

Abstract
The non-classical major histocompatibility complex molecule, human leukocyte antigen (HLA)-G, is thought to contribute to maternal immune tolerance and successful placentation during pregnancy. Genetic polymorphisms in HLA-G are known to influence expression levels as well as the relative expression of individual protein isoforms. As diminished or aberrant HLA-G expression patterns may contribute to the development of certain pregnancy complications, we sought to investigate the association between functional HLA-G polymorphisms and the risk of pre-eclampsia (PE) in African-American women. The association between maternal and fetal genotype at six HLA-G polymorphisms and risk of PE was assessed in 372 pregnancies (314 normotensive; 58 pre-eclamptic). We observed an elevated risk of PE (P = 0.00027) in pregnancies where the mother carried the 1597ΔC allele, a null allele that abolishes expression of full-length HLA-G isoforms. Furthermore, the frequency of the maternal 1597ΔC allele was highest in the subset of pre-eclamptic pregnancies that were delivered preterm, suggesting an association between the null allele and the severity of PE. We then replicated the association between higher maternal 1597ΔC allele frequency and increased severity of PE (P = 0.038) in an independent sample of 533 African-American women. Finally, to investigate the mechanistic basis of this association, we measured circulating soluble HLA-G (sHLA-G) concentrations in maternal serum collected during pregnancy in 51 healthy, normotensive African-American control women and found significantly lower levels in women carrying the 1597ΔC allele (P = 0.012). These results demonstrate that maternal HLA-G genotype is significantly associated with risk of PE in African-American women and is predictive of circulating sHLA-G levels during pregnancy.
Keywords: genetic predisposition, DNA variants, soluble HLA-G, toxemia of pregnancy, mutation
Introduction
Pre-eclampsia (PE) is a multisystem disorder characterized by the onset of gestational hypertension and proteinuria in the second half of pregnancy (Sibai, 2003; Steegers et al., 2010). PE occurs in 2–5% of all pregnancies in the USA (Wallis et al., 2008; Berg et al., 2009) and is a major contributor to maternal and fetal mortality and morbidity (Sibai et al., 2005). The precise etiology and pathogenesis of PE remains incompletely understood; however, it is generally accepted that the syndrome emerges from the action of multiple genetic, environmental and behavioral factors in the mother and the fetus (Roberts and Gammill, 2005; Redman and Sargent, 2005; Sibai et al., 2005). Furthermore, abnormal interactions between the maternal immune response and fetal-specific factors may contribute to the incidence and severity of PE (Roberts and Gammill, 2005; Steegers et al., 2010).
Familial inheritance of PE has been long recognized (Chesley et al., 1968), suggesting a genetic contribution to the syndrome. Indeed, both maternal and fetal (i.e. paternally derived) genetic factors have been associated with a predisposition to PE (Lie et al., 1998; Esplin et al., 2001). Candidate gene association studies have linked PE risk to genes involved in specific pathophysiological pathways, including the renin–angiotensin system, thrombophilia and hypofibrinolysis, oxidative stress and lipid metabolism, angiogenesis/anti-angiogenesis, endothelial dysfunction, intravascular inflammation and immune function (Redman and Sargent, 2005; Sibai et al., 2005; Mutze et al., 2008; Romero et al., 2008; Williams and Morgan, 2012). Among the genes involved in immunity, the human leukocyte antigen (HLA) gene, HLA-G, has been identified as a potential contributor to PE risk.
HLA-G is a non-classical HLA class Ib molecule with immunomodulatory properties that is thought to be involved in maternal tolerance of the semi-allogenic fetus. During pregnancy, HLA-G is highly expressed at the maternal–fetal interface by fetal extravillous cytotrophoblast cells (Kovats et al., 1990). Elevated levels of soluble HLA-G (sHLA-G) have been observed in the maternal peripheral blood during pregnancy (Hunt et al., 2000; Pfeiffer et al., 2000; Steinborn et al., 2007; Rizzo et al., 2009), and those levels likely reflect HLA-G expression from both fetal placental cells and maternal immune cells, such as monocytes (Alegre et al., 2007; Feger et al., 2007). A role for HLA-G in the pathogenesis of PE is supported by several lines of evidence. First, circulating sHLA-G levels during pregnancy were significantly lower in women who subsequently experienced PE compared with women with uncomplicated, successful pregnancies (Yie et al., 2005; Hackmon et al., 2007; Steinborn et al., 2007; Rizzo et al., 2009). Second, genetic variation in the HLA-G gene has been associated with variation in circulating sHLA-G levels during pregnancy (Hviid et al., 2004; Chen et al., 2008). Third, HLA-G polymorphisms, including those associated with circulating levels, have been associated with increased risk of PE in some studies (O’Brien et al., 2001; Moreau et al., 2008; Tan et al., 2008; Yie et al., 2008; Larsen et al., 2010), although not in others (Aldrich et al., 2000; Lin et al., 2006; Vianna et al., 2007; Iversen et al., 2008).
Despite evidence for HLA-G playing an important role during pregnancy, the precise relationship between genetic variation in HLA-G, levels of circulating sHLA-G and PE remains unresolved. To date, few studies have examined all the three in the same population. In addition, studies showing an association between HLA-G polymorphisms and sHLA-G levels have focused primarily on women of European ancestry. Consequently, they do not provide insight into the functional consequences of polymorphisms, such as the 1597ΔC null mutation, that are rare in Europeans but more common in other global populations (Matte et al., 2000; Aldrich et al., 2002). The focus on PE in women of European ancestry also fails to provide insight into the factors contributing to the disorder in other, more vulnerable populations. For example, few studies have focused on the genetic basis of PE in African-American women, despite rates of PE being consistently higher in that group (Samadi et al., 1996; Caughey et al., 2005; Tanaka et al., 2007; Gong et al., 2012).
To better understand the functional and clinical effects of HLA-G polymorphisms in an at-risk population, we examined HLA-G genetic variation in African-American mothers and infants enrolled in the Chicago Lying-In Pregnancy Program (CLIPP; Joseph et al., 2008; Neidich et al., 2008), and tested for an association between the observed genetic variation and risk and severity of PE. Next, we assessed the reproducibility of the observed genetic association in an independent population of African-American women with PE. Finally, we examined the functional consequences of the 1597ΔC allele, which abolishes expression of full-length transmembrane G1 and soluble G5 protein isoforms, on circulating sHLA-G levels in uncomplicated, term pregnancies. This study represents one of the most comprehensive studies of an important HLA-G polymorphism in an underrepresented minority population at high risk for PE.
Materials and Methods
Study subjects
Self-reported African-American women were identified through retrospective surveys of the CLIPP biobank (Joseph et al., 2008; Neidich et al., 2008). An initial sample of 69 African-American pre-eclamptic pregnancies was identified in the CLIPP delivery records, excluding women with chronic hypertension, pre-conceptual or gestational diabetes mellitus, multiple gestations or fetal anomalies. PE was defined as blood pressure of at least 140/90 mmHg with readings at least 6 h apart and proteinuria (300 mg/24 h or ≥30 mg/dl on random urine analysis; Gifford et al., 2000; Lindheimer et al., 2010). We then selected four to five women with normotensive term pregnancies (same exclusion criteria) for each woman with PE from the CLIPP database, matching for age (±3 years), race (African-American) and parity (0 versus ≥1). After confirming diagnoses and exclusions by medical record reviews and on the basis of availability of DNA samples, a total of 372 women remained: 58 with PE and 314 with a normotensive term delivery. In this final sample, there was a non-significant excess of nulliparous women among the pre-eclamptics compared with the controls (46.6 versus 33.0%, P = 0.068) (Table I). Among the pre-eclamptic pregnancies, 23 women delivered preterm and 35 women delivered at term. This study was approved by the University of Chicago Institutional Review Board and all participants gave written informed consent.
Table I.
Clinical characteristics of the CLIPP study participants, with PE cases stratified by preterm versus term delivery.
| Pre-eclampsia |
Controls (n = 314) | |||
|---|---|---|---|---|
| All (n = 58) | Preterm (n = 23) | Term (n = 35) | ||
| Mean age, years (SD) | 25.1 (6.0) | 24.4 (6.3) | 25.6 (5.7) | 25.6 (5.0) |
| % Nulliparous | 46.6 | 39.1 | 51.4 | 33.0 |
| Parity range | 0–4 | 0–3 | 0–4 | 0–6 |
| Mean gestational age at delivery, weeks (SD) | 36.1 (0.2) | 32.2 (4.1) | 38.7 (1.2) | 39.8 (1.1) |
| % Primigravida | 32.8 | 26.1 | 37.1 | 21.8 |
| Gravidity range | 1–8 | 1–7 | 1–8 | 1–10 |
| Infant sex, % male | 45.6 | 36.4 | 51.4 | 52.1 |
| Mean infant birthweight, g (SD) | 2625.9 (983.2) | 1731.9 (875.9) | 3214.0 (474.7) | 3277.9 (438.3) |
| Mean maximum systolic blood pressure in labor (SD), mmHg | 164.3 (13.2) | 170.3 (13.4) | 160.3 (11.6) | <140 |
| Mean maximum diastolic blood pressure in labor (SD), mmHg | 103.8 (11.9) | 108.8 (12.3) | 100.5 (10.5) | <90 |
To replicate our findings in the CLIPP Chicago population, HLA-G variation was examined in a second population of African-American women (also by self-report) with preterm PE (n = 98), term PE (n = 151) and term normotensive, uncomplicated (i.e. control) pregnancies (n = 284). These pregnancies were identified by searching the clinical database and the bank of biological specimens of the Perinatology Research Branch and Wayne State University, Detroit, MI. All the patients provided written informed consent for the collection and use of samples for research purposes (including DNA) under the protocols approved by the Institutional Review Boards of Wayne State University and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services (NICHD/NIH/DHHS).
Women were excluded from these studies if their pregnancies were associated with chronic hypertension, known major fetal or chromosomal anomaly or multiple gestations. All the women were enrolled at Hutzel Women's Hospital, Detroit, MI, and followed until delivery. PE was defined as a systolic blood pressure of at least 140 or a diastolic blood pressure of at least 90 mmHg on two separate readings at least 4 h apart and the presence of proteinuria (300 mg protein in 24 h or >30 mg/dl on two occasions at least 4 h apart; Sibai et al., 1997; Gifford et al., 2000). The control pregnancies were considered to be uncomplicated if there were no major medical, obstetrical or surgical complications and delivery occurred at term (>37 weeks) with an infant whose birthweight was appropriate for gestational age (10–90th percentile) according to the reference range for the USA (Alexander et al., 1996).
HLA-G genotyping
For the CLIPP subjects, maternal DNA was extracted from EDTA-anticoagulated whole blood using an isopropanol precipitation-based protocol using an Autogen AGF3000 DNA Extractor (Autogen, Inc., Holliston, MA). Infant DNA was extracted from either cord blood samples, using Gentra Puregene DNA isolation kits (Qiagen, Valencia, CA) or from placental villous tissue. In the latter case, ∼100 mg of placental villous tissue was obtained from the manual dissection of placental tissue under a microscope at 40× power. After isolating the placental villous tissue from the maternally derived tissues, it was washed twice in sterile Hank's balanced salt solution and then used for whole genome DNA extraction (Gentra Puregene kit). DNA concentration and purity were assessed by UV spectrophotometry and automated fluorimetric quantification (PicoGreen dsDNA Quantification Kit; Molecular Probes, Eugene, OR).
For the Detroit subjects, genomic DNA was isolated from 150 µl of buffy coat with an EZ1 DNA Blood 350 µl kit (Qiagen, USA) using an EZ1 instrument (Qiagen, USA). A DropSense 96 spectrophotometer (Trinean NV, Belgium) was used to quantify the amount of DNA prior to amplification. The whole genome amplification from genomic DNA was performed with REPLI-g Midi Kit (Qiagen, USA) according to the manufacturer's instruction. Briefly, 3 µl genomic DNA (>10 ng) from each specimen was mixed with 2 µl of Buffer D3 in individual wells in a 96-well plate and incubated at room temperature for 5 min to denature the DNA. Master mix (44 µl) that contained stop solution, water, REPLI-g Midi reaction buffer and REPLI-g Midi DNA polymerase was added to the denatured DNA. The mixture was incubated at 30°C for 8–16 h. After the incubation, the REPLI-g DNA polymerase was inactivated by heating the sample at 65°C for 3 min. The amplified DNA was stored at −20°C until further use.
DNA samples were genotyped for six HLA-G polymorphisms using a modification of the SNaPshot (Applied Biosystems, Carlsbad, CA) protocol described in Tan et al. (2008) that also included the 3′UTR +3142 (G/C) polymorphism from Tan et al. (2007). Polymorphisms were chosen on the basis of previously published associations with functional or clinical phenotypes and to capture the major haplotype structure of the gene. The six polymorphisms studied were: (i) the promoter variant −725 (G/C/T; rs1233334) associated with sporadic miscarriage (Ober et al., 2003) and variation in gene expression (Ober et al., 2006; Jassem et al., 2012); (ii) the +36 (G/A) polymorphism (rs1630185) in the untranslated first exon that differentiates the two major promoter clades (Tan et al., 2005); (iii) the 1597ΔC single-base insertion/deletion polymorphism (rs41557518) that prevents expression of full-length HLA-G protein isoforms (Ober et al., 1998) and has been associated with recurrent miscarriage (Aldrich et al., 2001; Pfeiffer et al., 2001); (iv) the non-conservative amino acid substitution (rs12722482) in exon 4 at codon 258 (Thr→Met) that has been associated with PE (Moreau et al., 2008; Tan et al., 2008); (v) the 14 bp insertion/deletion polymorphism (rs66554220) in the 3′UTR that has been associated with PE risk (Hylenius et al., 2004; Moreau et al., 2008; Larsen et al., 2010), HLA-G transcript levels (Hviid et al., 2003) and circulating sHLA-G levels in plasma (Chen et al., 2008) and (vi) the +3142 (G/C) polymorphism (rs1063320) in the 3′UTR that disrupts a micro(mi)RNA target site and influences HLA-G expression in the presence of specific miRNAs (Tan et al., 2007).
Detection of sHLA-G
sHLA-G protein (specifically, soluble G5 and shed transmembrane G1) was measured in serum from CLIPP participants using the EXBIO (Vestec, Czech Republic)/BioVendor (Brno, Czech Republic) ELISA kit, according to the manufacturer's instructions. For each well, absorbance at 450 nm was measured using a SpectraMax Plus384 Absorbance Microplate Reader (Molecular Devices, Sunnyvale, CA). sHLA-G concentrations of samples were determined using a calibration curve constructed from four-parameter logistic curve fitting of the mean absorbance of calibrators of known concentration. Each individual's serum sample was run in duplicate on the same ELISA plate; individuals who showed a coefficient of variation greater than 35% were excluded from further analyses. The mean sHLA-G concentration for each serum sample was used for subsequent analyses. The estimated mean sHLA-G concentration was below the limits of detection for eight women (five who did not carry the 1597ΔC allele and three who did carry it) and above the limits of detection for one individual. Because of this censoring, rank-based tests were used to subsequently assess the relationship between sHLA-G and clinical or genetic data. For the purposes of visualization and inclusion in the rank-based tests, the eight individuals with sHLA-G below the limit of detection were assigned a value of 0.00001 and the one above the limit of detection was assigned a value of 250 U/ml.
Statistical analyses
Two tests were used to assess HLA-G genetic associations with PE in CLIPP subjects. First, an allele test was used to compare allele counts in cases and controls using a 2 × 2 contingency table assessed by the Pearson chi-square test. Because the −725 polymorphism was tri-allelic (C/G/T), each of the two minor alleles was analyzed separately. Specifically, the P-value for the T allele corresponds to the difference in the number of T and non-T alleles in cases compared with controls, and the P-value for the G allele corresponds to the difference in the number of G and non-G alleles in cases versus controls. Second, a genotype test was used to compare genotype counts in PE cases and controls using a 2 × 3 contingency table assessed by the Pearson chi-square test or Fisher's exact probability text (one-tailed) when cell frequencies were equal to or less than five. At the tri-allelic −725 polymorphism, the six genotypes observed in mothers and five genotypes observed in infants were compared in 2 × 6 and 2 × 5 contingency tables, respectively.
The Cochran–Armitage linear trend test was used to test for an association between 1597ΔC genotype frequencies and increasing severity of PE. Genotype counts at 1597ΔC were compared across three groups ordered by severity (normotensive term pregnancy = 0, term pre-eclamptic pregnancy = 1, preterm pre-eclamptic pregnancy = 2). Because of the rarity of 1597ΔC allele homozygotes (only 6 homozygotes in 847 individuals), the trend test was performed in a 2 × 3 table comparing the CC genotype count with the pooled CT/TT genotype count. The trend test was implemented in the coin package (Hothorn et al., 2008) in R (version 2.15.0) using a standardized scalar test statistic, an alternative (one-sided) hypothesis and a null distribution approximated by Monte-Carlo resampling (2 000 000 random permutations). The association signals (P-values) obtained from the trend tests in the CLIPP and Detroit samples were combined using Stouffer's weighted Z-method (Whitlock, 2005), as implemented in the survcomp package in R.
The relationship between serum sHLA-G concentration and estimated gestational age and trimester at collection was assessed using the Wilcoxon rank sum test. The association between the presence/absence of the 1597ΔC allele in the mother and estimated serum sHLA-G concentration was also assessed using the Wilcoxon rank sum test, as implemented in the JMP software (SAS institute Inc., Cary, NC), version 10.0.0.
Results
Clinical characteristics of the CLIPP pregnancies diagnosed with PE and normotensive term pregnancies (i.e. controls) are described in Table I. PE cases and controls did not differ significantly in percent nulliparity, percent primigravida, maternal age or infant sex ratio (Table I). Not unexpectedly, PE pregnancies were delivered earlier than those of controls and, as a result, their infants were on average of lower birthweight. However, birthweights between term infants of PE and control pregnancies were not significantly different (P = 0.74).
HLA-G allele frequencies in PE cases and controls
Allele frequencies in the mothers did not differ in the 44 PE cases compared with the 271 controls for five of the six HLA-G polymorphisms; only the 1597ΔC polymorphism showed a significant difference between the two groups (P = 0.00027; Table II). Specifically, the frequency of the 1597ΔC allele was nearly three times higher in the PE cases (15.9%) compared with the controls (5.4%). The 1597ΔC allele frequency in the controls was similar to frequencies observed in previous studies of African-American individuals (Ishitani et al., 1999; Aldrich et al., 2002). The genotype frequency distribution for the 1597ΔC polymorphism also differed between the cases and the controls (P = 0.0029; Table III): women carrying one or two copies of the 1597ΔC allele had a significantly increased risk of PE [odds ratio (OR) = 3.39; 95% confidence interval (CI): 1.56–7.34].
Table II.
Allele frequencies for six HLA-G polymorphisms in 44 women with PE and 271 healthy, normotensive control women from the CLIPP study.
| Polymorphism | Allele | PE cases | Controls | P-value |
|---|---|---|---|---|
| −725C/G/T | C | 0.90 | 0.83 | |
| G | 0.049 | 0.065 | 0.58a | |
| T | 0.049 | 0.10 | 0.11a | |
| +36G/A | A | 0.61 | 0.55 | 0.36 |
| G | 0.39 | 0.45 | ||
| 1597ΔC | C | 0.84 | 0.95 | 0.00027 |
| Δ | 0.16 | 0.054 | ||
| Thr258Met | Thr | 0.99 | 0.99 | 0.53 |
| Met | 0.012 | 0.0077 | ||
| 14-bp indel | Del | 0.54 | 0.58 | 0.42 |
| Ins | 0.46 | 0.42 | ||
| +3142G/C | G | 0.64 | 0.66 | 0.76 |
| C | 0.36 | 0.34 |
aBecause the −725 polymorphism was tri-allelic, allele counts for each of the minor alleles were compared independently.
Table III.
HLA-G 1597ΔC genotype counts in 44 women with PE and 271 normotensive women with term deliveries (Controls) from the CLIPP study.
| Group | Number of each genotype |
P-value | ||
|---|---|---|---|---|
| C/C | C/Δ | Δ/Δ | ||
| PE cases (%) | 32 (72.7%) | 10 (22.7%) | 2 (4.5%) | 0.0029 |
| Controls (%) | 244 (90%) | 25 (9.2%) | 2 (0.74%) | |
HLA-G allele frequencies in the infants of 47 pre-eclamptic pregnancies and 281 control pregnancies did not differ for any of the six polymorphisms (Table IV). Although the 1597ΔC allele occurred at a higher frequency in the infants from PE pregnancies (11.7%) compared with infants from control pregnancies (6.6%), this difference was not significant (P = 0.078; Table IV) and could just reflect the higher allele frequency in the mothers with pre-eclamptic pregnancies.
Table IV.
Allele frequencies for six HLA-G polymorphisms in 47 infants from PE pregnancies and 281 infants from healthy, normotensive control pregnancies from the CLIPP study.
| Polymorphism | Allele | PE cases | Controls | P-value |
|---|---|---|---|---|
| −725C/G/T | C | 0.89 | 0.83 | |
| G | 0.063 | 0.057 | 0.84a | |
| T | 0.052 | 0.11 | 0.073a | |
| +36G/A | A | 0.63 | 0.54 | 0.086 |
| G | 0.37 | 0.46 | ||
| 1597ΔC | C | 0.88 | 0.93 | 0.078 |
| Δ | 0.12 | 0.066 | ||
| Thr258Met | Thr | 1.0 | 0.98 | 0.27 |
| Met | 0.00 | 0.018 | ||
| 14-bp indel | Del | 0.62 | 0.55 | 0.28 |
| Ins | 0.38 | 0.45 | ||
| +3142G/C | G | 0.68 | 0.65 | 0.51 |
| C | 0.32 | 0.35 |
aBecause the −725 polymorphism was tri-allelic, allele counts for each of the minor alleles were compared independently.
The 1597ΔC allele and severity of PE
Among the CLIPP women with PE, 39.7% delivered preterm (Table I). The women with preterm PE had significantly higher mean maximum systolic blood pressure (P = 0.0040) and mean maximum diastolic blood pressure during labor (P = 0.0075) compared with the women with PE who delivered at term (Table I).
We hypothesized that the frequency of the 1597ΔC allele would be further elevated in women with severe PE. As predicted, the 1597 deletion allele was present in six (37.5%) of the women with preterm PE (5 Δ/C, 1 Δ/Δ), six (21.4%) of the women with term PE (5 Δ/C, 1 Δ/Δ) and 27 (9.96%) of the women with normotensive pregnancies who delivered at term (25 Δ/C, 2 Δ/Δ) (Fig. 1). This association between an increasing frequency of the maternal 1597ΔC allele and increasing severity was statistically significant (Cochran–Armitage trend test, Z = 3.57, Ptrend = 0.0012). No such relationship was observed when considering infants' 1597ΔC allele frequency (not shown).
Figure 1.
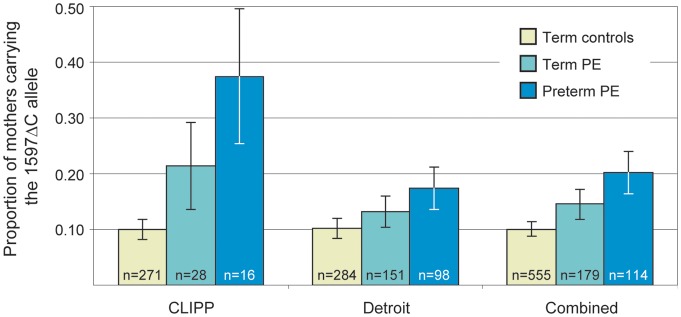
Proportion of women carrying the 1597ΔC allele in the CLIPP, Detroit and combined samples. The proportion of women carrying the 1597ΔC allele is shown separately for the term control, term PE and preterm PE pregnancies in each sample. The association between presence of the 1597ΔC allele and increased severity of PE was significant in the combined sample (Weighted Z-transform method, P = 0.0011). Error bars indicate the standard error of the proportion.
Replication of the 1597ΔC association with PE
The association between higher maternal 1597ΔC allele frequency and increased severity of PE was evaluated in an independent sample of African-American women from Detroit, Michigan. The proportion of Detroit women carrying the 1597ΔC allele was 17.4% in the preterm PE group, 13.3% in the term PE group and 10.2% in the controls (Fig. 1); this trend was statistically significant (Cochran–Armitage trend test, Z = 1.88, Ptrend = 0.038). No association was observed between the 1597ΔC allele frequency in the Detroit infants and severity of PE. The positive association between 1597ΔC allele frequency and increased severity of PE was significant in the combined sample of CLIPP and Detroit subjects (Fig. 1; weighted Z-transform method, P = 0.0011).
1597ΔC allele and sHLA-G levels during pregnancy
Serum samples collected during pregnancy were available for 56 of the healthy, normotensive control CLIPP pregnancies, but only for six of the women with PE. Therefore, associations between circulating sHLA-G levels and the 1597ΔC allele were assessed only in the controls. Five of the 56 control pregnancies were excluded from analysis because sHLA-G concentrations of their duplicate samples showed a coefficient of variation greater than 35%. For the remaining 51 samples, median sHLA-G concentrations were significantly lower in the 11 women who carried the 1597ΔC allele (10 heterozygotes, 1 homozygote) compared with the 40 women who did not carry this allele (Fig. 2; median sHLA-G = 12.5 U/ml for mothers carrying the 1597ΔC allele and 32.8 U/ml for mothers who did not carry the allele; Wilcoxon rank sum test, χ2 = 6.31, df = 1, P = 0.012). Consistent with the results of the aforementioned association studies, the 1597ΔC of the infant was not a significant predicator of serum sHLA-G levels in the mother (median sHLA-G = 15.1 U/ml for 12 infants carrying the 1597ΔC allele and 31.8 U/ml for 36 infants who did not carry the allele; Wilcoxon rank sum test, χ2 = 1.72, df = 1, P = 0.19). Circulating sHLA-G concentrations during pregnancy did not differ significantly as a function of the estimated gestational age (P = 0.50) or trimester (P = 0.53) in which the sample was collected (Supplementary data, Fig. S1).
Figure 2.

Concentrations of sHLA-G protein in maternal serum in uncomplicated, normotensive pregnancies as a function of maternal 1597ΔC. Box plots show the distribution of circulating sHLA-G in women who carried the 1597ΔC allele (Present) and those who did not carry the allele (Not present). Estimates of sHLA-G concentrations that were more than 1.5 interquartile ranges from the quartiles are shown individually as outliers (indicated by filled squares).
Discussion
Identification of immunogenetic factors that contribute to PE risk and severity will provide insight into the etiology of this multifactorial disorder, and may ultimately lead to improved methods for early diagnosis or treatment. In this study, we evaluated the association between genetic variation in the HLA-G gene and PE in two independent samples of African-American women. In both, we observed an increased risk of PE when the mother carried the 1597ΔC allele, which results from a single-base-pair deletion of a cytosine in exon 3 that abolishes the expression of the full-length HLA-G protein isoforms containing the α2 domain, i.e. HLA-G1 and -G5 (Ober et al., 1998). In contrast, the presence of the 1597ΔC allele in the fetus was not associated with PE risk. The observation that increasing frequency of the maternal, but not the fetal, 1597ΔC allele was associated with increasing severity of PE suggests that maternal-derived HLA-G may serve functions different from those of the placental-derived isoforms.
The lack of association between the 1597ΔC allele and PE in African-American infants is not new. We previously studied this polymorphism in 39 African-American infants with intrauterine growth restriction, 57 African-American infants from pre-eclamptic pregnancies and 111 African-American normal-weight term infants, ascertained from the same clinics as the CLIPP sample (Aldrich et al., 2000). This earlier study, which did not include the mothers, did not show an association with either disorder (Aldrich et al., 2000). We previously showed that the shorter HLA-G isoforms, G2 and G6, are expressed in the placenta of fetuses who are homozygous for the 1597ΔC allele, and suggested that those isoforms may compensate for reduced levels of HLA-G1 and -G5 in the placentas of fetuses carrying the 1597ΔC allele (Ober et al., 1998). These shorter HLA-G2 and -G6 isoforms also circulate in the maternal periphery during pregnancy (Hunt et al., 2000) and could explain why the presence of the 1597ΔC allele in the fetus is not associated with preeclampsia, as we have observed. Thus, the combined results of these studies support our suggestion that the effects of the 1597ΔC allele on PE risk is due to the maternal, but not the fetal, genotype.
To directly assess the effects of the maternal or fetal 1597ΔC allele on circulating sHLA-G levels, we measured concentrations of the HLA-G1 and -G5 isoforms in pregnancy serum from 51 control women. The maternal 1597ΔC allele was associated with lower circulating sHLA-G levels in these uncomplicated, normotensive pregnancies, indicating that maternal sHLA-G contributes to risk of PE but is not causal. This is also consistent with previous studies that reported lower levels of circulating sHLA-G during pregnancy in women with PE (Yie et al., 2005; Hackmon et al., 2007; Steinborn et al., 2007; Rizzo et al., 2009). In our study, we could not study circulating sHLA-G concentrations in the pre-eclamptic pregnancies, but the associations between 1597ΔC and PE risk, and between 1597ΔC and reduced levels of sHLA-G, are consistent with these earlier studies reporting reduced levels of sHLA-G being a risk factor for PE.
The results of the present study of African-American women did not confirm associations reported in previous HLA-G genetic association studies, such as those involving the 14 bp insertion/deletion polymorphism (rs66554220) or the Thr258Met variant (rs12722482). The presence of the 14 bp insertion in the fetal genome had been associated with increased risk of PE in women of European ancestry (Hylenius et al., 2004; Moreau et al., 2008; Larsen et al., 2010), but we did not observe differences in 14 bp insertion allele frequencies in the African-American PE cases compared with controls when considering either fetal or maternal genotype. The increased frequency of the fetal G*0106 haplotype (which is defined by the Thr258Met variant allele) in pre-eclamptic pregnancies reported previously (Moreau et al., 2008; Tan et al., 2009) was also not apparent in our African-American data, perhaps because of the fact that the Met allele occurred at very low frequencies (<2%) in the African-American women and infants in our study.
A potential limitation of our study is that we could not formally assess differences in local ancestry at the HLA-G locus in the case and control samples because we did not have genotype data for SNPs spanning this region. It is possible that the increased frequency of the 1597ΔC allele in the cases was due to increased African ancestry in those individuals, because that allele occurs at higher frequencies in populations of African descent (Aldrich et al., 2002). However, we think this is unlikely for three reasons. First, the −725T allele also occurs at significantly higher frequencies in African Americans compared with European Americans (10.2 versus 2.2%, respectively; Tan et al., 2005). Yet, the frequency of this allele was 4.9% in the pre-eclamptic cases and 10.0% in the normotensive term controls in the CLIPP sample. Second, we did not observe a significant difference in the 1597ΔC allele frequencies between infants born to pre-eclamptic and control pregnancies. Third, we demonstrated associations between the 1597ΔC allele and PE in two independent African-American samples, each with cases and controls recruited from within the same medical center, and it is unlikely that the effects of population stratification would be consistent in direction and magnitude in both populations (Thomas and Witte, 2002; Wacholder et al., 2002).
This study provides new insight into the genetic factors that contribute to PE risk. The differences between the current results and those reported for other populations further support the idea that PE is a multifactorial disorder that develops as a result of complex interactions between genetic and environmental factors, with potentially different contributions from maternal and fetal genes. Indeed, significant heterogeneity in the strength of associations has been observed in studies of candidate genes and PE risk. For studies of HLA-G, this heterogeneity may be due to the effects of small sample size, differences in population genetic structure or ancestry, differential impact of environmental influences or even maternal–fetal interactions (Tan et al., 2009). Thus, it is important for future studies of HLA-G and PE risk to be adequately powered in terms of sample size and genetic coverage and to include subjects of diverse ancestry to allow assessment of alleles that are rare in Europeans but common in other groups, such as the 1597ΔC allele described in this report.
Supplementary data
Supplementary data are available at http://molehr.oxfordjournals.org/.
Authors' roles
D.A.L., R.R. and C.O. were involved in study design, execution and presentation. K.M. and T.C. performed the patient consenting and acquisition of biological samples. D.A.L., C.B. and K.P. completed the genotyping and ELISA studies. D.A.L. and C.O. performed data processing and statistical analysis. D.A.L. and C.O. wrote and revised the manuscript. All the authors contributed critical discussion and manuscript review and gave final approval of the version to be published.
Funding
This work was supported by the National Institutes of Health (P01HD049480 to C.O., UL1RR024999 to the Institute for Translation Medicine at the University of Chicago) and the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS. D.A.L. was supported by NIH grants F32HL095268 and T32 HL007605.
Conflict of interest
None declared.
Supplementary Material
Acknowledgements
The authors thank Drs Joan Hunt, Margaret Petroff, Daniel Geraghty, J. Lee Nelson, D. Michael Nelson, Dale Abrahamson and David Albertini for helpful discussions on study design and interpretation of results; Dr Marshall Lindheimer for his clinical expertise; Shaneisha Allen for CLIPP recruitment and study coordination; the University of Chicago Institute of Medicine CTSA for core services, and the Chicago Lying-In Women's Board and the Department of OB/GYN for supporting the CLIPP biobank.
References
- Aldrich C, Verp MS, Walker MA, Ober C. A null mutation in HLA-G is not associated with pre-eclampsia or intrauterine growth retardation. J Reprod Immunol. 2000;47:41–48. doi: 10.1016/s0165-0378(00)00052-8. [DOI] [PubMed] [Google Scholar]
- Aldrich CL, Stephenson MD, Karrison T, Odem RR, Branch DW, Scott JR, Schreiber JR, Ober C. HLA-G genotypes and pregnancy outcome in couples with unexplained recurrent miscarriage. Mol Hum Reprod. 2001;7:1167–1172. doi: 10.1093/molehr/7.12.1167. [DOI] [PubMed] [Google Scholar]
- Aldrich CL, Wambebe C, Odama L, Di Rienzo A, Ober C. Linkage disequilibrium and age estimates of a deletion polymorphism (1597deltaC) in HLA-G suggest non-neutral evolution. Hum Immunol. 2002;63:405–412. doi: 10.1016/s0198-8859(02)00377-4. [DOI] [PubMed] [Google Scholar]
- Alegre E, Diaz-Lagares A, LeMaoult J, Lopez-Moratalla N, Carosella ED, Gonzalez A. Maternal antigen presenting cells are a source of plasmatic HLA-G during pregnancy: Longitudinal study during pregnancy. Hum Immunol. 2007;68:661–667. doi: 10.1016/j.humimm.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Alexander GR, Himes JH, Kaufman RB, Mor J, Kogan M. A United States national reference for fetal growth. Obstet Gynecol. 1996;87:163–168. doi: 10.1016/0029-7844(95)00386-X. [DOI] [PubMed] [Google Scholar]
- Berg CJ, MacKay AR, Qin C, Callaghan WM. Overview of maternal morbidity during hospitalization for labor and delivery in the United States 1993–1997 and 2001–2005. Obstet Gynecol. 2009;113:1075–1081. doi: 10.1097/AOG.0b013e3181a09fc0. [DOI] [PubMed] [Google Scholar]
- Caughey AB, Stotland NE, Washington AE, Escobar GJ. Maternal ethnicity, paternal ethnicity, and parental ethnic discordance—predictors of preeclampsia. Obstet Gynecol. 2005;106:156–161. doi: 10.1097/01.AOG.0000164478.91731.06. [DOI] [PubMed] [Google Scholar]
- Chen XY, Yan WH, Lin A, Xu HH, Zhang JG, Wang XX. The 14 bp deletion polymorphisms in HLA-G gene play an important role in the expression of soluble HLA-G in plasma. Tissue Antigens. 2008;72:335–341. doi: 10.1111/j.1399-0039.2008.01107.x. [DOI] [PubMed] [Google Scholar]
- Chesley LC, Annitto JE, Cosgrove RA. Familial factor in toxemia of pregnancy. Obstet Gynecol. 1968;32:303–311. [PubMed] [Google Scholar]
- Esplin MS, Fausett MB, Fraser A, Kerber R, Mineau G, Carrillo J, Varner MW. Paternal and maternal components of the predisposition to preeclampsia. N Engl J Med. 2001;344:867–872. doi: 10.1056/NEJM200103223441201. [DOI] [PubMed] [Google Scholar]
- Feger U, Tolosa E, Huang YH, Waschbisch A, Biedermann T, Melms A, Wiendl H. HLA-G expression defines a novel regulatory T-cell subset present in human peripheral blood and sites of inflammation. Blood. 2007;110:568–577. doi: 10.1182/blood-2006-11-057125. [DOI] [PubMed] [Google Scholar]
- Gifford RW, August PA, Cunningham G, Green LA, Lindheimer MD, McNellis D, Roberts JM, Sibai BM, Taler SJ Pro NHBPE. Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol. 2000;183:S1–S22. [PubMed] [Google Scholar]
- Gong J, Savitz DA, Stein CR, Engel SM. Maternal ethnicity and pre-eclampsia in New York City, 1995–2003. Paediatr Perinat Epidemiol. 2012;26:45–52. doi: 10.1111/j.1365-3016.2011.01222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackmon R, Koifman A, Hyobo H, Glickman H, Sheiner E, Geraghty DE. Reduced third-trimester levels of soluble human leukocyte antigen G protein in severe preeclampsia. Am J Obstet Gynecol. 2007;197:255.e1–255.e5. doi: 10.1016/j.ajog.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, van de Wiel MAV, Zeileis A. Implementing a class of permutation tests: the coin package. J Stat Softw. 2008;28:1–23. [Google Scholar]
- Hunt JS, Jadhav L, Chu W, Geraghty DE, Ober C. Soluble HLA-G circulates in maternal blood during pregnancy. Am J Obstet Gynecol. 2000;183:682–688. doi: 10.1067/mob.2000.106762. [DOI] [PubMed] [Google Scholar]
- Hviid TV, Hylenius S, Rorbye C, Nielson LG. HLA-G allelic variants are associated with differences in the HLA-G mRNA isoform profile and HLA-G mRNA levels. Immunogenetics. 2003;55:63–79. doi: 10.1007/s00251-003-0547-z. [DOI] [PubMed] [Google Scholar]
- Hviid TVF, Rizzo R, Christiansen OB, Melchiorri L, Lindhard A, Baricordi OR. HLA-G and IL-10 in serum in relation to HLA-G genotype and polymorphisms. Immunogenetics. 2004;56:135–141. doi: 10.1007/s00251-004-0673-2. [DOI] [PubMed] [Google Scholar]
- Hylenius S, Andersen AMN, Melbye M, Hviid TVF. Association between HLA-G genotype and risk of pre-eclampsia: a case-control study using family triads. Mol Hum Reprod. 2004;10:237–246. doi: 10.1093/molehr/gah035. [DOI] [PubMed] [Google Scholar]
- Ishitani A, Kishida M, Sageshima N, Yashiki S, Sonoda S, Hayami M, Smith AG, Hatake K. Re-examination of HLA-G polymorphism in African Americans. Immunogenetics. 1999;49:808–811. doi: 10.1007/s002510050555. [DOI] [PubMed] [Google Scholar]
- Iversen AC, Nguyen OTD, Tommerdal LF, Eide IP, Landsem VM, Acar N, Myhre R, Klungland H, Austgulen R. The HLA-G 14bp gene polymorphism and decidual HLA-G 14bp gene expression in pre-eclamptic and normal pregnancies. J Reprod Immunol. 2008;78:158–165. doi: 10.1016/j.jri.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Jassem RM, Shani WS, Loisel DA, Sharief M, Billstrand C, Ober C. HLA-G polymorphisms and soluble HLA-G protein levels in women with recurrent pregnancy loss from Basrah province in Iraq. Hum Immunol. 2012;73:811–817. doi: 10.1016/j.humimm.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Neidich AB, Ober C, Ross LF. Empirical data about women's attitudes toward a biobank focused on pregnancy outcomes. Am J Med Genet Part A. 2008;146A:305–311. doi: 10.1002/ajmg.a.32146. [DOI] [PubMed] [Google Scholar]
- Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, Demars R. A class-I antigen, HLA-G, expressed in human trophoblasts. Science. 1990;248:220–223. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- Larsen MH, Hylenius S, Andersen AMN, Hviid TVF. The 3′-untranslated region of the HLA-G gene in relation to pre-eclampsia: revisited. Tissue Antigens. 2010;75:253–261. doi: 10.1111/j.1399-0039.2009.01435.x. [DOI] [PubMed] [Google Scholar]
- Lie RT, Rasmussen S, Brunborg H, Gjessing HK, Lie-Nielsen E, Irgens LM. Fetal and maternal contributions to risk of pre-eclampsia: population based study. Br Med J. 1998;316:1343–1347. doi: 10.1136/bmj.316.7141.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Yan WH, Dai MZ, Chen XJ, Li BL, Chen BG, Fan LA. Maternal human leukocyte antigen-G polymorphism is not associated with pre-eclampsia in a Chinese Han population. Tissue Antigens. 2006;68:311–316. doi: 10.1111/j.1399-0039.2006.00667.x. [DOI] [PubMed] [Google Scholar]
- Lindheimer MD, Taler SJ, Cunningham FG. Hypertension in pregnancy. J Am Soc Hypertens. 2010;4:68–78. doi: 10.1016/j.jash.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Matte C, Lacaille J, Zijenah L, Ward B, Roger M, Roger M ZVITAMBO Study Group. HLA-G and HLA-E polymorphisms in an indigenous African population. Hum Immunol. 2000;61:1150–1156. doi: 10.1016/s0198-8859(00)00200-7. [DOI] [PubMed] [Google Scholar]
- Moreau P, Contu L, Alba F, Lai S, Simoes R, Orru S, Carcassi C, Roger M, Rabreau M, Carosella ED. HLA-G gene polymorphism in human placentas: possible association of G*0106 allele with preeclampsia and miscarriage. Biol Reprod. 2008;79:459–467. doi: 10.1095/biolreprod.108.068874. [DOI] [PubMed] [Google Scholar]
- Mutze S, Rudnik-Schoeneborn S, Zerres K, Rath W. Genes and the preeclampsia syndrome. J Perinat Med. 2008;36:38–58. doi: 10.1515/JPM.2008.004. [DOI] [PubMed] [Google Scholar]
- Neidich AB, Joseph JW, Ober C, Ross LF. Empirical data about women's attitudes towards a hypothetical pediatric biobank. Am J Med Genet Part A. 2008;146A:297–304. doi: 10.1002/ajmg.a.32145. [DOI] [PubMed] [Google Scholar]
- O'Brien M, McCarthy T, Jenkins D, Paul P, Dausset J, Carosella ED, Moreau P. Altered HLA-G transcription in pre-eclampsia is associated with allele specific inheritance: possible role of the HLA-G gene in susceptibility to the disease. Cell Mol Life Sci. 2001;58:1943–1949. doi: 10.1007/PL00000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Aldrich C, Rosinsky B, Robertson A, Walker MA, Willadsen S, Verp MS, Geraghty DE, Hunt JS. HLA-G1 protein expression is not essential for fetal survival. Placenta. 1998;19:127–132. doi: 10.1016/s0143-4004(98)90000-5. [DOI] [PubMed] [Google Scholar]
- Ober C, Aldrich CL, Chervoneva I, Billstrand C, Rahimov F, Gray HL, Hyslop T. Variation in the HLA-G promoter region influences miscarriage rates. Am J Hum Genet. 2003;72:1425–1435. doi: 10.1086/375501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Billstrand C, Kuldanek S, Tan Z. The miscarriage-associated HLA-G-725G allele influences transcription rates in JEG-3 cells. Hum Reprod. 2006;21:1743–1748. doi: 10.1093/humrep/del036. [DOI] [PubMed] [Google Scholar]
- Pfeiffer KA, Rebmann V, Passler M, van der Ven K, van der Ven H, Krebs D, Grosse-Wilde H. Soluble HLA levels in early pregnancy after in vitro fertilization. Hum Immunol. 2000;61:559–564. doi: 10.1016/s0198-8859(00)00123-3. [DOI] [PubMed] [Google Scholar]
- Pfeiffer KA, Fimmers R, Engels G, van der Ven H, van der Ven K. The HLA-G genotype is potentially associated with idiopathic recurrent spontaneous abortion. Mol Hum Reprod. 2001;7:373–378. doi: 10.1093/molehr/7.4.373. [DOI] [PubMed] [Google Scholar]
- Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- Rizzo R, Andersen AS, Lassen MR, Sorensen HC, Bergholt T, Larsen MH, Melchiorri L, Stignani M, Baricordi OR, Hviid TVF. Soluble human leukocyte antigen-G isoforms in maternal plasma in early and late pregnancy. Am J Reprod Immunol. 2009;62:320–338. doi: 10.1111/j.1600-0897.2009.00742.x. [DOI] [PubMed] [Google Scholar]
- Roberts JM, Gammill HS. Preeclampsia—recent insights. Hypertension. 2005;46:1243–1249. doi: 10.1161/01.HYP.0000188408.49896.c5. [DOI] [PubMed] [Google Scholar]
- Romero R, Nien JK, Espinoza J, Todem D, Fu W, Chung H, Kusanovic JP, Gotsch F, Erez O, Mazaki-Tovi S, et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J Matern Fetal Neonatal Med. 2008;21:9–23. doi: 10.1080/14767050701830480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadi AR, Mayberry RM, Zaidi AA, Pleasant JC, McGhee N, Rice RJ. Maternal hypertension and associated pregnancy complications among African-American and other women in the United States. Obstet Gynecol. 1996;87:557–563. doi: 10.1016/0029-7844(95)00480-7. [DOI] [PubMed] [Google Scholar]
- Sibai BM. Diagnosis and management of gestational hypertension and preeclampsia. Obstet Gynecol. 2003;102:181–192. doi: 10.1016/s0029-7844(03)00475-7. [DOI] [PubMed] [Google Scholar]
- Sibai BM, Ewell M, Levine RJ, Klebanoff MA, Esterlitz J, Catalano PM, Goldenberg RL, Joffe G. Risk factors associated with preeclampsia in healthy nulliparous women. The Calcium for Preeclampsia Prevention (CPEP) Study Group. Am J Obstet Gynecol. 1997;177:1003–1010. doi: 10.1016/s0002-9378(97)70004-8. [DOI] [PubMed] [Google Scholar]
- Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365:785–799. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- Steegers EAP, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- Steinborn A, Varkonyi T, Scharf A, Bahlmann F, Klee A, Sohn C. Early detection of decreased soluble HLA-G levels in the maternal circulation predicts the occurrence of preeclampsia and intrauterine growth retardation during further course of pregnancy. Am J Reprod Immunol. 2007;57:277–286. doi: 10.1111/j.1600-0897.2007.00475.x. [DOI] [PubMed] [Google Scholar]
- Tan Z, Shon AM, Ober C. Evidence of balancing selection at the HLA-G promoter region. Hum Mol Genet. 2005;14:3619–3628. doi: 10.1093/hmg/ddi389. [DOI] [PubMed] [Google Scholar]
- Tan Z, Randall G, Fan J, Camoretti-Mercado B, Brockman-Schneider R, Pan L, Solway J, Gern JE, Lemanske RF, Nicolae D, et al. Allele-specific targeting of microRNAs to HLA-G and risk of asthma. Am J Hum Genet. 2007;81:829–834. doi: 10.1086/521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CY, Ho JFV, Chong YS, Loganath A, Chan YH, Ravichandran J, Lee CG, Chong SS. Paternal contribution of HLA-G*0106 significantly increases risk for pre-eclampsia in multigravid pregnancies. Mol Hum Reprod. 2008;14:317–324. doi: 10.1093/molehr/gan013. [DOI] [PubMed] [Google Scholar]
- Tan CY, Chong YS, Loganath A, Chan YH, Ravichandran J, Lee CG, Chong SS. Possible gene-gene interaction of KIR2DL4 with its cognate ligand HLA-G in modulating risk for preeclampsia. Reprod Sci. 2009;16:1135–1143. doi: 10.1177/1933719109342280. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Jaamaa G, Kaiser M, Hills E, Soim A, Zhu MT, Shcherbatykh IY, Samelson R, Bell E, Zdeb M, et al. Racial disparity in hypertensive disorders of pregnancy in New York State: a 10-year longitudinal population-based study. Am J Public Health. 2007;97:163–170. doi: 10.2105/AJPH.2005.068577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Witte JS. Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev. 2002;11:505–512. [PubMed] [Google Scholar]
- Vianna P, Dalmaz CA, Veit TD, Tedoldi C, Roisenberg I, Chies JAB. Immunogenetics of pregnancy: role of a 14-bp deletion in the maternal HLA-G gene in primiparous pre-eclamptic Brazilian women. Hum Immunol. 2007;68:668–674. doi: 10.1016/j.humimm.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N. Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:513–520. [PubMed] [Google Scholar]
- Wallis AB, Saftlas AF, Hsia J, Atrash HK. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am J Hypertens. 2008;21:521–526. doi: 10.1038/ajh.2008.20. [DOI] [PubMed] [Google Scholar]
- Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J Evol Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Williams PJ, Morgan L. The role of genetics in pre-eclampsia and potential pharmacogenomic interventions. Pharmacogenetics Personalized Med. 2012;5:37–51. doi: 10.2147/PGPM.S23141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yie SM, Taylor RN, Librach C. Low plasma HLA-G protein concentrations in early gestation indicate the development of preeclampsia later in pregnancy. Am J Obstet Gynecol. 2005;193:204–208. doi: 10.1016/j.ajog.2004.11.062. [DOI] [PubMed] [Google Scholar]
- Yie SM, Li LH, Xiao R, Librach CL. A single base-pair mutation in the 3′-untranslated region of HLA-G mRNA is associated with pre-eclampsia. Mol Hum Reprod. 2008;14:649–653. doi: 10.1093/molehr/gan059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.