

Abstract
OBJECTIVE
Taspoglutide is a long-acting glucagon-like peptide 1 receptor agonist developed for treatment of type 2 diabetes. The efficacy and safety of once-weekly taspoglutide was compared with twice-daily exenatide.
RESEARCH DESIGN AND METHODS
Overweight adults with inadequately controlled type 2 diabetes on metformin ± a thiazolidinedione were randomized to subcutaneous taspoglutide 10 mg weekly (n = 399), taspoglutide 20 mg weekly (n = 398), or exenatide 10 µg twice daily (n = 392) in an open-label, multicenter trial. The primary end point was change in HbA1c after 24 weeks.
RESULTS
Mean baseline HbA1c was 8.1%. Both doses of taspoglutide reduced HbA1c significantly more than exenatide (taspoglutide 10 mg: –1.24% [SE 0.09], difference –0.26, 95% CI –0.37 to –0.15, P < 0.0001; taspoglutide 20 mg: –1.31% [0.08], difference –0.33, –0.44 to –0.22, P < 0.0001; exenatide: –0.98% [0.08]). Both taspoglutide doses reduced fasting plasma glucose significantly more than exenatide. Taspoglutide reduced body weight (taspoglutide 10 mg, –1.6 kg; taspoglutide 20 mg, –2.3 kg) as did exenatide (–2.3 kg), which was greater than with taspoglutide 10 mg (P < 0.05). HbA1c and weight effects were maintained after 52 weeks. More adverse events with taspoglutide 10 and 20 mg than exenatide developed over time (nausea in 53, 59, and 35% and vomiting in 33, 37, and 16%, respectively). Allergic and injection-site reactions were more common with taspoglutide. Discontinuations were greater with taspoglutide. Antitaspoglutide antibodies were detected in 49% of patients.
CONCLUSIONS
Once-weekly taspoglutide demonstrated greater glycemic control than twice-daily exenatide with comparable weight loss, but with unacceptable levels of nausea/vomiting, injection-site reactions, and systemic allergic reactions.
Glucagon-like peptide 1 (GLP-1) receptor agonists have emerged as antihyperglycemic medications with added therapeutic value beyond glucose-lowering properties. Exenatide,a twice-daily GLP-1 mimetic, and liraglutide, a once-daily GLP-1 analog, are currently licensed for the treatment of type 2 diabetes. In randomized clinical trials, these subcutaneously administered compounds have demonstrated antihyperglycemic and weight loss effects with a low risk of hypoglycemia (1). The most common adverse events with exenatide and liraglutide are gastrointestinal disturbances such as nausea (8–44 and 8–35%, respectively) and vomiting (4–13 and 7–12%, respectively), which have limited their use and adherence in clinical practice (2–5).
The investigational GLP-1 receptor agonist taspoglutide has 93% homology with endogenous GLP-1 and was considered to have potency equivalent to GLP-1 (6). In short-term phase 2 clinical studies, once-weekly taspoglutide demonstrated meaningful antihyperglycemic and weight loss effects (7,8). Conceivably, weekly administration of a GLP-1 receptor agonist, such as taspoglutide, could result in beneficial effects on glycemic control as well as greater acceptability by patients, enhancing treatment compliance.
The American Diabetes Association/European Association for the Study of Diabetes consensus statement, which includes the use of GLP-1 receptor agonists as a secondary option to add to metformin, recommends head-to-head comparative studies to assess the value of new agents to achieve the currently recommended glycemic goals and their safety profiles (9). Accordingly, we designed a long-term study (T-emerge 2) to compare the efficacy and safety of once-weekly taspoglutide with twice-daily exenatide in patients with type 2 diabetes inadequately controlled with metformin, thiazolidinedione, or a combination of metformin and thiazolidinedione. Prior to the completion of the long-term extension arm of this study, the taspoglutide phase 3 clinical trials were terminated because of a significantly increased rate of unwanted adverse events. Nevertheless, we believe that transparent reporting of the T-emerge 2 study results will provide important information to help put in perspective important safety issues related to current and future trials with GLP-1 receptor agonists. We report the key efficacy results from the 24-week, open-label, active-controlled core phase and the 28-week, open-label extension phase. We are also presenting the cumulative safety data for the entire study up to the last dose administered (week 104).
RESEARCH DESIGN AND METHODS
Eligible participants were 18–75 years of age with type 2 diabetes, HbA1c between 7 and 10%, and BMI ≥25 kg/m2 (>23 kg/m2 for Asians) and ≤45 kg/m2 (with stable body weight [±5%] for 3 months), and were receiving a stable dose of antihyperglycemic medication (metformin ≥1,500 mg/day, a thiazolidinedione [either rosiglitazone ≥4 mg/day or pioglitazone ≥30 mg/day], or both) for ≥3 months prior to screening. Key exclusion criteria were advanced diabetes complications, gastrointestinal disease, previous bariatric surgery, pancreatitis, cardiovascular disease, or previous exposure to GLP-1 receptor agonists.
The trial was conducted in accordance with the Declaration of Helsinki and national regulations, and the protocol was approved by local independent ethics committees or institutional review boards. All participants provided written consent prior to any procedure.
Study design and interventions
T-emerge 2 was a randomized, open-label, active-comparator, parallel-group, phase 3 trial with a 24-week core phase, a 28-week extension phase, and an optional 104-week, long-term extension phase; the trial was conducted at 189 sites in 23 countries. Participants were randomly assigned (1:1:1) to receive subcutaneous injections of taspoglutide 10 mg weekly, taspoglutide 10 mg weekly for the initial 4 weeks followed by 20 mg weekly, or exenatide (Byetta; Amylin Pharmaceuticals, San Diego, CA) 5 µg twice daily for the initial 4 weeks followed by 10 µg twice daily. Participants who completed the initial 24 weeks of treatment entered the 28-week extension phase. At week 52, patients were invited to participate in the 104-week, long-term extension phase maintaining the same randomized study agent.
Taspoglutide was administered subcutaneously before breakfast once a week. Exenatide was injected as per prescribing information within a 60-min period before the morning and evening meals. All patients received self blood glucose monitoring devices (ACCU-CHEK; Roche Diagnostics, Indianapolis, IN). During the study, background antihyperglycemic treatment was maintained at prestudy doses. If glycemic control deteriorated (fasting plasma glucose >13.3 mmol/L [>240 mg/dL] between weeks 4 and 8, >12.2 mmol/L [>220 mg/dL] between weeks 8 and 12, and >11.1 mmol/L [>200 mg/dL] between weeks 12 and 24), additional antihyperglycemic rescue medication was prescribed (first choice was a sulfonylurea), and patients continued in the study.
End points and assessments
The primary efficacy end point was the absolute change from baseline in HbA1c (%) after 24 weeks of treatment. Secondary efficacy end points included changes in HbA1c (%), fasting plasma glucose, and body weight during 52 weeks of treatment and changes in fasting proinsulin, fasting proinsulin/insulin ratio, and homeostasis model assessment of β-cell function after 52 weeks of treatment. Exploratory end points included changes in lipid profile, high-sensitivity C-reactive protein, and blood pressure after 52 weeks of treatment and the proportion of patients who received rescue medication. Samples were assayed by a central laboratory (Covance Central Laboratory, Geneva, Switzerland).
Safety assessments included adverse events, vital signs, physical examinations, clinical laboratory tests, electrocardiograms, and testing for antitaspoglutide antibodies (only in patients receiving taspoglutide).
Randomization and statistical analysis
Randomization was stratified by baseline HbA1c (<8.0 or ≥8.0%) and background antidiabetic treatment. Randomization was performed centrally using either a telephone- or web-based system. Investigators and sponsor were masked to the results of efficacy assessments during the study. Approximately 330 subjects per arm were needed to provide at least 80% power with an α = 0.05 for the noninferiority test of taspoglutide versus exenatide, assuming a noninferiority limit of 0.3% (this margin used for sample size calculation only), a 0% difference from exenatide in HbA1c change from baseline, and a standard deviation of 1.2. Analyses of efficacy outcomes at 24 and 52 weeks were based on the intention-to-treat population, which consisted of all randomized patients who received at least one dose of study drug and had an evaluable baseline and at least one evaluable postbaseline measurement of HbA1c. The per-protocol population was used as sensitivity analysis to check the robustness of the results. The primary end point was determined using ANOVA, with treatment, region, and background antihyperglycemic treatment as variables and baseline value of the end point as covariate. Missing values were imputed as the last observation carried forward. HbA1c was tested in each of the two active arms versus exenatide for noninferiority first (if the upper limit of the two-sided 95% CI for the treatment difference was <0.4%, a prespecified noninferiority margin). The Hochberg procedure was used to control for the two comparisons. Superiority (if the upper limit of the two-sided CI limit was <0) was tested under a gatekeeping test procedure only if noninferiority was met in both arms. Changes in fasting plasma glucose and body weight were analyzed similarly with the testing sequence. The other continuous secondary and exploratory end points were assessed using ANCOVA but were not part of the testing sequence. HbA1c response rates and related 95% CIs were calculated according to Pearson–Clopper. Patients were included in the safety analysis if they received at least one dose of the study drug and had at least one safety follow-up (or reported an adverse event).
RESULTS
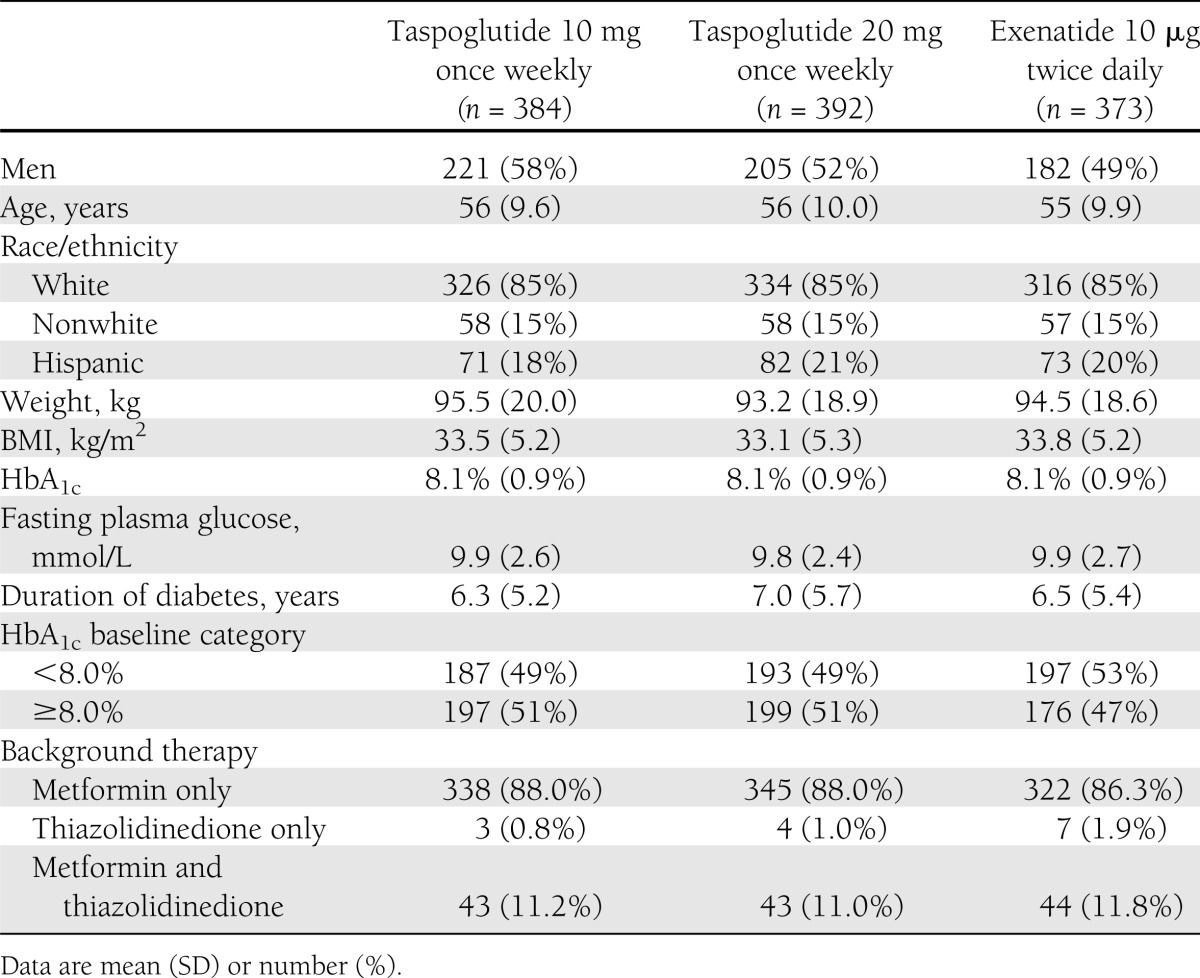
The first participant was enrolled on 25 July 2008, and the final visit for any subject was 22 December 2010. Of the 1,189 patients randomized, 1,173 patients were included in the safety population and 1,149 patients were included in the intention-to-treat population of the 24-week core phase. Baseline demographic and disease characteristics for the intention-to-treat population were well balanced between treatment groups (Table 1). Of the 784 patients who completed the core and extension phases of the study up to 52 weeks, 664 patients entered the long-term extension phase of the study. During the first 24-week study period, withdrawal rates were similar between groups (16, 22, and 16% of patients receiving taspoglutide 10 mg, taspoglutide 20 mg, or exenatide, respectively), but were clearly higher in the taspoglutide groups at study end (26, 34, and 16%, respectively) (see Supplementary Fig. 1 for complete description of patient disposition).
Table 1.
Baseline demographic and disease characteristics (intention-to-treat population, n = 1,149)
Efficacy
After 24 weeks of treatment, least squares mean change in HbA1c was –1.24% (SE 0.09), –1.31% (0.08), and –0.98% (0.08) in the taspoglutide 10-mg, taspoglutide 20-mg, and exenatide groups, respectively, from a mean baseline HbA1c of 8.1% (0.9). Reduction in HbA1c with both doses of taspoglutide was significantly greater than with exenatide (estimated treatment difference of –0.26 [95% CI –0.37 to –0.15], P < 0.0001, and –0.33 [–0.44 to –0.22], P < 0.0001, for taspoglutide 10 and 20 mg, respectively), which met noninferiority and then superiority criteria. These reductions in HbA1c persisted through 52 weeks of treatment, with a change in HbA1c of –1.16% (SE 0.09), –1.18% (0.09), and –0.94% (0.09) in the taspoglutide 10-mg, taspoglutide 20-mg, and exenatide groups, respectively (Fig. 1A and B). Reduction in HbA1c with both doses of taspoglutide was significantly greater than with exenatide (estimated treatment difference of –0.22 [95% CI –0.34 to –0.11], P < 0.0005, and –0.25 [–0.37 to –0.13], P < 0.0001, for taspoglutide 10 and 20 mg, respectively), sustaining the superiority criterion.
Figure 1.

Glycemic control and body weight. A: HbA1c values from baseline to week 52. B: Change in HbA1c values from baseline to weeks 24 and 52. C: Fasting plasma glucose concentrations from baseline to week 52. D: Change in fasting plasma glucose concentrations from baseline to weeks 24 and 52. E: Body weight from baseline to week 52. F: Change in body weight from baseline to weeks 24 and 52. A, C, and E: Open circle, taspoglutide 10 mg once weekly (n = 384), baseline 8.1%; closed circle, taspoglutide 20 mg once weekly (n = 392), baseline 8.1%; open square, exenatide 10 μg twice daily (n = 373), baseline 8.1%. B, D, and F: White bar, taspoglutide 10 mg; black bar, taspoglutide 20 mg; striped bar, exenatide.
Taspoglutide reduced fasting plasma glucose significantly more than exenatide at 24 weeks, with changes of –2.18 (0.2) mmol/L, –2.48 (0.2) mmol/L, and –1.81 (0.19) mmol/L from baseline 9.9 (0.13) mmol/L, 9.8 (0.13) mmol/L, and 9.0 (0.13) mmol/L in the taspoglutide 10-mg, taspoglutide 20-mg, and exenatide groups, respectively (estimated treatment difference vs. exenatide of –0.37 [95% CI –0.63 to –0.11], P < 0.01, and –0.67 [–0.94 to –0.41], P < 0.0001, for taspoglutide 10 and 20 mg, respectively). Reductions persisted after 52 weeks of treatment, and taspoglutide reduced fasting plasma glucose more than exenatide (estimated treatment difference vs. exenatide of –0.31 [–0.62 to –0.01], P = 0.054, and –0.34 [–0.64 to –0.03], P = 0.034, for taspoglutide 10 and 20 mg, respectively) (Fig. 1C and D). Taspoglutide reduced body weight in a dose-dependent manner at week 24, with changes from baseline of –1.6 (0.4) kg, –2.3 (0.4) kg, and –2.3 (0.4) kg in patients receiving taspoglutide 10 mg, taspoglutide 20 mg, or exenatide, respectively. At week 52, weight loss was maintained in all groups and met the prespecified criteria for noninferiority versus exenatide (limit of 3 kg) in the taspoglutide 10- and 20-mg groups; however, weight loss with taspoglutide 10 mg was significantly less than with exenatide (P = 0.01) (Fig. 1E and F).
Only taspoglutide (both doses) significantly increased homeostasis model assessment of β-cell function from baseline; the differences were significantly better than with exenatide. No significant differences were observed in fasting insulin from baseline or between treatment groups. All treatments significantly decreased the proinsulin/insulin ratio to a similar degree (see Supplementary Table 1 for changes in indices of islet function and cardiovascular risk).
Safety and tolerability
During the entire study (core, extension, and long-term extension phases), adverse events were reported among 92, 94, and 89% of patients treated with taspoglutide 10 mg, taspoglutide 20 mg, and exenatide, respectively (Table 2). Among taspoglutide-treated patients experiencing severe adverse events, 34% were gastrointestinal disorders, 6% were injection-site reactions, and 4% were hypersensitivity reactions. In the exenatide group, nausea (9%) and nephrolithiasis (5%) were the most common severe adverse events. A total of 121 patients experienced serious adverse events. Those attributed to study treatment by the investigators included anaphylactic reaction (n = 2), anaphylactoid reaction (n = 1), hypersensitivity (n = 1), and dyspepsia (n = 1) for taspoglutide 10 mg; hypersensitivity (n = 3), anaphylactoid reaction (n = 2), abdominal pain (n = 1), pancreatitis (n = 1), and acute myocardial infarction (n = 1) for taspoglutide 20 mg; and hypoglycemia (n = 1) for exenatide. Four patients died during the study. Causes of death were completed suicide (n = 1) and hemorrhagic stroke (n = 1) in the taspoglutide 10-mg group, myocardial infarction (n = 1) in the taspoglutide 20-mg group, and bleeding varicose vein (n = 1) in the exenatide group. These deaths occurred during the extension phase (n = 3) and the long-term extension phase (n = 1) and were deemed not related to treatment by the investigators.
Table 2.
Summary of adverse events and withdrawals during the entire study (up to 104 weeks)
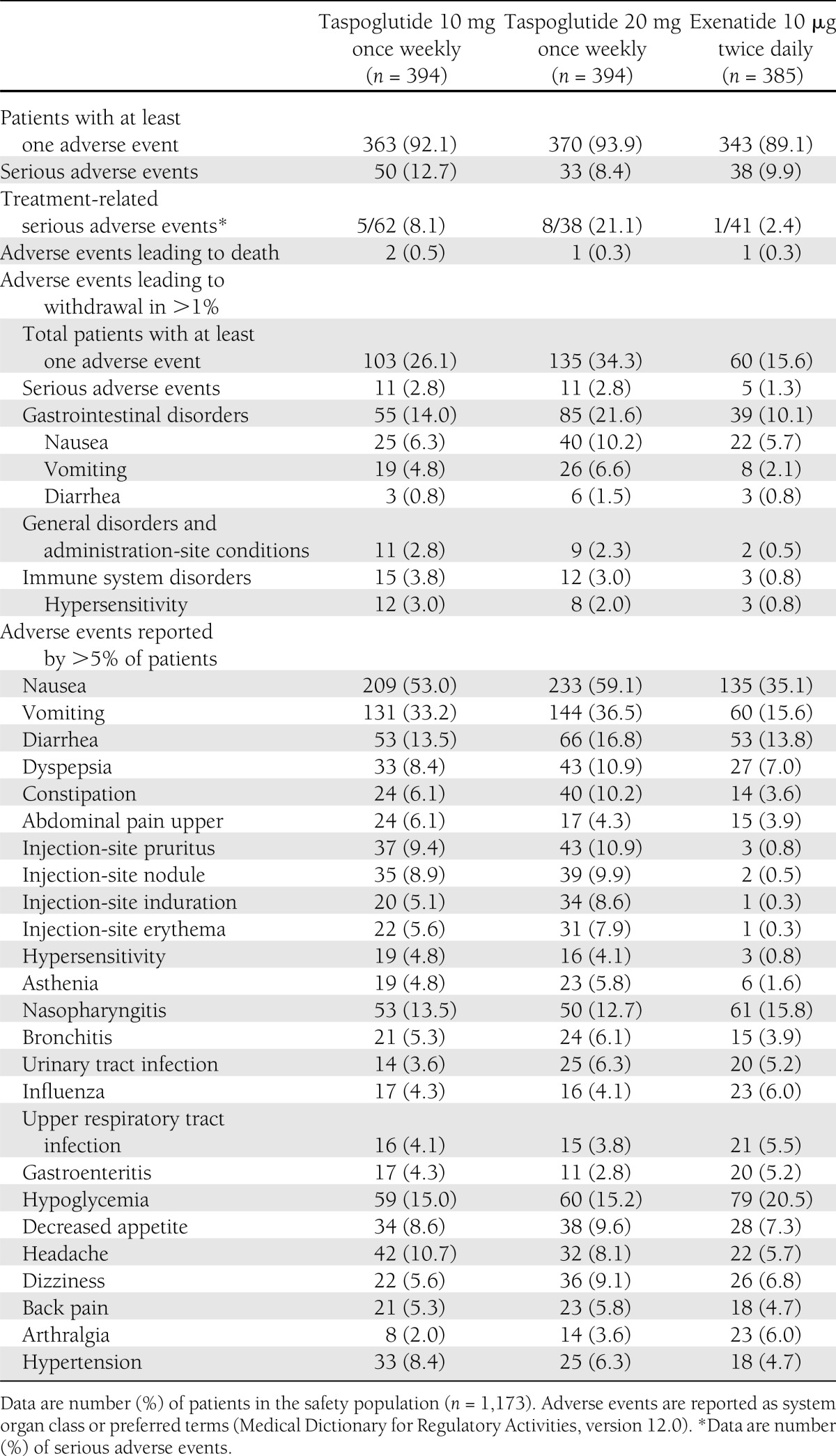
Withdrawal due to adverse events was most commonly due to nausea and vomiting (taspoglutide 10 mg [6.3 and 4.8%, respectively], taspoglutide 20 mg [10.2 and 6.6%], and exenatide [5.7 and 2.1%]) (Table 2). Immune system disorders (including hypersensitivity, anaphylactoid, or anaphylactic reactions) and injection-site adverse events were also reasons for withdrawal more often in the taspoglutide groups than in the exenatide group. One case of pancreatitis of severe intensity was reported in the taspoglutide 20-mg group during the long-term extension phase of the study. Although it resolved without sequelae, it led to study withdrawal.
The most frequent adverse events were gastrointestinal disorders, with higher incidences in the taspoglutide 10-mg (68%) and 20-mg (72%) groups than in the exenatide (57%) group (Table 2). Injection-site reactions were more frequently reported with taspoglutide 10 mg (35%) and taspoglutide 20 mg (41%) than with exenatide (6%) (Table 2).
Systemic allergic reactions were reported in 51 patients, occurring in 25 and 23 patients treated with taspoglutide 10 and 20 mg (each 6%), respectively, versus 3 (1%) with exenatide. Hypersensitivity was the most common systemic allergic reaction reported in 19 (5%) and 16 (4%) patients in the taspoglutide 10- and 20-mg groups, respectively, and in 3 (1%) patients in the exenatide group. Serious systemic allergic reaction adverse events occurred in four patients treated with taspoglutide 10 mg (anaphylactic reaction [n = 2], anaphylactoid reaction [n = 1], and hypersensitivity [n = 1]) and five patients treated with taspoglutide 20 mg (hypersensitivity [n = 2], anaphylactoid reaction [n = 2], and type I hypersensitivity [n = 1]). Study withdrawal due to systemic allergic reaction adverse events occurred in 18 (5%) patients in the taspoglutide 10-mg group, 13 (3%) patients in the taspoglutide 20-mg group, and 2 (0.5%) patients in the exenatide group.
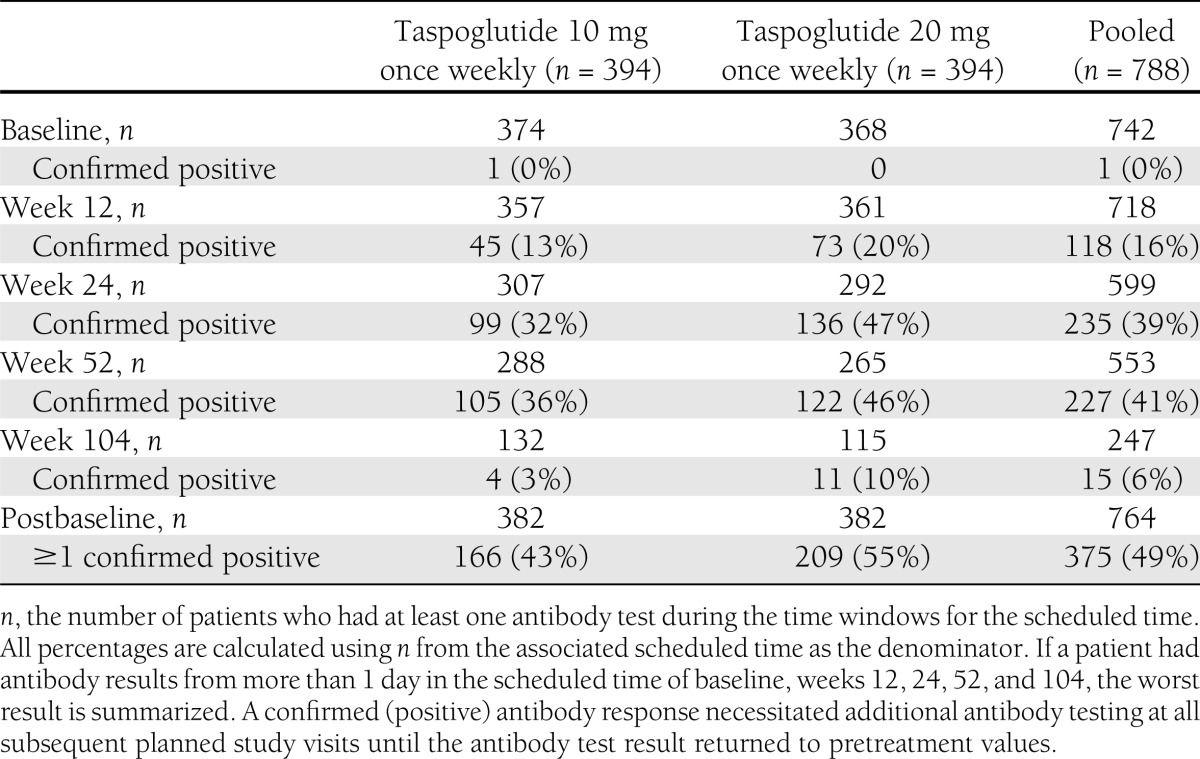
Among those patients with a postbaseline antitaspoglutide antibody test result, 43% in the taspoglutide 10-mg group and 55% in the taspoglutide 20-mg group had at least one positive test result (Table 3). A confirmed positive result >230 ng-eq/mL was reported for 31% of patients. The proportion of patients with a confirmed positive antitaspoglutide antibody test increased from 16% at week 12 to 39% at week 24, with no further increase noted at week 52. As a result of the implemented risk-mitigation plan, patients with confirmed positive antitaspoglutide antibody test >230 ng-eq/mL were discontinued during the long-term extension phase of the study, resulting in a substantial withdrawal rate. Consequently, no interpretation of the data can be made for week 104 results.
Table 3.
Summary of confirmed antitaspoglutide antibody results (safety population, n = 788)
Confirmed hypoglycemia (plasma glucose <3.1 mmol/L [<55 mg/dL]) occurred in 5 (1.3%), 14 (3.6%), and 15 (3.9%) of the taspoglutide 10-mg, taspoglutide 20-mg, and exenatide groups by week 52, respectively. There were no cases of severe hypoglycemia.
Thyroid neoplasm–related adverse events were reported in 22 patients (7 [2%], taspoglutide 10 mg; 9 [2%], taspoglutide 20 mg; and 6 [2%], exenatide). These data were based on a Roche-selected list of Medical Dictionary for Regulatory Activities preferred terms that were reported. No cases of medullary thyroid hyperplasia or carcinoma were detected.
CONCLUSIONS
This head-to-head study demonstrated that both once-weekly taspoglutide and the twice-daily exenatide significantly reduced HbA1c, fasting plasma glucose, and body weight from baseline after 24 weeks of treatment, with no severe hypoglycemia. Noninferiority and superior HbA1c reductions with both taspoglutide 10 and 20 mg compared with exenatide were demonstrated at 24 weeks. The reductions in HbA1c were observed at as early as 4 weeks, continued to decrease until 16 weeks, and were maintained up to 52 weeks.
Despite a nonsignificant difference in HbA1c reduction between the two doses of taspoglutide, greater weight loss was seen with the 20-mg dose; this suggests that doses higher than necessary for glycemic control may further reduce body weight, as has been seen with liraglutide (10).
However, the overall safety profile of taspoglutide was clearly worse than exenatide with respect to gastrointestinal tolerability, systemic allergic reactions, and injection-site reactions. The greater proportion of taspoglutide-treated patients who experienced nausea and/or vomiting compared with exenatide may reflect pharmacokinetic differences between once-weekly and twice-daily formulations, as episodes tended to occur more frequently on the day of injection for taspoglutide, which is probably related to the initial higher maximum plasma concentration of taspoglutide, an effect that may be related to the specific nature of the taspoglutide formulation tested (data not shown).
As a consequence of the higher incidence of adverse events observed in both taspoglutide groups, almost twice as many patients receiving taspoglutide (34%) withdrew from the study than patients taking exenatide (16%). The higher than expected discontinuation rates primarily due to gastrointestinal tolerability observed in the analyses of the 52-week data factored into the decision to terminate the clinical study. The withdrawal rate was further increased during the long-term extension phase of the study as a result of the risk-mitigation plan requiring discontinuation of patients with confirmed positive antitaspoglutide antibody test >230 ng-eq/mL regardless of the presence or absence of allergic adverse events.
Systemic allergic reactions were more common in the taspoglutide-treated patients than in exenatide-treated patients and resulted in withdrawal from the study of 31 of the 51 patients having a reaction in the taspoglutide groups. Most cases occurred after the 24-week core study period. Although allergic reactions are possible with protein-based therapies, the rate of this adverse event with taspoglutide was higher than anticipated and has rarely been reported with other GLP-1 receptor agonists (11).
Antitaspoglutide antibodies were confirmed positive in 49% of taspoglutide-treated patients. Antibody production has been reported with exenatide and liraglutide, although rates appear to be lower with liraglutide (11). For exenatide, the long-acting, once-weekly formulation may have higher rates of antibody formation, with 74% reported in one study (12).
Injection-site reactions were mainly mild or moderate and were reported with higher frequency in taspoglutide-treated patients; some reactions may be related to the subcutaneous depot produced by the long-acting formulation, and similar types of reactions have been reported with other weekly GLP-1 receptor agonists (12,13).
The absence of severe hypoglycemia is consistent with the glucose-dependent insulinotropic mechanism of GLP-1 receptor agonists. The rates of confirmed hypoglycemia were similar between the taspoglutide 20-mg and exenatide groups, and the absolute number of hypoglycemic events was slightly lower in patients receiving taspoglutide 10 mg.
Although acute pancreatitis has been associated with exenatide in postmarketing reports, only one case of pancreatitis was reported in this study in a patient treated with taspoglutide 20 mg. It was considered treatment related and resulted in study withdrawal. A recent observational study found a threefold increased risk of pancreatitis in patients with type 2 diabetes (14), making any assessment of causality difficult in this relatively small study.
Once-weekly taspoglutide was superior to twice-daily exenatide for glycemic control, while offering similar weight loss but an increased rate of nausea or vomiting, injection-site reactions, and hypersensitivity reactions. Clearly, GLP-1 receptor agonists may not be appropriate for all patients with type 2 diabetes, as they are limited by gastrointestinal intolerance with rates of nausea in the 25–45% range and vomiting in the 8–15% range with different compounds, depending on study populations; however, the greater frequency of vomiting seen with the taspoglutide formulation tested in phase 3, compounded by the allergic reactions and high discontinuation rate, made this formulation clinically unacceptable. (In September 2010, Roche decided to stop dosing patients in the taspoglutide phase 3 trials because higher than expected discontinuation rates of taspoglutide-treated patients were observed, mainly due to gastrointestinal tolerability and the implementation of the risk-mitigation plan to address serious hypersensitivity reactions. Since this time, Roche has worked on the root cause analysis and on the modified taspoglutide formulations with the input of Ipsen. After further analysis, Roche has now made the decision to stop the development of taspoglutide and to return the product to the originator, Ipsen, which is currently pursuing further investigations.) Although there are no head-to-head comparisons between taspoglutide and other weekly GLP-1 agonists, the efficacy and safety profile of taspoglutide as revealed by this study could be of relevance for other long-acting GLP-1 agonists, such as the currently available weekly exenatide and the other weekly GLP-1 receptor agonists in development (e.g., albiglutide and dulaglutide).
There seems to be a wide range of individual responses to GLP-1 receptor agonists. Not all patients have satisfactory glucose-lowering responses with meaningful weight loss, but some have robust efficacy responses with good tolerance; more research is clearly needed to help identify those responders in future trials.
Supplementary Material
Acknowledgments
The T-emerge 2 study was funded by F. Hoffmann-La Roche Ltd. Support for third-party writing assistance for the manuscript, furnished by Giles Brooke and Susan Kaup (UBC-Envision Group, Horsham, U.K., and Philadelphia, PA), was provided by F. Hoffmann-La Roche Ltd. J.R. has served on advisory boards and received honorarium or consulting fees from F. Hoffmann-La Roche, Pfizer, sanofi-aventis, Novo Nordisk, Eli Lilly, MannKind, GlaxoSmithKline, Takeda, Daiichi Sankyo, Forest, Johnson & Johnson, Novartis, Boehringer Ingelheim, and Amylin Pharmaceuticals and has received research grants from F. Hoffmann-La Roche, Merck, Pfizer, sanofi-aventis, Novo Nordisk, Bristol-Myers Squibb, Eli Lilly, Forest, GlaxoSmithKline, Takeda, Novartis, AstraZeneca, Amylin Pharmaceuticals, Johnson & Johnson, Daiichi Sankyo, MannKind, and Boehringer Ingelheim. B.C. has served as a consultant to Takeda and has received travel expenses and lecture fees from F. Hoffmann-La Roche, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, GlaxoSmithKline, Merck, Novo Nordisk, sanofi-aventis, and Takeda. R.R. has served as a consultant to Amylin Pharmaceuticals, GlaxoSmithKline, Novo Nordisk, and sanofi-aventis, has received grant support from GlaxoSmithKline, Novo Nordisk, sanofi-aventis, and Boehringer Ingelheim, and has stock ownership in Merck, Johnson & Johnson, and Abbott. B.B. is an employee of F. Hoffman-La Roche. M.B. is an employee and stockholder of F. Hoffman-La Roche. R.B. was employed by F. Hoffmann-La Roche at the time the study was conducted and is a stockholder. No other potential conflicts of interest relevant to this article were reported.
J.R. and B.B. researched data, contributed to the discussion, and wrote, reviewed, and edited the manuscript. B.C., G.B.B., R.R., and R.B. researched data, contributed to the discussion, and reviewed and edited the manuscript. M.B. researched data, reviewed and edited the manuscript, and was responsible for the statistical analyses. All authors were involved in planning the manuscript, its critical review and editing, subsequent revisions, and approval for submission. J.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented at the 70th Scientific Sessions of the American Diabetes Association, Orlando, Florida, 25–29 June 2010; the 46th Annual Meeting of the European Association for the Study of Diabetes, Stockholm, Sweden, 20–24 September 2010; and the 28th Annual Scientific Meeting of the Obesity Society, 8–12 October 2010, San Diego, California.
The authors thank the study investigators, their staff, clinical trial personnel, and the study participants. The authors also thank Neena Sarkar (F. Hoffmann-La Roche) for assistance with statistical analyses.
Footnotes
Clinical trial reg. no. NCT00717457, clinicaltrials.gov.
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc12-0709/-/DC1.
R.B. is currently affiliated with Eli Lilly, Erl Wood Manor, Windlesham Surry, U.K.
A complete list of the T-emerge 2 study investigators can be found in the Supplementary Data online.
References
- 1.Monami M, Marchionni N, Mannucci E. Glucagon-like peptide-1 receptor agonists in type 2 diabetes: a meta-analysis of randomized clinical trials. Eur J Endocrinol 2009;160:909–917 [DOI] [PubMed] [Google Scholar]
- 2.Byetta: exenatide injection [package insert]. San Diego, CA, Amylin Pharmaceuticals, 2005. Available from http://pi.lilly.com/us/byetta-pi.pdf Accessed 27 September 2010
- 3.Moretto TJ, Milton DR, Ridge TD, et al. Efficacy and tolerability of exenatide monotherapy over 24 weeks in antidiabetic drug-naive patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2008;30:1448–1460 [DOI] [PubMed] [Google Scholar]
- 4.Victoza: liraglutide (rDNA origin) injection [package insert]. Princton, NJ, Novo Nordisk, 2010. Available from http://www.victozapro.com/pdf/Victoza_ComboPI_5.24.pdf Accessed 27 September 2010
- 5.Montanya E, Sesti G. A review of efficacy and safety data regarding the use of liraglutide, a once-daily human glucagon-like peptide 1 analogue, in the treatment of type 2 diabetes mellitus. Clin Ther 2009;31:2472–2488 [DOI] [PubMed] [Google Scholar]
- 6.Sebokova E, Christ AD, Wang H, et al. Taspoglutide, an analog of human glucagon-like peptide-1 with enhanced stability and in vivo potency. Endocrinology 2010;151:2474–2482 [DOI] [PubMed] [Google Scholar]
- 7.Nauck MA, Ratner RE, Kapitza C, Berria R, Boldrin M, Balena R. Treatment with the human once-weekly glucagon-like peptide-1 analog taspoglutide in combination with metformin improves glycemic control and lowers body weight in patients with type 2 diabetes inadequately controlled with metformin alone: a double-blind placebo-controlled study. Diabetes Care 2009;32:1237–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratner R, Nauck M, Kapitza C, Asnaghi V, Boldrin M, Balena R. Safety and tolerability of high doses of taspoglutide, a once-weekly human GLP-1 analogue, in diabetic patients treated with metformin: a randomized double-blind placebo-controlled study. Diabet Med 2010;27:556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathan DM, Buse JB, Davidson MB, et al. American Diabetes Association. European Association for Study of Diabetes Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009;32:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Astrup A, Rössner S, Van Gaal L, et al. NN8022-1807 Study Group Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 2009;374:1606–1616 [DOI] [PubMed] [Google Scholar]
- 11.Aroda VR, Ratner R. The safety and tolerability of GLP-1 receptor agonists in the treatment of type 2 diabetes: a review. Diabetes Metab Res Rev 2011;27:528–542 [DOI] [PubMed] [Google Scholar]
- 12.Drucker DJ, Buse JB, Taylor K, et al. DURATION-1 Study Group Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet 2008;372:1240–1250 [DOI] [PubMed] [Google Scholar]
- 13.Rosenstock J, Reusch J, Bush M, Yang F, Stewart M, Albiglutide Study Group Potential of albiglutide, a long-acting GLP-1 receptor agonist, in type 2 diabetes: a randomized controlled trial exploring weekly, biweekly, and monthly dosing. Diabetes Care 2009;32:1880–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009;32:834–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.