

Abstract
Hydrogen sulfide (sulfide, H2S) is a colorless, water-soluble gas with a typical smell of rotten eggs. In the past, it has been investigated for its role as a potent toxic gas emanating from sewers and swamps or as a by-product of industrial processes. At high concentrations, H2S is a powerful inhibitor of cytochrome c oxidase; in trace amounts, it is an important signaling molecule, like nitric oxide (NO) and carbon monoxide (CO), together termed “gasotransmitters.” This review will cover the physiological role and the pathogenic effects of H2S, focusing on ethylmalonic encephalopathy, a human mitochondrial disorder caused by genetic abnormalities of sulfide metabolism. We will also discuss the options that are now conceivable for preventing genetically driven chronic H2S toxicity, taking into account that a complete understanding of the physiopathology of H2S has still to be achieved.
In ethylmalonic encephalopathy, sulfide intermediates are not degraded properly. Sulfides build up and inhibit cytochrome c oxidase, the last enzyme in the mitochondrial electron-transport chain.
Because of its fame as a deadly gas, the physiological role of H2S had been long overlooked, until Abe and Kimura in 1996 (Abe and Kimura 1996) demonstrated that H2S is a neuromodulator that facilitates the induction of long-term potentiation (LTP) by enhancing the activity of N-methyl-d-aspartate (NMDA) receptors in hippocampal pyramidal neurons. H2S was later shown to induce relaxation of smooth muscle, to regulate the release of insulin, and to act as a mediator of inflammation.
The physiological importance of H2S has further been supported by demonstrating that H2S is endogenously produced in mammals by two pyridoxal-5′-phosphate-dependent enzymes, cystathionine β-synthase (CBS, EC 4.2.1.22) and cystathionine γ-lyase (CSE, EC 4.4.1.1), which both utilize l-cysteine as a substrate (Stipanuk and Beck 1982; Hosoki et al. 1997; Zhao et al. 2001). l-cysteine is taken up with the diet, extracted from endogenous proteins, or synthesized endogenously via trans-sulfuration of serine by l-methionine. In humans, the end products of cysteine sulfur catabolism are sulfate (which accounts for 77%–92% of the total sulfur excreted in the urine), sulfate esters (7%–9%), and taurine (2%–6%) (Stipanuk 2004) (Fig. 1). In some tissues, both CBS and CSE can generate H2S, whereas in others only one enzyme is present (Diwakar and Ravindranath 2007). For instance, CBS is predominantly expressed in the nervous system but is also active in the liver and kidney (Enokido et al. 2005). CSE is mainly expressed in liver, as well as vascular and nonvascular smooth muscle (e.g., the small intestine and stomach [Awata et al. 1995; Ishii et al. 2004]).
Figure 1.

Sulfide production. Methionine, derived from alimentary sources, is converted into S-adenosylmethionine by methionine adenosyltransferase (MAT). S-adenosylmethionine is hydrolyzed to homocysteine by glycine N-methyltransferase (GNMT). Cystathionine β-synthase (CBS) transfers serine to homocysteine and produces cystathionine. Cystathionine γ-lyase (CSE) converts cystathionine to cysteine (Cys). One pathway of cysteine metabolism involves its oxidation to cysteine sulfinate by cysteine deoxygenase (CDO), which is then converted to hypotaurine by cysteine sulfinate decarboxylase (CSD); hypotaurine is eventually oxidated to taurine by hypotaurine dehydrogenase (HDH). In mitochondria, cysteine can be converted to 3-mercaptopyruvate by aspartate aminotransferase (AAT), which can then be converted to H2S by 3-mercaptopyruvate sulfur transferase. The conversion of cysteine sulfinate to sulfinyl pyruvate by AAT, followed by a nonenzymatic reaction, can also yield sulfite, which is then oxidized to sulfate by sulfite oxidase (SUOX) in a glutathione (GSH)-dependent process. The H2S produced by either AAT or CBS/CSE can be further oxidized by a sulfur dioxygenase (SDO/ETHE1)-dependent pathway (see Fig. 2). The metabolic pathway carried out by Moco-dependent enzymes is encircled in red (see text for details).
Both CBS and CSE activities increase in rat brain after birth, reaching adult levels at week four (Awata et al. 1995).
An important additional source of H2S is the enterobacterial anaerobic flora. The intestinal epithelium expresses specialized enzyme systems that efficiently convert H2S into thiosulfate and sulfate, to both prevent the local increase of H2S to toxic levels and its entry through the portal vein system in the liver and other organs. Excessive production and absorption of H2S, as well as reduced detoxification by colonic epithelial cells (colonocytes), is thought to play an important role in the mucosal damage of ulcerative colitis (Rowan et al. 2009).
A third, however marginal, source of H2S is the nonenzymatic reduction of elemental sulfur, which is present in traces in human (Westley and Westley 1991) and mouse (Buzaleh et al. 1991) blood, into H2S by electrons provided through glycolysis (Searcy and Lee 1998).
The main catabolic pathway of H2S takes place in mitochondria and consists of a series of oxidative reactions ultimately yielding sulfate (SO42−), with thiosulfate (S2O32−) and sulfite (SO32−) as intermediate compounds (Beauchamp et al. 1984; Szabò 2007). The existence of such a pathway has been known for more than 20 years (Powell and Somero 1986), although the enzymatic actors and detailed steps have been elucidated (Hildebrandt and Grieshaber 2008) and further refined (Jackson et al. 2012) only recently. The pathway proposed by Hildebrandt (2008), later modified by Jackson, is shown in Figure 2. The pathway consists of (1) a mitochondrial inner-membrane-bound sulfide quinone reductase (SQR) (Theissen and Martin 2008), which fixes H2S to sulfite to produce thiosulfate; (2) mitochondrial thiosulfate sulfur transferase, also known as rhodanese, which reconstitutes sulphite from thiosulfate by fixating the sulfane sulfur of the latter onto an –SH-containing substrate, for instance glutathione, GSH, to form a persulfide (R-S-SH) species; (3) a mitochondrial matrix sulfur dioxygenase (SDO), which oxidizes the sulfur atom extracted from persulfide, converting it again into sulfite (SO32−); and (4) mitochondrial sulfite oxidase, which further oxidizes sulfite into sulfate (SO42−). Thiosulfate can also be converted into two molecules of sulfate by the combined action of a thiosulfate reductase (Müller et al. 2004) and sulfite oxidase (Feng et al. 2007). Additional mechanisms of detoxification rely on the fixation of H2S to methemoglobin, which is formed from hemoglobin by oxidation of the Fe center, for instance by sodium nitrate, to produce sulfo-methemoglobin, or on the administration of oxidized glutathione (GSSG) (Beauchamp et al. 1984), which is able to fixate H2S to produce glutathione persufide (GSSH), thus preventing the interaction of H2S with critical enzymatic activities. Additional sources of sulfite are known, for instance by the cysteine dioxygenase/aspartate aminotransferase (CDO/AAT) system (Cooper et al. 2002; Stipanuk and Ueki 2010), converting cysteine into β-sulfinylpyruvate, which then decomposes into pyruvate and sulfite.
Figure 2.
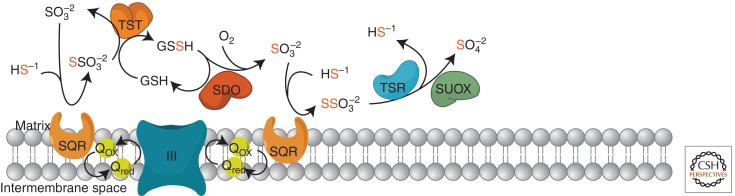
Proposed pathway for sulfide oxidation. H2S is produced from cysteine (see Fig. 1) and is initially fixed by a membrane-bound sulfide quinone oxidoreductase (SQR). The electrons extracted from H2S enter the respiratory chain at the level of the quinone pool (UQH/UQ), are then transferred to complex III (cIII), and are finally fixed by cytochrome c oxidase (complex IV, cIV) to molecular oxygen with the formation of water. A sulfur dioxygenase (SDO/ETHE1), present as an Fe-binding dimer in the mitochondrial matrix, oxidizes the persulfide to sulfite (H2SO3) in a reaction that includes molecular oxygen and water. See text for details. SUOX, sulfite oxidase; TST, thiosulfate sulfur transferase; TSR, thiosulfate reductase.
H2S: PHYSICAL PROPERTIES
Sulfur, an essential element for life, occurs in different oxidation states, from –2 (as in sulfide, S2−) to +6 (as in sulfate, SO42−). Sulfur is present in living organisms as a constituent of proteins (mainly in sulfurated amino acids such as cysteine and methionine), in coenzymes (for instance CoA, biotine, and thiamine), and as a component of iron-sulfur clusters in metalloproteins. From a physical–chemical standpoint, H2S is the sulfur analog of water, but because its intermolecular bonds are weaker than H2O, at room temperature and atmospheric pressure, it is a gas. In mammals, at a physiological pH of 7.4, approximately one-third of H2S exists as the undissociated form and two-thirds as the hydrosulfide anion (HS–) (Reiffenstein et al. 1992). In the undissociated form, H2S can easily penetrate the plasma membranes of cells because of its high solubility in lipids.
PHYSIOLOGICAL ACTIONS OF H2S
A preliminary question, however crucial for understanding the pathophysiology of sulfide metabolism, is how much free H2S is present in tissues and body fluids of mammalian organisms. Free H2S is difficult to measure, mainly because of its high volatility and the interference of cysteine residues contained in proteins, which hamper the results of the most common analytical methods based on spectrophotometric assays from tissue extracts. Levels as high as 30–150 µm have been reported in different tissues in normal conditions (Goodwin et al. 1989; Mitchell et al. 1993; Ogasawara et al. 1994; Wang 2002; Fiorucci et al. 2006;), a remarkable overestimation since these values are well above the olfactory threshold for H2S and are clearly associated with severe toxicity in vivo, as shown in both cells and mammalian organisms. Using a gas-chromatographic assay that avoids interference by cysteines, Furne et al. (2008) have recently shown that the concentration of free sulfide in mouse liver and brain is 15 nm, that is 10−3 − 10−4 less than the values obtained by spectrophotometric assays.
Recent studies have demonstrated for H2S a multiplicity of physiological functions, including up-regulation of antioxidant systems and amplification of the effects produced by known antioxidants such as N-acetylcysteine (NAC) and glutathione (Whiteman 2004; Yan 2006); up-regulation of anti-inflammatory and cytoprotective genes including heme oxygenase (HO1, also known as HMOX1) in pulmonary smooth-muscle cells (Qingyou et al. 2004) and macrophages (Oh et al. 2006). In the CNS, low concentrations of H2S are sufficient to increase the production of cAMP, enhance NMDA-receptor-mediated responses, and facilitate the induction of long-term potentiation in hippocampal neurons (Kimura and Kimura 2004).
H2S displays a number of different effects on myocardial cells and vascular smooth muscle. By activating KATP channels, H2S increases the membrane potential of vascular smooth-muscle cells that become hyperpolarized, therefore causing vasorelaxation (Zhao et al. 2001; Elsey et al. 2010). Accordingly, hypertension is a consistent feature of CSE knockout mice, confirming that H2S plays a role as a smooth-muscle relaxant and suggesting that it may physiologically regulate blood pressure (Yang et al. 2008).
This effect could, however, be mediated by a quenching action of H2S on nitric oxide (NO), possibly via the formation of a nitrothiol intermediate (Whiteman et al. 2006). A counteracting action by H2S on vasodilation induced by NO could explain why low concentrations of H2S (10–100 µm) induce vasoconstriction, whereas concentrations over 100 µm induce vasorelaxation (Ali et al. 2006). A second mechanism of interaction between H2S and NO may involve transcriptional regulation of CSE, which is induced by NO (Zhao et al. 2001). It is thus possible that the vasorelaxant effect of H2S may be mediated, at least in part, by a local regulatory effect on the NO concentration.
H2S induces the proliferation of smooth-muscle cells by activating the PI3 Kinase/Akt pathway, thus stimulating angiogenesis (Cai et al. 2007) and apoptosis by activating the mitogen-activated protein kinase (MAPK) pathway (Yang et al. 2004).
Finally, H2S can directly modulate the function of the heart, with negative inotropic and chronotropic effects (Ferdinandy et al. 2007), which are mediated by activation of KATP channels. This mechanism results in the hyperpolarization of the membrane potential, closure of L-type calcium channels, and consequent reduction of contraction strength dependent on calcium-activated calcium release. In addition, the activation of KATP channels depresses the activity of the sinoatrial pacemaker, thus reducing the heart rate (Xu et al. 2008).
The several effects that H2S exerts on the cardiovascular system suggest that it may have a protective role during ischemia/reperfusion injury. During ischemia, cells are in hypoxic conditions, and subsequent reperfusion and reoxygenation lead to inflammation and cell death by increasing reactive oxygen species (ROS) production, increasing mitochondrial Ca2+ overload, and opening the mitochondrial permeability transition pore (mPTP) (Ferdinandy et al. 2007). In experimental ischemia/reperfusion tests, the treatment with H2S is protective, either before the induction of ischemia (preconditioning) or before the reperfusion phase (postconditioning). These effects are mediated by activation of the cardioprotective PI3K/Akt pathway, activation of KATP channels, and increase of the free thiol pool, which acts as ROS scavenger, thus inhibiting the opening of mPTP. The protective effects of H2S in ischemia/reperfusion experiments have been further confirmed in a mouse model characterized by a twofold increase of H2S in the myocardium due to transgenic overexpression of CSE (Elrod et al. 2007).
TOXIC EFFECTS OF H2S
Whereas at physiological (nanomolar) concentrations, Although H2S acts as a cytoprotective agent, at micromolar concentrations, it can interfere with a variety of cellular functions, including mitochondrial respiration, via inhibition of cytochrome c oxidase (COX, EC 1.9.3.1) (Hill et al. 1984). COX activity is inhibited by the formation of a covalent bond between H2S and the Fe atom coordinated by heme a, which is exquisitely sensitive to the toxic action of H2S (and cyanide as well). The chronic exposure and binding of H2S to COX causes accelerated degradation of its protein subunits (Di Meo et al. 2011), thus reducing the amount of fully assembled and functionally active enzyme.
H2S is also an inhibitor of carbonic anhydrase (EC 4.2.1.1) (Coleman 1967), an effect that could explain the alterations of ventilatory dynamics in response to inhaled H2S due to changes in the reactivity and distribution of intrapulmonary CO2 receptors (Klentz and Fedde 1978).
A further effect of H2S is inhibition of enzymatic activity of short-chain acylCoA dehydrogenase (SCAD), which can cause dicarboxylic aciduria (Pedersen et al. 2003).
Acute exposure to gaseous H2S causes anoxic brain injury, pulmonary edema, and death (Yalamanchili and Smith 2008). Neurological impairment is consistent with prolonged brain hypoxia, whereas respiratory insufficiency and pulmonary edema are consistent with acute respiratory syndrome. These two injuries are the most likely cause of death after acute exposure to H2S, whereas chronic exposure to low levels determines irritant effects including upper airway inflammation, conjunctivitis, and wheezing. A direct effect of H2S on the respiratory center in the brainstem, resulting in apnea, is likely to be caused by inhibition of COX activity (Yalamanchili and Smith 2008).
H2S IN ETHYLMALONIC ENCEPHALOPATHY (EE)
Ethylmalonic encephalopathy (EE) (Online Mendelian Inheritance in Man [OMIM] #602473) (Burlina et al. 1991) is a severe mitochondrial disease of early infancy, clinically characterized by a combination of progressive encephalopathy, vascular lesions determining bouts of petechial purpura and orthostatic acrocyanosis (Fig. 3A), and chronic hemorrhagic diarrhea. Neurological symptoms include psychomotor regression, spastic tetraparesis, axial hypotonia, dystonia, and seizures, reflecting the presence of patchy, bilateral lesions in the basal nuclei and brainstem gray matter (Fig. 3B). The onset is in the first months after birth; children usually die within the first decade.
Figure 3.

Ethylmalonic encephalopathy: clinical and molecular features.
(A) Skin areas with petechial hemorrhages are indicated by arrows. The insets show the acrocyanosis. (B) T2-weighted MRI images of a transverse section of the brain: symmetrical, patchy, high-intensity signals are present in the head of nucleus caudatus and putamen (arrows). (C) COX activity is virtually absent in skeletal muscle of an EE patient as compared to normal reactivity present in control. (D) Gas-chromatographic profile of urinary organic acids in EE. The abnormal peak of ethylmalonic acid (EMA) is indicated in red. Asterisks indicate internal standards. (E) ETHE1 mutations identified in our patients. Missense (gray) and nonsense (white) mutations are indicated along the schematic representation of the ETHE1 cDNA subdivided in the corresponding exons (indicated by numbers). The black areas represent the 5′ and 3′ UTRs. The genomic organization is shown below the cDNA.
Biochemically, EE is characterized by the unusual combination of severe deficiency of COX, the terminal component of the mitochondrial respiratory chain, in muscle (Fig. 3C) and brain, leading to high levels of lactate in blood, and accumulation of ethylmalonate (ethylmalonic acid, EMA) in urines (Fig. 3D). EMA, a dicarboxylic organic acid produced by the carboxylation of butyrate, is a hallmark of enzymatic defects of β-oxidation of fatty acids and branched-chain amino acids, for instance defects of the short-chain and branched-chain acylCoA dehydrogenase (SCAD, BCAD) (Burlina et al. 1991; Gregersen et al. 2000; Korman 2006). Like in SCAD and BCAD deficiency, accumulation of C4- and C5-acylcarnitines has been documented in blood of EE patients. However, SCAD and BCAD activities are normal in EE fibroblasts (Nowaczyk et al. 1998) and muscle, therefore excluding them from being the primary abnormality in this disease. For still unexplained reasons, in virtually all cases, the origin of these EE families is from or could be traced to different regions of the Mediterranean basin or the Arabic peninsula.
In 2004, we demonstrated the presence of several pathogenic mutations in a gene dubbed ETHE1 for EE 1 (Tiranti et al. 2004). We have reported a series of 29 patients, presenting a fairly homogeneous clinical and biochemical presentation, in spite of a wide spectrum of ETHE1 mutations including missense, nonsense, frame-shift and deletion of single exons or of the entire gene (Tiranti et al. 2004, 2006) (Fig. 3E, shows only part of the mutations identified). Mutations in the ETHE1 gene have been found in 32 patients affected by EE worldwide (Di Rocco et al. 2006; Merinero et al. 2006; Zafeiriou et al. 2007; Mineri et al. 2008). A total of 18 nonsense (Tiranti et al. 2004, 2006; Merinero et al. 2006) and nine missense (Tiranti et al. 2004, 2006; Di Rocco et al. 2006; Zafeiriou et al. 2007) mutations has so far been described. To date, a total of 70 mutant patients have been identified in our laboratory (V Tiranti and M Zeviani, unpubl.). Western-blot analysis of the Ethe1 protein (Ethe1p) indicated that some of the missense mutations are associated with the presence of the protein, suggesting that the corresponding wild-type amino acid residues have a catalytic function (Tiranti et al. 2006). In fact, a 3D model of the Ethe1p makes it possible to analyze the spatial location of the amino acid changes predicted by missense mutations and to classify the mutation as catalytic versus structural (Tiranti et al. 2006). More recently, the availability of the crystal structure of the Ethe1p from Arabidopsis thaliana (Holdorf et al. 2008) allowed us to demonstrate that the predicted 3D model of human Ethe1p was a faithful in silico reconstruction of the presumed real structure of the native protein (Mineri et al. 2008).
The Ethe1p, a 30 kDa polypeptide located in the mitochondrial matrix, functions in vivo as a homodimeric, Fe-containing sulfur dioxygenase (SDO) activity (Tiranti et al. 2009), which we have already mentioned as one of the reactions involved in the catabolic oxidation of sulfide to sulfate.
Impaired activity of ETHE1-SDO leads to the accumulation of H2S in critical tissues, including colonic mucosa, liver, muscle, and brain, up to concentrations that inhibit SCAD and COX activities, therefore accounting for EMA aciduria and high levels of C4 and C5 acylcarnitines, EMA, and lactate in plasma (Tiranti et al. 2009). Chronic COX inhibition by accumulated H2S leads to accelerated degradation of the COX protein backbone (Di Meo et al. 2011). Although ETHE1 activity is measurable in all organs, muscle and brain of Ethe1-less (Ethe1−/−) mice show both defective COX activity and reduced COX amount, whereas liver does not. This may reflect the presence of alternative and highly effective metabolic pathways protecting hepatocytes from H2S toxicity.
In addition to H2S, which is a highly unstable, difficult-to-measure gas, its stable derivative thiosulfate is also present at very high concentrations in tissues and body fluids of Ethe1−/− mice and EE patients, and can well be considered a specific biomarker of the disease. The pathway outlined in Figure 1 explains these findings: H2S is converted into thiosulfate by the SQR activity. In normal conditions, rhodanese-TST catalyzes the transfer of the sulfane sulfur from thiosulfate to GSH (or other R-SH substrate) to form persulfide, which is the substrate of the Ethe1-SDO activity, but since the latter is missing, the disposal of the sulfane sulfur is also blocked and consequently both thiosulfate and sulfide accumulate.
We have demonstrated that the ablation of Ethe1 restricted to muscle or brain is clearly associated with an isolated COX deficiency in the targeted tissue but not in other, Ethe1-competent, tissues (Di Meo et al. 2011). This data unequivocally demonstrates that failure to neutralize the endogenous production of H2S is sufficient for COX activity to decrease, but not for the animals to become sick or for thiosulfate to increase. This observation suggests that multiorgan accumulation of H2S and diffusion of exogenously released H2S from the bacterial flora are both needed to determine the severe metabolic impairment and the fatal clinical course of Ethe1-less mice and humans.
Besides COX and SCAD deficiency, other symptoms of EE are explained by accumulation of H2S, including damage of endothelial cells and vasodilation, which account for the petechiae and the acrocyanosis. Since a substantial amount of H2S derives from the anaerobic bacterial flora residing in the intestinal lumen, COX activity is markedly reduced in the luminal colonocytes of Ethe1−/− mice, whereas it is normal in cryptal colonocytes that are relatively secluded from the luminal surface. This difference is likely to reflect different exposure of the two cell populations to the inhibitory action of exogenous H2S. Excessive production and absorption of H2S, as well as reduced detoxification by colonocytes, is regarded to play an important role in the mucosal damage of ulcerative colitis. A similar mechanism can well account for the severe chronic diarrhea afflicting EE patients.
THERAPEUTIC APPROACHES IN EE
In principle, H2S production from the anaerobes of the large intestine can be attenuated by reducing the bacterial contingent, and the excess of free H2S can be buffered by suitable compounds in critical tissues. For instance, metronidazole is a drug widely used to combat anaerobic infections by reducing the bacterial load in the large intestine (Kang et al. 2008). Metronidazole is a nitroimidazole anti-infective compound that is in fact a pro-drug, being activated in anaerobic organisms by the redox enzyme pyruvate-ferredoxin oxidoreductase. Metronidazole is selectively taken up by anaerobic bacteria and sensitive protozoal organisms (Samuelson 1999) because of the unique ability of these organisms to reduce uptaken metronidazole to its active form within their cell bodies. The nitro group of metronidazole is chemically reduced by ferredoxin (or a ferredoxin-linked metabolic process), and the products are responsible for disrupting the DNA helical structure, thus inhibiting nucleic-acid synthesis (Eisenstein and Schaechter 2007). NAC is a cell-permeable precursor of glutathione (GSH), an abundant compound in mitochondria (Marí et al. 2009), where it acts as one of the physiological acceptors of the sulfur atom of H2S operated by SQR. This reaction leads to the formation of GSSH persulfide, which acts in turn as a substrate for the Ethe1-SDO. If Ethe1-SDO activity is missing, GSH can buffer, at least in part, accumulating H2S by forming GSSH, which unlike free H2S, is a nontoxic compound (Atkuri et al. 2007).
Combined exposure to metronidazole and NAC has been effective in prolonging the survival of Ethe1−/− mice and in improving the main symptoms in a pilot study on EE patients (Viscomi et al. 2010), including marked attenuation or disappearance of the vascular lesions and diarrhea, as well as amelioration of some neurological abnormalities. These encouraging results suggest that the invariably fatal clinical course of EE could be modified by a pharmacological protocol based on the off-label use of low-cost, relatively safe drugs.
More recently, Hildebrandt (2011) demonstrated that sulfide toxicity can be modified by the addition of dehydroascorbic acid (DHA) in rat mitochondria. DHA significantly reduced the inhibitory effect of sulfide on COX, resulting in higher rates of respiration and sulfide oxidation. For this purpose, DHA is more suitable than ascorbic acid, the main functionally active form of vitamin C, because its concentration is lower and more flexible. The fraction of oxidized vitamin C reflects the redox state of the cell or organelle and can therefore induce a specific modification of sulfide metabolism. This observation has important clinical implications because DHA could be used during the treatment of sulfide poisoning either after accidental environmental exposure or in patients with EE.
Additional useful information, potentially relevant in the treatment of EE, was recently obtained by Palmfeldt and colleagues (2011), who performed a proteomics study on mitochondria derived from cultured patient fibroblast cells and compared it with healthy controls. Sulfide:quinone oxidoreductase (SQRDL), which takes part in the same sulfide pathway as ETHE1, was also underrepresented in EE patients. Two proteins, the mitochondrial superoxide dismutase (SOD2) and aldehyde dehydrogenase X (ALDH1B) involved in pathways of detoxification and oxidative/reductive stress, were down-regulated in EE. These authors propose that redox perturbation could be an additional factor in the molecular mechanism of EE and an additional therapeutic target.
Another approach to scavenge circulating sulfide coming from the gut could be restoration of the ETHE1-SDO activity in liver of constitutive Ethe1−/− individuals by using adeno-associated virus (AAV)-based gene replacement. Because liver is the main checkpoint organ against toxic compounds from the gut, AAV-mediated liver-specific ETHE1 expression may be beneficial to Ethe1−/− organisms. This very strategy has recently been successful in markedly prolonging the survival of constitutive Ethe1−/− mice (Di Meo et al. 2012).
A complementary strategy, based on the same rationale, is bone marrow transplantation (BMT). BMT has emerged as a major therapeutic option in the treatment of malignant diseases and has also become the treatment of choice for a number of nonmalignant disorders. As for liver-targeted, AAV-based gene replacement, the rationale of BMT-based therapy in the context of EE is to provide a substantial contingent of Ethe1-SDO proficient cells able to clear the excess of H2S from plasma and other body fluids. A similar strategy has recently been proposed for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), due to loss of activity of thymidine phosphorylase (TP), resulting in high plasma and tissue levels of thymidine (Thd) and deoxyuridine (dUrd), which become mutagenic on mitochondrial DNA (Halter et al. 2011).
CONCLUDING REMARKS
Because of the multiple functions of mitochondria, not only does OXPHOS impairment determine various degrees of energy failure, but so also do a host of additional effects that account for the intricacy and complexity of the pathophysiology of mitochondrial disorders.
The recent elucidation of the pathogenetic basis of EE has provided evidence for the existence of yet another mechanism of mitochondrial energy impairment, namely the genetically determined failure to detoxify OXPHOS poisons, such as H2S. In principle, accumulation of a harmful compound can be contrasted more effectively than intrinsic failure of OXPHOS energy metabolism. Therefore, we believe that the specific toxic mechanism underpinning EE makes effective therapy a realistic goal. The results of our effort will constitute a fundamental step toward the development of a cure for this fatal mitochondrial disease in humans.
Footnotes
Editors: Douglas C. Wallace and Richard J. Youle
Additional Perspectives on Mitochondria available at www.cshperspectives.org
REFERENCES
- Abe K, Kimura H 1996. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16: 1066–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, Moore PK 2006. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol 149: 625–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA 2007. N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7: 355–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awata S, Nakayama K, Suzuki I, Sugahara K, Kodama H 1995. Changes in cystathionine γ-lyase in various regions of rat brain during development. Biochem Mol Biol Int 35: 1331–1338 [PubMed] [Google Scholar]
- Beauchamp RO Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA 1984. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol 13: 25–97 [DOI] [PubMed] [Google Scholar]
- Burlina A, Zacchello F, Dionisi-Vici C, Bertini E, Sabetta G, Bennet MJ, Hale DE, Schmidt-Sommerfeld E, Rinaldo P 1991. New clinical phenotype of branched-chain acyl-CoA oxidation defect. Lancet 338: 1522–1523 [DOI] [PubMed] [Google Scholar]
- Buzaleh AM, Vazquez ES, del Carmen Battle AM 1990. Cyanide intoxication—III: On the analogous and different effects provoked by non-lethal and lethal challenged doses. Gen Pharmacol 21: 27–32 [DOI] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC 2007. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res 76: 29–40 [DOI] [PubMed] [Google Scholar]
- Coleman JE 1967. Mechanism of action of carbonic anhydrase: Subtrate, sulfonamide, and anion binding. J Biol Chem 242: 5212–5219 [PubMed] [Google Scholar]
- Cooper AJ, Bruschi SA, Iriarte A, Martinez-Carrion M 2002. Mitochondrial aspartate aminotransferase catalyses cysteine S-conjugate β-lyase reactions. Biochem J 368: 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo I, Fagiolari G, Prelle A, Viscomi C, Zeviani M, Tiranti V 2011. Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid Redox Signal 15: 353–362 [DOI] [PubMed] [Google Scholar]
- Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M 2012. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med 4: 1008–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rocco M, Caruso U, Briem E, Rossi A, Allegri AE, Buzzi D, Tiranti V 2006. A case of ethylmalonic encephalopathy with atypical clinical and biochemical presentation. Mol Genet Metab 89: 395–397 [DOI] [PubMed] [Google Scholar]
- Diwakar L, Ravindranath V 2007. Inhibition of cystathionine γ-lyase leads to loss of glutathione and aggravation of mitochondrial dysfunction mediated by excitatory amino acid in the CNS. Neurochem Int 50: 418–426 [DOI] [PubMed] [Google Scholar]
- Eisenstein BI, Schaechter M 2007. DNA and chromosome mechanics. In Schaechter’s mechanisms of microbial disease (ed. NC Engleberg, et al. ), p. 28 Lippincott Williams & Wilkins: Hagerstown, MD [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al. 2007. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci 104: 15560–15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF 2010. Regulation of cardiovascular cell function by hydrogen sulfide (H2S). Cell Biochem Funct 28: 95–106 [DOI] [PubMed] [Google Scholar]
- Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H 2005. Cystathionine β-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J 19: 1854–1856 [DOI] [PubMed] [Google Scholar]
- Feng C, Tollin G, Enemark JH 2007. Sulfite oxidizing enzymes. Biochim Biophys Acta 1774: 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF 2007. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59: 418–458 [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL 2006. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 131: 259–271 [DOI] [PubMed] [Google Scholar]
- Furne J, Saeed A, Levitt MD 2008. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol 295: R1479–R1485 [DOI] [PubMed] [Google Scholar]
- Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW 1989. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: Postmortem studies and two case reports. J Anal Toxicol 13: 105–109 [DOI] [PubMed] [Google Scholar]
- Gregersen N, Andresen BS, Bross P 2000. Prevalent mutations in fatty acid oxidation disorders: diagnostic considerations. Eur J Pediatr 159: S213–S218 [DOI] [PubMed] [Google Scholar]
- Halter J, Schüpbach WM, Casali C, Elhasid R, Fay K, Hammans S, Illa I, Kappeler L, Krähenbühl S, Lehmann T, et al. 2011. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): A consensus conference proposal for a standardized approach. Bone Marrow Transplant 46: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt TM 2011. Modulation of sulfide oxidation and toxicity in rat mitochondria by dehydroascorbic acid. Biochim Biophys Acta 1807: 1206–1213 [DOI] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK 2008. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275: 3352–3361 [DOI] [PubMed] [Google Scholar]
- Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ 1984. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem J 224: 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdorf MM, Bennett B, Crowder MW, Makaroff CA 2008. Spectroscopic studies on Arabidopsis ETHE1, a glyoxalase II-like protein. J Inorg Biochem 102: 1825–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H 1997. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237: 527–531 [DOI] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H 2004. Murine cystathionine γ-lyase: Complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 381: 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Melideo SL, Jorns MS 2012. Human sulfide: Quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51: 6804–6815 [DOI] [PubMed] [Google Scholar]
- Kang SS, Bloom SM, Norian LA, Geske MJ, Flavell RA, Stappenbeck TS, Allen PM 2008. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Kimura H 2004. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18: 1165–1167 [DOI] [PubMed] [Google Scholar]
- Klentz RD, Fedde MR 1978. Hydrogen sulfide: Effects on avian respiratory control and intrapulmonary CO2 receptors. Respir Physiol 32: 355–367 [DOI] [PubMed] [Google Scholar]
- Korman SH, Inborn errors of isoleucine degradation: A review. 2006. Mol Genet Metab 89: 289–99 [DOI] [PubMed] [Google Scholar]
- Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC 2009. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 11: 2685–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merinero B, Pérez-Cerdá C, Ruiz Sala P, Ferrer I, García MJ, Martínez Pardo M, Belanger-Quintana A, de la Mota JL, Martin-Hernández E, Vianey-Saban C, et al. 2006. Persistent increase of plasma butyryl/isobutyrylcarnitine concentrations as marker of SCAD defect and ethylmalonic encephalopathy. J Inherit Metab Di 29: 685. [DOI] [PubMed] [Google Scholar]
- Mineri R, Rimoldi M, Burlina AB, Koskull S, Perletti C, Heese B, von Döbeln U, Mereghetti P, Di Meo I, Invernizzi F, et al. 2008. Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy. J Med Genet 45: 473–478 [DOI] [PubMed] [Google Scholar]
- Mitchell TW, Savage JC, Gould DH 1993. High-performance liquid chromatography detection of sulfide in tissues from sulfide-treated mice. J Appl Toxicol 13: 389–394 [DOI] [PubMed] [Google Scholar]
- Müller FH, Bandeiras TM, Urich T, Teixeira M, Gomes CM, Kletzin A 2004. Coupling of the pathway of sulphur oxidation to dioxygen reduction: Characterization of a novel membrane-bound thiosulphate:quinone oxidoreductase. Mol Microbiol 53: 1147–1160 [DOI] [PubMed] [Google Scholar]
- Nowaczyk MJ, Lehotay DC, Platt BA, Fisher L, Tan R, Phillips H, Clarke JT 1998. Ethylmalonic and methylsuccinic aciduria in ethylmalonic encephalopathy arise from abnormal isoleucine metabolism. Metabolism 47: 836–839 [DOI] [PubMed] [Google Scholar]
- Ogasawara Y, Isoda S, Tanabe S 1994. Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biol Pharm Bull 17: 1535–1542 [DOI] [PubMed] [Google Scholar]
- Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, Jeon SB, Jeon WK, Chae HJ, Chung HT 2006. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW2647 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med 41: 106–119 [DOI] [PubMed] [Google Scholar]
- Palmfeldt J, Vang S, Stenbroen V, Pavlou E, Baycheva M, Buchal G, Monavari AA, Augoustides-Savvopoulou P, Mandel H, Gregersen N 2011. Proteomics reveals that redox regulation is disrupted in patients with ethylmalonic encephalopathy. J Proteome Res 10: 2389–2396 [DOI] [PubMed] [Google Scholar]
- Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N 2003. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J Biol Chem 278: 47449–47458 [DOI] [PubMed] [Google Scholar]
- Powell MA, Somero GN 1986. Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science 233: 563–566 [DOI] [PubMed] [Google Scholar]
- Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, Chunyu Z 2004. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 317: 30–37 [DOI] [PubMed] [Google Scholar]
- Reiffenstein RJ, Hulbert WC, Roth SH 1992. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 32: 109–134 [DOI] [PubMed] [Google Scholar]
- Rowan FE, Docherty NG, Coffey JC, O’Connell PR 2009. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br J Surg 96: 151–158 [DOI] [PubMed] [Google Scholar]
- Searcy DG, Lee SH 1998. Sulfur reduction by human erythrocytes. J Exp Zool 282: 310–322 [DOI] [PubMed] [Google Scholar]
- Samuelson J 1999. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother 43: 1533–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH 2004. Role of the liver in regulation of body cysteine taurine levels: A brief review. Neurochem Res 29: 105–110 [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW 1982. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 206: 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH, Ueki I 2010. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 34: 17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C 2007. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6: 917–935 [DOI] [PubMed] [Google Scholar]
- Theissen U, Martin W 2008. Sulfide : quinone oxidoreductase (SQR) from the lugworm Arenicola marina shows cyanide- and thioredoxin-dependent activity. FEBS J 275: 1131–1139 [DOI] [PubMed] [Google Scholar]
- Tiranti V, D’Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva MT, Vollmer B, et al. 2004. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet 74: 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Briem E, Lamantea E, Mineri R, Papaleo E, De Gioia L, Forlani F, Rinaldo P, Dickson P, Abu-Libdeh B, et al. 2006. ETHE1 mutations are specific to ethylmalonic encephalopathy. J Med Genet 43: 340–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, et al. 2009. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 15: 200–205 [DOI] [PubMed] [Google Scholar]
- Viscomi C, Burlina AB, Dweikat I, Savoiardo M, Lamperti C, Hildebrandt T, Tiranti V, Zeviani M 2010. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med 16: 869–871 [DOI] [PubMed] [Google Scholar]
- Wang R 2002. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J 16: 1792–1798 [DOI] [PubMed] [Google Scholar]
- Westley AM, Westley J 1991. Biological sulfane sulfur. Biochemistry 195: 63–67 [DOI] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK 2004. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite “scavenger?” J Neurochem 90: 765–768 [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK 2006. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun 343: 303–310 [DOI] [PubMed] [Google Scholar]
- Xu M, Wu YM, Li Q, Wang X, He RR 2008. Electrophysiological effects of hydrogen sulfide on pacemaker cells in sinoatrial nodes of rabbits. Sheng Li Xue Bao 60: 175–180 [PubMed] [Google Scholar]
- Yalamanchili C, Smith MD 2008. Acute hydrogen sulfide toxicity due to sewer gas exposure. Am J Emerg Med 26: 518.e5–7 [DOI] [PubMed] [Google Scholar]
- Yan SK, Chang T, Wang H, Wu L, Wang R, Meng QH 2006. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem Biophys Res Commun 361: 485–491 [DOI] [PubMed] [Google Scholar]
- Yang G, Sun X, Wang R 2004. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J 18: 1782–1784 [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. 2008. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafeiriou DI, Augoustides-Savvopoulou P, Haas D, Smet J, Triantafyllou P, Vargiami E, Tamiolaki M, Gombakis N, van Coster R, Sewell AC, et al. 2007. Ethylmalonic encephalopathy: Clinical and biochemical observations. Neuropediatrics 38: 78–82 [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R 2001. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 20: 6008–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]