, 19.6 g, 69.5 mmol) gave the crude product as a clear orange oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc, 9:1) afforded the desired product as a clear pale yellow oil (14.7 g, 80%): 1H NMR (300 MHz, CDCl3) δ 0.99 (d, J = 6.6 Hz, 6H), 1.98 (dp, J = 13.6, 6.8, 1H), 2.27 (d, J = 7.2 Hz, 2H), 3.80 (s, 3H), 5.76 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 21.9, 25.8, 43.7, 51.9, 112.4, 118.4 (q, J = 318.0 Hz, CF3), 158.3, 162.8; HRMS (ES+) m/z calcd for C9H14F3O5S [M + H]+ 291.0514, found 291.0519.
General Procedure C: Synthesis of (E)-Triflates
A mixture of Me4N+OH– (25% in H2O, 5.0 equiv) and water (72.5 mL) was added dropwise over 30 min to a vigorously stirred solution of the keto ester (1.0 equiv) in heptane (300 mL) at 0 °C. After the addition was complete, the mixture was stirred at 0 °C for 15 min before triflic anhydride (2.5 equiv) was added dropwise over 1 h. After the addition was complete, the reaction was stirred for another 1 h at 0 °C before being quenched by careful addition of water (75 mL). The mixture was allowed to warm to rt, and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with water (75 mL) and brine (75 mL), dried over Na2SO4, and concentrated under reduced pressure to yield the crude product.
(E)-Ethyl 3-(Trifluoromethylsulfonyloxy)but-2-enoate13
Using general procedure C, Me4N+OH– (25% in H2O, 137 mL, 140 g, 384 mmol), water (72.5 mL), ethyl acetoacetate (9.7 mL, 10.0 g, 76.8 mmol) in heptanes (300 mL), and triflic anhydride (32.3 mL, 54.2 g, 192.1 mmol) gave the crude product as a clear colorless oil (14.4 g, 71%), which was used without further purification: 1H NMR (400 MHz, CDCl3) δ 1.31 (t, J = 7.0 Hz, 3H), 2.51 (s, 3H), 4.22 (q, J = 7.0 Hz, 2H), 5.95 (s, 1H).
(E)-Ethyl 3-(Trifluoromethylsulfonyloxy)pent-2-enoate13
Using general procedure C, Me4N+OH– (25% in H2O, 124 mL, 26.4 g, 347 mmol), water (72.5 mL), ethyl 3-oxopentanoate (10.0 g, 69.4 mmol) in heptanes (300 mL), and triflic anhydride (29.2 mL, 48.9 g, 173.4 mmol) gave the crude product as a clear pale yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc, 19:1) afforded the desired product as a clear colorless oil (10.6 g, 55%): 1H NMR (400 MHz, CDCl3) δ 1.21 (t, J = 7.5 Hz, 3H), 1.31 (t, J = 7.0 Hz, 3H), 2.94 (q, J = 7.5 Hz, 2H), 4.22 (q, J = 7.0 Hz, 2H), 5.92 (s, 1H).
(E)-Ethyl 3-(Trifluoromethylsulfonyloxy)hex-2-enoate36
Using general procedure C, Me4N+OH– (25% in H2O, 113 mL, 115 g, 316.1 mmol), water (72.5 mL), ethyl 3-oxohexanoate (10.0 g, 63.2 mmol) in heptanes (300 mL), and triflic anhydride (26.6 mL, 44.6 g, 158.0 mmol) gave the crude product as a clear yellow oil (13.8 g, 75% yield), which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 0.99 (t, J = 7.5 Hz, 3H), 1.30 (t, J = 7.0 Hz, 3H), 1.59–1.73 (m, 2H), 2.90 (t, J = 7.5 Hz, 2H), 4.22 (q, J = 7.0 Hz, 2H), 5.95 (s, 1H).
(E)-Ethyl 5-Methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate34
A 20 L jacketed reactor was charged with hexane (7.83 L) and ethyl 3-oxo-5-methylhexanoate (261.0 g, 1 equiv) under an inert atmosphere. The solution was cooled to 5 °C and stirred vigorously while a mixture of 25% aq tetramethylammonium hydroxide (2.76 kg, 2.76 L, 5 equiv) and water (1.89 L) was added over 30 min at 15 °C. The biphasic mixture was stirred vigorously for 10 min at 5–10 °C, and then triflic anhydride (1.07 kg, 637 mL, 2.5 equiv) was added dropwise to the mixture over 1.5 h at <10 °C. The mixture was stirred vigorously at 5 °C for a further 1 h, agitation was stopped, and the layers were allowed to settle before the upper organic layer was sampled. The mixture was then allowed to warm from 5 to 25 °C over 1 h while being quenched with water (1.97 L) and then stirred for 5 min. The mixture was allowed to settle, and layers were separated. The aqueous layer was extracted with EtOAc (3.92 L). The combined organic layers were washed with water (1.97 L) and then 20% brine (1.97 L), stirred with MgSO4 (∼300g) for 15–30 min, and then filtered. The residue was washed with EtOAc (260 mL), and the filtrate was concentrated under vacuum at ≤40 °C to give the E-vinyl triflate as a brown oil (overall yield of 378 g, 82%; corrected for 8.4% w/w EtOAc content, 75.5% area purity by GC). NMR analysis indicated that the isomeric ratio was 6:1: 1H NMR (400 MHz, CDCl3): δ 0.92 (6H, d, J = 6.7 Hz), 1.24 (3H, t, J = 7.2 Hz), 1.95 (1H, hept, J = 6.8 Hz), 2.17 (0.33H, d, J = 7.2 Hz), 2.76 (2H, d, J = 7.3 Hz), 4.14 (2H, q, J = 7.2 Hz), 4.18 (0.28H, q, J = 7.2 Hz), 5.66 (0.14H, s), 5.91 (1H, s).
(E)-Methyl 5-Methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate
Using general procedure C, Me4N+OH– (25% in H2O, 113 mL, 115 g, 316 mmol), water (72.5 mL), ethyl 3-oxo-5-methylhexanoate (10.0 g, 63.2 mmol) in heptane (300 mL), and triflic anhydride (26.6 mL, 44.6 g, 158 mmol) gave the crude product as a clear pale yellow oil. Purification by flash chromatography (silica gel, cyclohexane–EtOAc, 19:1) afforded the desired product as a clear colorless oil (10.0 g, 54%): 1H NMR (400 MHz, CDCl3) δ 0.99 (d, J = 6.5 Hz, 6H), 2.02 (m, 1H), 2.83 (d, J = 7.5 Hz, 2H), 3.76 (s, 3H), 5.99 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 21.8, 22.1, 26.5, 39.8, 51.9, 112.9, 118.4 (q, J = 319.8 Hz, CF3), 164.5, 165.1; HRMS (ES+) m/z calcd for C9H14F3O5S [M + H]+ 291.0514, found 291.0516.
General Procedure D: Synthesis of α,β-Dehydro Cyanoesters
The triflate (1.0 equiv) and Zn(CN)2 (0.6 equiv) were added to dry DMF (75 mL). The mixture was purged with N2 for 15 min before Pd(PPh3)4 (0.01 equiv) was added, and the mixture was heated at 70 °C for 15 h. The reaction mixture was allowed to cool to rt, and toluene (125 mL) was added. The mixture was washed with aqueous ammonium hydroxide (20%, 2 × 50 mL), and the aqueous washings were extracted with toluene (50 mL). The combined organic extracts were washed with brine (50 mL), dried over Na2SO4, and concentrated under reduced pressure to give the crude product.
(Z)-Ethyl 3-Cyanobut-2-enoate [(Z)-1a]37
Using general procedure D, (Z)-ethyl 3-(trifluoromethylsulfonyloxy)but-2-enoate (12.3 g, 46.8 mmol), Zn(CN)2 (3.30 g, 28.1 mmol) in DMF (75 mL), and Pd(PPh3)4 (0.54 g, 0.47 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1) afforded the desired product as a clear pale yellow oil (4.23 g, 65%): 1H NMR (400 MHz, CDCl3) δ 1.33 (t, J = 7.0 Hz, 3H), 2.16 (s, 3H), 4.28 (q, J = 7.0 Hz, 2H), 6.32 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.0, 61.5, 116.4, 122.8, 133.1, 162.7.
(E)-Ethyl 3-Cyanobut-2-enoate [(E)-1a]37
Using general procedure D, (E)-ethyl 3-(trifluoromethylsulfonyloxy)but-2-enoate (12.3 g, 46.8 mmol), Zn(CN)2 (3.30 g, 28.1 mmol) in dry DMF (75 mL), and Pd(PPh3)4 (0.54 g, 0.47 mmol) gave the crude product as a clear red oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc, 9:1) afforded the desired product as a clear pale yellow oil (4.52 g, 69%): 1H NMR (400 MHz, CDCl3): δ 1.31 (t, J = 7.0 Hz, 3H), 2.35 (s, 3H), 4.23 (q, J = 7.0 Hz, 2H), 6.41 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 14.1, 17.3, 61.2, 118.8, 125.9, 132.7, 163.8.
(Z)-Ethyl 3-Cyanopent-2-enoate [(Z)-2a)]
Using general procedure D, (Z)-ethyl 3-(trifluoromethylsulfonyloxy)pent-2-enoate (17.66 g, 63.9 mmol), Zn(CN)2 (4.50 g, 38.4 mmol) in dry DMF (75 mL), and Pd(PPh3)4 (0.74 g, 0.64 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1 afforded the desired product as a clear pale yellow oil (7.27 g, 74%): 1H NMR (400 MHz, CDCl3) δ 1.24 (t, J = 7.5 Hz, 3H), 1.34 (t, J = 7.1 Hz, 3H), 2.45 (qd, J = 7.4, 1.5 Hz, 2H), 4.29 (q, J = 7.1 Hz, 2H), 6.33 (t, J = 1.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 12.0, 14.0, 29.3, 61.5, 115.9, 129.2, 131.4, 162.9; HRMS (ESI) m/z calcd for C8H11NO2Na (M + Na+) 176.0682, found 176.0680.
(E)-Ethyl 3-Cyanopent-2-enoate [(E)-2a]
Using general procedure D, (E)-ethyl 3-(trifluoromethyl sulfonyloxy)pent-2-enoate (10.62 g, 38.4 mmol), Zn(CN)2 (2.71 g, 23.1 mmol) in dry DMF (75 mL), and Pd(PPh3)4 (0.44 g, 0.38 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1) afforded the desired product as a clear pale yellow oil (4.9 g, 84%): 1H NMR (400 MHz, CDCl3) δ 1.23 (t, J = 7.6 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 2.81 (qd, J = 7.6, 1.2 Hz, 2H), 4.24 (q, J = 7.1 Hz, 2H) 6.39 (t, J = 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 12.4, 14.1, 23.6, 61.2, 118.0, 131.8, 132.6, 163.7; HRMS (EI) m/z calcd for C8H11NO2 (M+) 153.0790, found 153.0798.
(Z)-Ethyl 3-Cyanohex-2-enoate [(Z)-3a]
Using general procedure D, (Z)-ethyl 3-(trifluoromethylsulfonyloxy)hex-2-enoate (13.6 g, 46.8 mmol), Zn(CN)2 (3.30 g, 28.1 mmol) in dry DMF (75 mL), and Pd(PPh3)4 (0.54 g, 0.47 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1, product from first column was repurified on a second silica column) afforded the desired product as a clear pale yellow oil (4.56 g, 58%): 1H NMR (300 MHz, CDCl3) δ 0.98 (t, J = 7.4 Hz, 3H), 1.34 (t, J = 7.1 Hz, 3H), 1.68 (m, 2H), 2.37 (td, J = 7.5, 1.5 Hz, 2H), 4.29 (q, J = 7.1 Hz, 2H), 6.32 (t, J = 1.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.1, 14.0, 20.8, 37.7, 61.5, 115.9, 127.8, 132.3, 162.9; HRMS (ESI) m/z calcd for C9H13NO2Na (M + Na+) 190.0839, found 190.0838.
(E)-Ethyl 3-Cyanohex-2-enoate [(E)-3a]
Using general procedure D, (E)-ethyl 3-(trifluoromethylsulfonyloxy)hex-2-enoate (13.6 g, 46.8 mmol), Zn(CN)2 (3.30 g, 28.1 mmol) in dry DMF (75 mL), and Pd(PPh3)4 (0.54 g, 0.47 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc, 19:1, product from first column was repurified on second silica column) afforded the desired product as a clear pale yellow oil (4.02 g, 51%): 1H NMR (400 MHz, CDCl3) δ 1.00 (t, J = 7.4 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.67 (dq, J = 14.8, 7.4 2H), 2.77 (td, J = 7.6, 1.1 Hz, 2H), 4.23 (q, J = 7.0 Hz, 2H), 6.42 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 14.1, 21.3, 31.8, 61.2, 118.2, 131.2, 132.4, 163.7; HRMS (EI) m/z calcd for C9H13NO2 (M+) 167.0946, found 167.0952.
(Z)-Ethyl 5-Methyl-3-cyanohex-2-enoate [(Z)-4a]34
Using general procedure D, (Z)-ethyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate (271.38 g, 1 equiv), Zn(CN)2 (62.8 g, 0.6 equiv) in dry DMF (1.63 L), and Pd(PPh3)4 (10.31 g, 0.01 equiv) gave the crude product as a yellow oil. Purification by chromatography on a Biotage apparatus (25.3g per run using a 65i cartridge (350 g silica) and 1–6% EtOAc in cyclohexane) gave the product as a yellow oil (67 g, 60% yield): 1H NMR (400 MHz, CDCl3) δ 0.90 (d, J = 6.6 Hz, 6H), 1.27 (t, J = 7.2 Hz, 3H), 1.99 (hept, J = 6.8 Hz, 1H), 2.19 (dd, J = 7.2 Hz, 1.2 Hz, 2H), 4.22 (q, J = 7.2 Hz, 2H), 6.21 (t, J = 1.2 Hz, 1H).
(E)-Ethyl 5-Methyl-3-cyanohex-2-enoate [(E)-4a]34
Using general procedure D, (E)-ethyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate (378 g, 1 equiv), Zn(CN)2 (87.6 g, 0.6 equiv) in dry DMF (2.27 L), and Pd(PPh3)4 (14.4g; 0.01 equiv) gave the crude product as a red oil (244 g, 108%). A portion of the crude product (111 g) was purified by chromatography on a Biotage apparatus using a 65i cartridge (350 g silica) and 1–6% EtOAc in cyclohexane to give the product as an yellow-orange oil (55 g, 53%): 1H NMR (400 MHz, CDCl3) δ 0.91 (d, J = 6.7 Hz, 6H), 1.24 (t, J = 7.1 Hz, 3H), 1.95 (hept, J = 6.8 Hz, 1H), 2.61 (dd, J = 7.3, 1.1 Hz, 2H), 4.15 (q, J = 7.1 Hz, 2H), 6.37 (t, J = 1.1 Hz, 1H).
(Z)-Methyl 5-Methyl-3-cyanohex-2-enoate [(Z)-5a]38
Using general procedure D, (Z)-methyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate (14.7 g, 50.8 mmol) and Zn(CN)2 (3.58 g, 30.5 mmol) in dry DMF (75 mL) and Pd(PPh3)4 (0.59 g, 0.51 mmol) gave the crude product as a clear yellow oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1) afforded the desired product as a clear pale yellow oil (7.32 g, 86%): 1H NMR (400 MHz, CDCl3) δ 0.96 (d, J = 6.5 Hz, 6H), 2.05 (m, 1H), 2.20 (dd, J = 7.0, 1.0 Hz, 2H), 3.82 (s, 3H), 6.29 (t, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 27.1, 44.8, 52.3, 116.0, 127.5, 132.6, 163.2.
(E)-Methyl 5-Methyl-3-cyanohex-2-enoate [(E)-5a]34
Using general procedure D, (E)-methyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate (9.96 g, 34.3 mmol), Zn(CN)2 (2.42 g, 20.6 mmol) in dry DMF (75 mL) and Pd(PPh3)4 (0.40 g, 0.34 mmol) gave the crude product as a clear red oil. Purification by flash chromatography (silica gel, cyclohexane/EtOAc 9:1, product from first column was repurified on a second silica column) afforded the desired product as a clear pale yellow oil (4.24 g, 74%): 1H NMR (400 MHz, CDCl3) δ 0.98 (d, J = 6.5 Hz, 6H), 2.02 (m, 1H), 2.69 (dd, J = 7.5, 1.0 Hz, 2H), 3.77 (s, 3H), 6.45 (t, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.0, 28.0, 38.5, 52.0, 118.3, 131.1, 132.5, 164.2.
Synthesis and Characterization of Substrates (Z)-6a and (E)-7a
General Procedure E: Synthesis of α,β-Dehydro Cyanoesters (Z)-6a and (E)-7a
To a solution of starting ester (1 mmol) in alcohol (methanol or n-propanol, 25 mL) in a round-bottom flask (100 mL) equipped with a reflux condenser was added hydrochloric acid (conc, 0.5 mL). The resulting mixture was stirred under reflux until all starting material was consumed. After the reaction was complete, a solution of NaHCO3 (saturated, 15 mL) was added, and the mixture was extracted three times with ethyl acetate. The combined organic phases were dried over Na2SO4 and yielded the crude product after the solvent was removed under reduced pressure.
(Z)-Methyl 3-Cyanohex-2-enoate [(Z)-6a]
Using general procedure E, (Z)-ethyl 3-cyanohex-2-enoate [(Z)-3a] (300 mg, 1.79 mmol) in methanol (40 mL) and hydrochloric acid (conc, 0.5 mL) gave the product after 5 days reaction time. Purification by flash chromatography (silica gel, petroleum ether/EtOAc 20:1) afforded the product (173 mg, 63%): 1H NMR (300 MHz, CDCl3) δ 6.33 (s, 1H), 3.83 (s, 3H), 2.38 (dd, J = 10.8, 4.1, 2H), 1.84–1.60 (m, 2H), 0.98 (t, J = 7.4, 3H); 13C NMR (75 MHz, CDCl3) δ 163.3, 131.9, 128.2, 115.9, 52.3, 37.7, 20.8, 13.1; HRMS (EI) m/z calcd for C8H11NO2 (M+) 153.0790, found 153.0786.
(E)-Propyl 3-Cyano-5-methylhex-2-enoate [(E)-7a]
Using general procedure E, (E)-ethyl 3-cyano-5-methylhex-2-enoate [(E)-5a] (150 mg, 0.82 mmol) in n-propanol (30 mL) and hydrochloric acid (conc., 0.5 mL) gave the pure product (120 mg, 75%) after 5 days reaction time: 1H NMR (300 MHz, CDCl3) δ 6.47 (s, 1H), 4.14 (t, J = 6.7, 2H), 2.70 (dd, J = 7.3, 1.0, 2H), 2.17–1.94 (m, 1H), 1.79–1.64 (m, 2H), 1.00 (d, J = 6.0, 6H), 0.98 (t, J = 9.0, 3H); 13C NMR (75 MHz, CDCl3) δ 163.9, 133.0, 130.6, 118.4, 66.8, 38.5, 28.0, 22.0, 21.8, 10.4; HRMS (EI) m/z calcd for C11H17NO2 (M+) 195.1259, found 195.1277.
Synthesis and Characterization of Racemic Reference Compounds rac-1b–7b
General Procedure F: Synthesis of Racemic Reference Compounds rac-1b–6b
The starting acrylic ester (0.5 mmol) was dissolved in THF (5 mL) and hydrogenated under H2 at atmospheric pressure at room temperature employing 10% Pd/C (5 mg) as catalyst by stirring overnight at room temperature. Afterward, the reaction mixture was filtered through Celite and concentrated to yield the pure hydrogenated product in quantitative conversion.
rac-Ethyl 3-Cyanobutanoate (rac-1b)10
Using general procedure F, hydrogenation of (Z)-ethyl 3-cyanobut-2-enoate [(Z)-1a] (51 mg, 0.39 mmol) gave the pure product (97%, 50.3 mg, 0.35 mmol): 1H NMR (300 MHz, CDCl3) δ 4.22 (dq, J = 14.3, 7.2, 2H), 3.30–2.98 (m, 1H), 2.63 (ddd, J = 56.4, 16.6, 7.2, 2H), 1.39 (d, J = 7.1, 3H), 1.29 (t, J = 7.1, 3H); 13C NMR (75 MHz, CDCl3) δ 169.6, 121.8, 61.3, 38.2, 21.7, 17.7, 14.1.
rac-Ethyl 3-Cyanopentanoate (rac-2b)10
Using general procedure F, hydrogenation of (Z)-ethyl 3-cyanopent-2-enoate [(Z)-2a] (50 mg, 0.33 mmol) gave the pure product (97%, 49.5 mg, 0.32 mmol): 1H NMR (300 MHz, CDCl3) δ 4.20 (q, J = 7.1, 2H), 3.14–2.86 (m, 1H), 2.63 (ddd, J = 23.3, 16.5, 7.2, 2H), 1.84–1.57 (m, 2H), 1.29 (t, J = 7.1, 3H), 1.12 (t, J = 7.4, 3H); 13C NMR (75 MHz, CDCl3) δ 169.8, 121.0, 61.3, 36.4, 29.1, 25.2, 14.1, 11.4.
rac-Ethyl 3-Cyanohexanoate (rac-3b)10
Using general procedure F, hydrogenation of (Z)-ethyl 3-cyanohex-2-enoate [(Z)-3a] (51 mg, 0.31 mmol) gave the pure product (80%, 42 mg, 0.25 mmol): 1H NMR (300 MHz, CDCl3) δ 4.21 (q, J = 7.1, 2H), 3.03 (m, 1H), 2.82–2.46 (m, 2H), 1.82–1.34 (m, 4H), 1.34 (t, J = 9, 3H), 0.98 (t, J = 6.8, 3H); 13C NMR (75 MHz, CDCl3) δ 169.8, 121.1, 61.3, 36.8, 33.8, 27.4, 20.2, 14.1, 13.4.
rac-Methyl 3-Cyano-5-methylhexanoate (rac-5b)34
Using general procedure F, hydrogenation of (Z)-methyl 3-cyano-5-methylhex-2-enoate [(Z)-5a] (90 mg, 0.54 mmol) gave the pure product (93%, 85 mg, 0.50 mmol). 1H NMR (300 MHz, CDCl3): δ 3.75 (s, 3H), 3.16–2.96 (m, 1H), 2.63 (ddd, J = 23.3, 16.6, 7.1, 2H), 1.98–1.76 (m, 1H), 1.65 (ddd, J = 13.5, 10.7, 4.8, 1H), 1.40–1.28 (m, 1H), 0.97 (dd, J = 6.6, 3.5, 6H); 13C NMR (75 MHz, CDCl3): δ 170.2, 121.1, 52.3, 40.7, 36.9, 26.1, 25.8, 22.9, 21.2.
rac-Methyl 3-Cyanohexanoate (rac-6b)39
Using general procedure F, hydrogenation of (Z)-methyl 3-cyano-hex-2-enoate [(Z)-6a] (40 mg, 0.24 mmol) gave the pure product (92%, 38 mg, 0.22 mmol): 1H NMR (300 MHz, CDCl3) δ 3.76 (s, 3H), 3.16–2.94 (m, 1H), 2.66 (ddd, J = 23.4, 16.6, 7.2, 2H), 1.73–1.53 (m, 4H), 0.99 (t, J = 6.9, 3H); 13C NMR (75 MHz, CDCl3) δ 170.3, 121.1, 52.2, 36.6, 33.8, 27.4, 20.2, 13.4.
Preparation of rac-Ethyl 3-Cyano-5-methylhexanoate (rac-4b)34
A 1:1 mixture of (R)- and (S)-ethyl 3-cyano-5-methylhexanoate (4b) was prepared.
Synthesis of rac-Propyl 3-cyano-5-methylhexanoate (rac-7b)
Using general procedure E, rac-ethyl 3-cyano-5-methylhexanoate (rac-4b) (30 mg, 0,17 mmol) in n-propanol (20 mL) and hydrochloric acid (conc, 0.2 mL) gave the pure product (88%, 29 mg, 0,15 mmol) after 24 h reaction time: 1H NMR (300 MHz, CDCl3) δ 4.11 (t, J = 6.7, 2H), 3.07 (dt, J = 12.1, 7.0, 1H), 2.63 (ddd, J = 48.8, 16.5, 7.1, 2H), 1.98–1.56 (m, 4H), 1.41–1.28 (m, 1H), 1.04–0.85 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 169.9, 121.2, 66.9, 40.8, 37.2, 26.1, 25.8, 22.9, 21.9, 21.2, 10.3; HRMS (EI) m/z calcd for C11H18NO2 ([M – H]+) 196.1338, found 196.1329.
Preparative-Scale Bioreduction of β-Cyanoacrylate Esters and Chiral Characterization of (S)-1b–3b
General Procedure for Preparative-Scale Bioreductions of β-Cyanoacrylate Esters20
Biotransformations of (E)-1a, (E)-2a, and (E)-3a were carried out in 100 mL volumes at 30 °C for 19–25 h. Reactions contained alkene substrate (10 mmol), 2-propanol (26 mmol), Lactobacillus brevis alcohol dehydrogenase (3200 U), NADP+ (1 mmol), OYE2 (4–8 g wet E. coli cells; OYE2 was constructed in pET28b and expressed in BL21 E. coli cells), and 100 mM potassium phosphate buffer (pH 7). Reactions were monitored by GC until conversion of alkene substrates were complete. The reaction mixtures were centrifuged and the resulting supernatants were extracted with EtOAc. The organic layers were dried over MgSO4 and concentrated under reduced pressure, and the resulting crude products were purified by Kugelrohr distillation.
(S)-Ethyl 3-Cyanobutanoate [(S)-1b]10
Bioreduction of (E)-1a afforded 1.17 g (83%) of (S)-1b as a clear colorless liquid (100%). Spectroscopic data were in agreement with data for rac-1b; [α]23D +30.7 (c 1.07, MeOH); ee >99% determined by GC analysis (G-TA, 135–165 °C temperature gradient), tR 5.09 min. For determination of absolute configuration, (S)-1b (0.73 g, 5.2 mmol) was converted to the methyl ester by transesterification40 with Ti(OEt)4 (0.5 g, 2.2 mmol) and ethylene glycol (0.07 g, 1.1 mmol) in refluxing MeOH (25 mL) for 118 h. The reaction mixture was concentrated (∼5 mL) under reduced pressure, diluted with 10 mL of 4 N aqueous HCl, and extracted with EtOAc. The organic extract was washed with water, dried over magnesium sulfate, concentrated under reduced pressure and Kugelrohr distilled to give 0.46 g (60%) of (S)-methyl 3-cyanobutanoate as a clear liquid (97%, contaminated with ∼3% (S)-1b). Spectral data and specific rotation for (S)-methyl 3-cyanobutanoate were consistent with literature values:12 [α]23D +33.2 (c 1.11, MeOH); ee >99% determined by GC analysis (γ-TA, 135–165 °C temperature gradient) tR 4.79 min; lit.41 [α]23D +35.3 (c 1.05, MeOH), 93% ee.
(S)-Ethyl 3-Cyanopentanoate [(S)-2b]10
Bioreduction of (E)-2a afforded 1.0 g (64%) of (S)-2b as a clear colorless liquid (>99%). Spectroscopic data were in agreement with data for racemic 2b. [α]23D +12.7 (c 1.03, MeOH); ee >99% determined by GC analysis (G-TA 135–165 °C temperature gradient), tR 6.16 min. For determination of absolute configuration, (S)-2b (0.73 g, 4.7 mmol) was converted to the methyl ester by transesterification according to the procedure described for transesterification of (S)-1b. 0.56 g (84%) of (S)-methyl 3-cyanopentanoate was obtained as a clear liquid (∼97%, contaminated with ∼3% (S)-2b). Spectral data and specific rotation for (S)-methyl 3-cyanopentanoate were consistent with literature values:41 [α]23D +14.9 (c 1.06, MeOH); ee >99% determined by GC analysis (G-TA, 135–165 °C temperature gradient) tR 5.82 min [lit.41 [α]24D +11.8 (c 1.00, MeOH), 98% ee].
(S)-Ethyl 3-Cyanohexanoate [(S)-3b]10
Bioreduction of (E)-3a afforded 1.05 g (60%) of (S)-3b as a clear colorless liquid (98%). Spectroscopic data were in agreement with data for racemic 3b: [α]23D +2.2 (c 1.03, MeOH); ee >99% determined by GC analysis (G-TA, 135 °C - 165 °C temperature gradient) tR = 7.18 min. For determination of absolute configuration, (S)-3b (0.66 g, 3.9 mmol) was converted to the corresponding carboxylic acid by hydrolysis with 50 wt % NaOH (0.25 mL) in MeOH (2.5 mL) at room temperature for 3 h. The reaction mixture was acidified with 2 mL of 4 N HCl, diluted with 10 mL of water and extracted with EtOAc. The organic extract was dried over Na2SO4 and concentrated under reduced pressure to give 0.5 g (91%) of (S)-3-cyanohexanoic acid as a viscous liquid. Spectral data for (S)-3-cyanohexanoic acid were consistent with literature values for (R)-3-cyanohexanoic acid and specific rotation was consistent with literature value in magnitude but opposite in direction:39 [α]23D −3.1 (c 1.18, CHCl3). For chiral GC analysis, the acid (0.25 mg in 0.5 mL EtOAc) was converted to the methyl ester by treatment with TMS-diazomethane (0.02 mL) and MeOH (0.02 mL) for 30 min at room temperature; ee >99% determined by GC analysis (G-TA, 135–165 °C temperature gradient) tR 6.66 min [lit.13 [α]25D +1.1 (c 1.1, CHCl3), 98% ee].
Synthesis and Characterization of Chiral Reference Compounds (R)-4b and (S)-4b–7b
(S)-Ethyl 3-Cyano-5-methylhexanoate [(S)-4b]
Reference material for (S)-4b was independently prepared from diethyl 2-(1-cyano-3-methylbutyl)malonate according to the published procedure11 and purified by Kugelrohr distillation for characterization: 98.3% ee determined by GC analysis (G-TA, 135–165 °C temperature gradient) tR = 7.53 min; [α]20D −10.9 (c 1.09, MeOH). Spectral data for (S)-4b were in agreement with data for rac-4b and with previously reported data.42
(R)-Ethyl 3-Cyano-5-methylhexanoate [(R)-4b]
Preparation of (S)-4b also gave (R)-diethyl 2-(1-cyano-3-methylbutyl)malonate as a byproduct of the enzymatic hydrolysis step.11 For preparation of (R)-4b, the published procedure was modified by running the enzymatic hydrolysis step to >50% conversion to obtain (R)-diethyl 2-(1-cyano-3-methylbutyl)malonate with higher enantiomeric purity. Partial hydrolysis of (R)-diethyl 2-(1-cyano-3-methylbutyl)malonate was carried out with KOH in EtOH and H2O. Aqueous KOH (8.8 wt %, 13.6 g, 21.3 mmol) was slowly added over 1 h to an ice-cold solution of (R)-diethyl 2-(1-cyano-3-methylbutyl)malonate (5.0 g, Kugelrohr distilled, 19.6 mmol, >99% ee) in EtOH (5.9 mL). The ice bath was removed and the reaction was stirred for 18 h at rt. The reaction mixture was extracted with toluene (3 × 10 mL) and the toluene layers discarded. The remaining aqueous layer was stirred at 100 °C for 4 h. The reaction mixture was extracted with EtOAc, dried over MgSO4 and concentrated under reduced pressure to give the crude product. Purification of the crude product by Kugelrohr distillation afforded (R)-4b as a clear colorless liquid (>99% by GC, 1.18 g, 32.8%); 89.8% ee determined by GC analysis (G-TA, 135–165 °C temperature gradient) tR 7.34 min; [α]20D +10.9 (c 1.08, MeOH). Spectral data for (R)-4b were in agreement with data for rac-4b.
(S)-Methyl 3-Cyano-5-methylhexanoate [(S)-5b]
Using general procedure E, (S)-ethyl 3-cyano-5-methylhexanoate [(S)-4b] (52 mg, 0.28 mmol) in methanol (5 mL) and hydrochloric acid (conc, 0.2 mL) gave the pure product (73%, 35 mg, 0.21 mmol) after 24 h reaction time. Spectral data for (S)-5b were in agreement with data for rac-5b.
(S)-Methyl 3-Cyanohexanoate [(S)-6b]
Using general procedure E, (S)-ethyl 3-cyanohexanoate [(S)-3b] (11 mg, 0.07 mmol) in methanol (5 mL) and hydrochloric acid (conc, 0.1 mL) gave the pure product (54%, 7 mg, 0.04 mmol) after 24 h reaction time. Spectral data for (S)-6b were in agreement with data for rac-6b.
(S)-Propyl 3-Cyano-5-methylhexanoate [(S)-7b]
Using general procedure E, (S)-ethyl 3-cyano-5-methylhexanoate [(S)-4b] (30 mg, 0.16 mmol) in n-propanol (20 mL) and hydrochloric acid (conc, 0.5 mL) gave the pure product (99%, 32 mg, 0.16 mmol) after 24 h reaction time. Spectral data for (S)-7b were in agreement with data for rac-7b.
Acknowledgments
We acknowledge Almac Sciences (Nick Tyrrell & Scott Wharry, Craigavon, Northern Ireland) and Celtic Catalyts (Johan Granander, Dublin, Ireland) for assistance in the preparation of analogues 1a–5a. Financial support by the Austrian Science Fund (FWF, Vienna, project P22722) is gratefully acknowledged. Klaus Zangger (Graz) is cordially thanked for his help in NMR spectroscopy. Brian Jones and Robert Saf are thanked for their help in obtaining HRMS analyses.
Supporting Information Available
13C and 1H NMR for new compounds: (Z)-ethyl 3-(trifluoromethylsulfonyloxy)hex-2-enoate, (Z)-methyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate, (E)-methyl 5-methyl-3-(trifluoromethylsulfonyloxy)hex-2-enoate, rac-1b-7b, (E)-2a, (Z)-2a, (E)-3a, (Z)-3a, (Z)-6a, (E)-7a, and (S)-4b and chiral analytical methods. Cloning and purification of EBP1 from Candida albicans and of Lycopersicon esculentum OPR1 variants Cys20Tyr, Ile287His, and His245Asp. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Winkler C. K.; Tasnadi G.; Clay D.; Hall M.; Faber K. J. Biotechnol. 2012, 162, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuermer R.; Hauer B.; Hall M.; Faber K. Curr. Opin. Chem. Biol. 2007, 11, 203–213. [DOI] [PubMed] [Google Scholar]
- Toogood H. S.; Gardiner J. M.; Scrutton N. S. ChemCatChem 2010, 2, 892–914. [Google Scholar]
- Winkler C. K.; Stueckler C.; Mueller N. J.; Pressnitz D.; Faber K. Eur. J. Org. Chem. 2010, 6354–6358. [Google Scholar]
- Stueckler C.; Winkler C. K.; Bonnekessel M.; Faber K. Adv. Synth. Catal. 2010, 352, 2663–2666. [Google Scholar]
- Stueckler C.; Winkler C. K.; Hall M.; Hauer B.; Bonnekessel M.; Zangger K.; Faber K. Adv. Synth. Catal. 2011, 353, 1169–1173. [Google Scholar]
- Swiderska M. A.; Stewart J. D. Org. Lett. 2006, 8, 6131–6133. [DOI] [PubMed] [Google Scholar]
- Kosjek B.; Fleitz F. J.; Dormer P. G.; Kuethe J. T.; Devine P. N. Tetrahedron: Asymmetry 2008, 19, 1403–1406. [Google Scholar]
- Stueckler C.; Mueller N. J.; Winkler C. K.; Glueck S. M.; Gruber K.; Steinkellner G.; Faber K. Dalton Trans. 2010, 39, 8472–8476. [DOI] [PubMed] [Google Scholar]
- Mukherjee H.; Martinez C. A. ACS Catal. 2011, 1, 1010–1013. [Google Scholar]
- Martinez C. A.; Hu S.; Dumond Y.; Tao J.; Kelleher P.; Tully L. Org. Process Res. Dev. 2008, 12, 392–398. [Google Scholar]
- Dunn P. J.; Hettenbach K.; Kelleher P.; Martinez C. In Green Chemistry in the Pharmaceutical Industry; Dunn P. J., Wells A., Williams M., Eds.; Wiley-VCH: Weinheim, 2010; p 161. [Google Scholar]
- Babinski D.; Soltani O.; Frantz D. E. Org. Lett. 2008, 10, 2901–2904. [DOI] [PubMed] [Google Scholar]
- Selnick H. G.; Smith G. R.; Tebben A. J. Synth. Commun. 1995, 25, 3255–3261. [Google Scholar]
- Stueckler C.; Hall M.; Ehammer H.; Pointner E.; Kroutil W.; Macheroux P.; Faber K. Org. Lett. 2007, 9, 5409–5411. [DOI] [PubMed] [Google Scholar]
- Brenna E.; Fronza G.; Fuganti C.; Monti D.; Parmeggiani F. J. Mol. Catal. B Enzym. 2011, 73, 17–21. [Google Scholar]
- Brenna E.; Gatti F. G.; Manfredi A.; Monti D.; Parmeggiani F. Org. Process Res. Dev. 2011, 16, 262–268. [Google Scholar]
- Tasnádi G.; Winkler C. K.; Clay D.; Sultana N.; Fabian W. M. F.; Hall M.; Ditrich K.; Faber K. Chem.—Eur. J. 2012, 18, 10362–10367. [DOI] [PubMed] [Google Scholar]
- Tasnadi G.; Winkler C. K.; Clay D.; Hall M.; Faber K. Catal. Sci. Technol. 2012, 2, 1548–1552. [Google Scholar]
- Tauber K.; Hall M.; Kroutil W.; Fabian W. M. F.; Faber K.; Glueck S. M. Biotechnol. Bioeng. 2011, 108, 1462–1467. [DOI] [PubMed] [Google Scholar]
- Bougioukou D. J.; Kille S.; Taglieber A.; Reetz M. T. Adv. Synth. Catal. 2009, 351, 3287–3305. [Google Scholar]
- Padhi S. K.; Bougioukou D. J.; Stewart J. D. J. Am. Chem. Soc. 2009, 131, 3271–3280. [DOI] [PubMed] [Google Scholar]
- Hall M.; Stueckler C.; Kroutil W.; Macheroux P.; Faber K. Angew. Chem., Int. Ed. 2007, 46, 3934–3937. [DOI] [PubMed] [Google Scholar]
- Breithaupt C.; Kurzbauer R.; Lilie H.; Schaller A.; Strassner J.; Huber R.; Macheroux P.; Clausen T. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14337–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzing K.; Fitzpatrick T. B.; Wilken C.; Sawa J.; Bourenkov G. P.; Macheroux P.; Clausen T. J. Biol. Chem. 2005, 280, 27904–27913. [DOI] [PubMed] [Google Scholar]
- Muller A.; Hauer B.; Rosche B. Biotechnol. Bioeng. 2007, 98, 22–29. [DOI] [PubMed] [Google Scholar]
- Hall M.; Stueckler C.; Hauer B.; Stuermer R.; Friedrich T.; Breuer M.; Kroutil W.; Faber K. Eur. J. Org. Chem. 2008, 1511–1516. [Google Scholar]
- Mueller N. J.; Stueckler C.; Hauer B.; Baudendistel N.; Housden H.; Bruce Neil C.; Faber K. Adv. Synth. Catal. 2010, 352, 387–394. [Google Scholar]
- Durchschein K.; Ferreira-da Silva B.; Wallner S.; Macheroux P.; Kroutil W.; Glueck S. M.; Faber K. Green Chem. 2010, 12, 616–619. [Google Scholar]
- Yanto Y.; Winkler C. K.; Lohr S.; Hall M.; Faber K.; Bommarius A. S. Org. Lett. 2011, 13, 2540–2543. [DOI] [PubMed] [Google Scholar]
- Mangan D.; Miskelly I.; Moody T. S. Adv. Synth. Catal. 2012, 354, 2185–2190. [Google Scholar]
- de Souza L. C.; dos Santos A. F.; Sant’Ana A. E. G.; de Oliveira Imbroisi D. Bioorg. Med. Chem. 2004, 12, 865–869. [DOI] [PubMed] [Google Scholar]
- Xuefei Z.; Xiaofei J.; Xiaoxia W.; Guanqun X. Synth. Commun. 2007, 37, 1617–1625. [Google Scholar]
- Burrell A. J. M.; Martinez C. A.; McDaid P. O.; O’Neill P. M.; Wong J. W. PCT Int. Appl. WO 2012/025861 A1;; Chem. Abstr. 2012, 156, 336927. [Google Scholar]
- Davis J. B.; Bailey J. D.; Sello J. K. Org. Lett. 2009, 11, 2984–2987. [DOI] [PubMed] [Google Scholar]
- Wada A.; Fukunaga K.; Ito M.; Mizuguchi Y.; Nakagawa K.; Okano T. Bioorg. Med. Chem. 2004, 12, 3931–3942. [DOI] [PubMed] [Google Scholar]
- Mowry D. T.; Rossow A. G. J. Am. Chem. Soc. 1945, 67, 926–928. [Google Scholar]
- Hirata Y.; Yada A.; Morita E.; Nakao Y.; Hiyama T.; Ohashi M.; Ogoshi S. J. Am. Chem. Soc. 2010, 132, 10070–10077. [DOI] [PubMed] [Google Scholar]
- Sammis G. M.; Jacobsen E. N. J. Am. Chem. Soc. 2003, 125, 4442–4443. [DOI] [PubMed] [Google Scholar]
- Schnurrenberger P.; Züger M. F.; Seebach D. Helv. Chim. Acta 1982, 65, 1197–1201. [Google Scholar]
- Enders D.; Niemeier O. Heterocycles 2005, 66, 385–403. [Google Scholar]
- Hoekstra M. S.; Sobieray D. M.; Schwindt M. A.; Mulhern T. A.; Grote T. M.; Huckabee B. K.; Hendrickson V. S.; Franklin L. C.; Granger E. J.; Karrick G. L. Org. Process Res. Dev. 1997, 1, 26–38. [Google Scholar]
- Breithaupt C.; Strassner J.; Breitinger U.; Huber R.; Macheroux P.; Schaller A.; Clausen T. Structure 2001, 9, 419–429. [DOI] [PubMed] [Google Scholar]
- Morley K. L.; Kazlauskas R. J. Trends Biotechnol. 2005, 23, 231–237. [DOI] [PubMed] [Google Scholar]
- Mangan D.; Miskelly I.; Moody T. S. Adv. Synth. Catal. 2012, 354, 2185–2190. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.