

Abstract
Cyclin-dependent kinase 9/cyclin T, the protein kinase heterodimer that constitutes positive transcription elongation factor b, is a well-validated target for treatment of several diseases, including cancer and cardiac hypertrophy. In order to aid inhibitor design and rationalize the basis for CDK9 selectivity, we have studied the CDK-binding properties of six different members of a 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile series that bind to both CDK9/cyclin T and CDK2/cyclin A. We find that for a given CDK, the melting temperature of a CDK/cyclin/inhibitor complex correlates well with inhibitor potency, suggesting that differential scanning fluorimetry (DSF) is a useful orthogonal measure of inhibitory activity for this series. We have used DSF to demonstrate that the binding of these compounds is independent of the presence or absence of the C-terminal tail region of CDK9, unlike the binding of the CDK9-selective inhibitor 5,6-dichlorobenzimidazone-1-β-d-ribofuranoside (DRB). Finally, on the basis of 11 cocrystal structures bound to CDK9/cyclin T or CDK2/cyclin A, we conclude that selective inhibition of CDK9/cyclin T by members of the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile series results from the relative malleability of the CDK9 active site rather than from the formation of specific polar contacts.
Introduction
Positive transcription elongation factor b (P-TEFb), a key regulator of transcription in eukaryotic cells, has been identified as a drug target for several pathologies including cardiac hypertrophy and certain cancers.1 The P-TEFb complex is composed of cyclin dependent kinase 9 (CDK9) associated with cyclin T1 or T2.2 Several CDK9 inhibitory chemotypes have been identified in the course of drug discovery targeting cell-cycle regulatory CDK-cyclin complexes. These CDK inhibitors have been found to induce apoptosis in cancer cells through inhibition of P-TEFb, thereby reducing levels of RNA transcripts that promote cell growth and cell survival.3−7 Several CDK9 inhibitors, including flavopiridol, N-[5-[[[5-(1,1-dimethylethyl)-2-oxa(zolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarbox)amide (9, SNS-032; Chart 1),8 and 2-phenyl-5,7-dihydroxy-8-(2-(hydroxymethyl)-1-methylpyrrolidin-3-yl)-4H-chromen-4-ones (10, P276-00),9 are currently under evaluation in clinical trials against cancer (http://clinicaltrials.gov/). Although their cellular effects are consistent with achieving inhibition of CDK9 at growth inhibitory concentrations, they demonstrate activity against a range of cyclin-dependent kinases:1,9 flavopiridol inhibits CDK9 with a Ki of 3 nM but is a submicromolar inhibitor of CDK1 (Ki = 70 nM) and CDK2 (Ki = 190 nM); 9 inhibits CDK9 with a Ki of 4 nM but is also potent against CDK2 (Ki = 38 nM) and somewhat active against CDK1 and CDK4 (Ki values of 480 and 925 nM, respectively); 10 inhibits each of CDK1, CDK2, and CDK4, with Ki values of less than 100 nM. While results obtained with these inhibitors speak to the potential of CDK9 as a therapeutic target, inhibitors that exhibit higher specificity for CDK9 are needed to establish final target validation.
Chart 1.

The P-TEFb complex is required for the synthesis of the majority of RNA polymerase II (Pol II) transcripts.10 P-TEFb promotes transcription elongation by phosphorylating the C-terminal domain (CTD) of Pol II, as well as the pause inducing factors NELF and DSIF.4,11 In addition to overcoming the paused state of Pol II, these activities promote the further processing of nascent transcripts. Inhibition of CDK9 selectively reduces the transcription of short-lived mRNAs,5 including those that encode regulators of proliferation and apoptosis such as c-Myc and Mcl-1. These proteins are up-regulated in many cancers, leading to the inhibition of apoptosis and the promotion of proliferation. The therapeutic potential of interfering with these processes has been highlighted by promising preclinical and clinical data gathered for inhibitors of CDK9 and its regulators.12−16
The discovery of the 2,4-disubstituted pyrimidine CDK inhibitors has been reported previously.17,18 Lead compounds from this series induced apoptosis in cancer cells and exhibited potent in vivo antitumor activity.19 This antitumor activity was further shown to be due to inhibition of CDK9-dependent transcription of antiapoptotic proteins.19−21 More recently, we have identified a novel class of 2,4,5-trisubstituted pyrimidine CDK inhibitors, as described in ref (22). 12c, one of the lead compounds described in ref (22), induces apoptosis in cancer cells more readily than in noncancerous cells and was able to selectively kill patient-derived chronic lymphocytic leukemia (CLL) cells.21 Taken together, these results support the view that the N-phenyl-4-heteroarylpyrimidine-2-amine scaffold might be optimized to yield a clinically useful CDK9 inhibitor.
In order to achieve this goal, we have conducted crystallographic studies to identify the inhibitor–target interactions that promote CDK9-specific inhibition. As part of this investigation, we have explored the behavior of analogues of 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile in differential scanning fluorimetry (DSF) experiments with P-TEFb and used DSF experiments to explore whether the CDK9/cyclin T affinity of such analogues is dependent on the presence of the CDK9 tail region, as observed elsewhere for other CDK9-selective inhibitors.23
Here and in ref (22), we present the crystal structures of six analogues of 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile bound to CDK9/cyclin T and CDK2/cyclin A and analyze the similarities and differences in inhibitor binding to these enzymes. Our analysis provides the most extensive investigation of the CDK9 ATP binding site to date and allows for a fuller interpretation of inhibitor binding to CDK9. The data confirm that the ATP binding site of CDK9 presents a highly malleable environment capable of accommodating a range of chemically diverse moieties attached to the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine scaffold.
Results
Potent Inhibitors Thermally Stabilize CDK9/Cyclin T and CDK2/Cyclin A
We used DSF to evaluate CDK9 inhibitors for their ability to bind to the enzyme. This technique identifies the melting temperature (Tm) of a protein and the change in melting temperature (ΔTm) induced by the binding of inhibitors.24 The relatively high-throughput nature of DSF allows rapid screening of both a large number of inhibitors and analysis of different protein constructs.
All but two of the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines tested confer an increase in the thermal stability of CDK9/cyclin T. The values measured ranged from 2.9 to 4.7 °C, indicating strong inhibitor binding (Table 1, Figure 1A,B, and Supporting Information Figure S1). The two exceptions were 7 and 27a. This result correlates with the observed Ki values: strongly stabilizing inhibitors display Ki values in the range 6–43 nM, while 7 and 27a have substantially lower inhibitory activity against CDK9/cyclin T, with Ki values of 6.19 and 9.83 μM, respectively (Table 1). This correlation between ΔTm and inhibitory Ki extends beyond the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines (Figure 1A, Table 1). Flavopiridol is the most potent CDK9 inhibitor studied here (Ki = 3 nM) and gives rise to the largest increase in melting temperature (ΔTm = 5.03 ± 0.13 °C). 8 (DRB)25 is much less potent than flavopiridol and stabilizes the complex by only 1.22 ± 0.32 °C. Similar correlations between potency and effect on Tm have been reported previously for inhibitors of other kinases including the PIM-1 kinase and AAK1.24,26
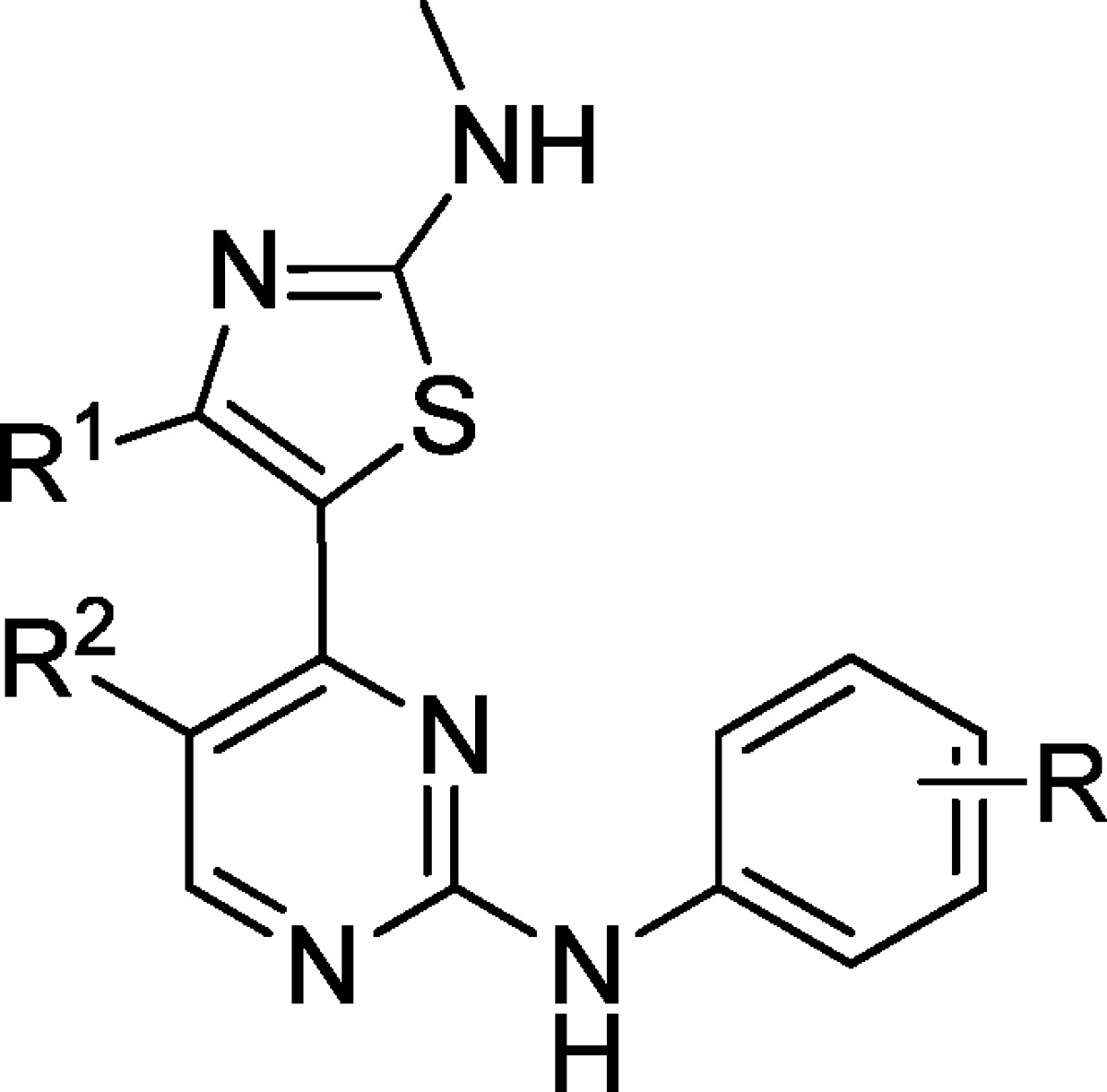
Table 1. Ki and ΔTm for 2-Amino-4-heteroarylpyrimidine and Other CDK9 Inhibitors.
|
Ki (μM)a |
ΔTm (°C) |
||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R | CDK9 | CDK2 | CDK9 | CDK2 |
| 12a | Me | CN | m-NO2 | 0.006 | 0.001 | 4.68 ± 0.29 | 8.88 ± 0.36 |
| 12c | Me | CN | m-SO2NH2 | 0.006 | 0.004 | 4.12 ± 0.07 | 10.53 ± 0.18 |
| 12t | Me | CN | m-acetylhomopiperazine | 0.007 | 0.131 | 3.50 ± 0.30 | 2.77 ± 0.87 |
| 12u | Me | CN | m-homopiperazine | 0.007 | 0.568 | 3.92 ± 0.32 | 4.20 ± 0.87 |
| 1 | Me | CN | p-SO2-morph | 0.008 | 0.003 | 3.69 ± 0.05 | NA |
| 2 | Me | CN | p-SO2NH2 | 0.008 | 0.0002 | 4.09 ± 0.65 | NA |
| 3 | Me | CN | m-OH | 0.011 | 0.012 | 4.12 ± 0.23 | 6.91 ± 0.90 |
| 4 | Me | CN | m-SO2-morph, p-Me | 0.022 | 0.123 | 3.00 ± 0.06 | NA |
| 5 | Me | CN | p-OH | 0.026 | 0.003 | 4.04 ± 0.09 | NA |
| 6 | Me | CN | m-CO-morph | 0.043 | 0.147 | 2.93 ± 0.08 | 1.96 ± 0.43 |
| 7 | Ph | H | m-SO2-morph, p-Me | 6.185 | >5 | 0.154 ± 0.04 | –0.61 ± 0.07 |
| 27a | Ph | H | m-NO2 | 9.835 | >5 | –1.68 ± 0.21 | –2.49 ± 0.36 |
| staurosporine | 0.007 | 0.004 | 3.60 ± 0.02 | 6.22 ± 0.08 | |||
| flavopiridol | 0.003 | 0.19 | 5.03 ± 0.13 | NA | |||
| (S)-CR8b | 0.110 | 0.08 | 3.29 ± 0.08 | 2.81 ± 0.73 | |||
| 8 | 0.340 | 65 | 1.22 ± 0.32 | –1.37 ± 0.20 | |||
| 11 | 0.35 | 69 | 1.57 ± 0.10 | 0.152 ± 0.05 | |||
| roscovitine | 0.79 | 0.25 | 1.57 ± 0.02 | 2.56 ± 1.58 | |||
Apparent inhibition constants (Ki) were calculated from IC50 values and the appropriate Km (ATP) values for CDK9 and CDK2.
(S)-CR8 is 2-(S)-(1-ethyl-2-hydroxyethylamino)-6-(4-(2-pyridyl)benzyl)-9-isopropylpurine.
Figure 1.

Stabilization of CDK9/cyclin T and CDK2/cyclin A by 2-amino-4-heteroarylpyrimidines. (A, B) ΔTm values for CDK9/cyclin T (A) or CDK2/cyclin A (B) bound to a set of selected inhibitors are plotted against the corresponding IC50 values, showing a strong correlation between ΔTm and IC50. A log–linear trend line for CDK2 has an R2 of 0.79, while the equivalent line for CDK9 has an R2 of 0.91. (C) ΔTm values induced by inhibitor binding to CDK9/cyclin T (solid bars) and CDK2/cyclin A (open bars) are compared. DRB and CAN508 correspond to 8 and 11. (D) ΔTm values induced by inhibitor binding to full length CDK9/cyclin T (solid bars) and CDK9330/cyclin T (open bars) are compared.
To compare inhibitor binding to CDK9/cyclin T with inhibitor binding to CDK2/cyclin A, we next evaluated the ΔTm induced by the binding of these inhibitors to CDK2/cyclin A. This DSF analysis shows that ΔTm and Ki also correlate for inhibitor binding to CDK2/cyclin A (Figure 1B). However, the ΔTm values induced by inhibitor binding to CDK2/cyclin A are systematically higher than those induced by inhibitor binding to CDK9/cyclin T. For this reason, the absolute ΔTm values induced by a given inhibitor binding to the two different CDK/cyclin complexes do not directly predict the relative selectivity of that inhibitor.
Nevertheless, DSF provides a straightforward method to identify compounds that are potent against one or more CDKs. For example, compound 3 induces a thermal shift of >4 °C for CDK9/cyclin T and >6.5 °C for CDK2/cyclin A, reflecting its activity as a potent inhibitor of both kinases (Table 1 and Figure 1C). By contrast 12t increases the Tm of CDK9/cyclin T by an amount greater than it does that of CDK2/cyclin A, confirming the Ki data that identify this compound as being CDK9 selective. However, the generally greater effect of inhibitor binding on the Tm of CDK2 means that it is not justified to infer CDK2 selectivity over CDK9 from a relatively higher ΔTm: 12u, which stabilizes CDK2 more than it stabilizes CDK9, turns out to be a more potent inhibitor of CDK9.
Potency of the 4-(Thiazol-5-yl)-2-(phenylamino)pyrimidines toward CDK9 Is Not Dependent on the Presence of the CDK9 C-Terminal Tail
Previous studies have shown that the C-terminus of CDK9 contributes to the binding affinity of the CDK9-selective inhibitor 8.23 To determine if the C-terminal tail influences the binding of the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines, we measured the stabilization induced by these inhibitors on a CDK9/cyclin T complex containing a version of CDK9 truncated at the C-terminus (CDK9330/cyclin T) and compared the results to those observed for the full-length CDK9/cyclin T complex (Figure 1D). This comparative analysis showed that, in contrast to 8, the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines show little ability to discriminate between CDK9/cyclin T and CDK9330/cyclin T. This result suggests that the C-terminal tail neither forms nor significantly modulates interactions between the CDK9 active site and this family of inhibitors.
4-(Thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile CDK Inhibitors Adopt the Same Binding Mode within the ATP Binding Sites of CDK9 and CDK2
The detailed structure–activity relationship (SAR) of this chemical series has been discussed in ref (22). In brief, different functional groups attached to the pyrimidinyl or aniline ring system have a dramatic effect on CDK potency and selectivity. In order to rationalize the observed SAR, we solved the crystal structures of six representative inhibitors bound to both CDK9 and CDK2 (Table 2, Figure S2, and Table S1), described here and in ref (22).
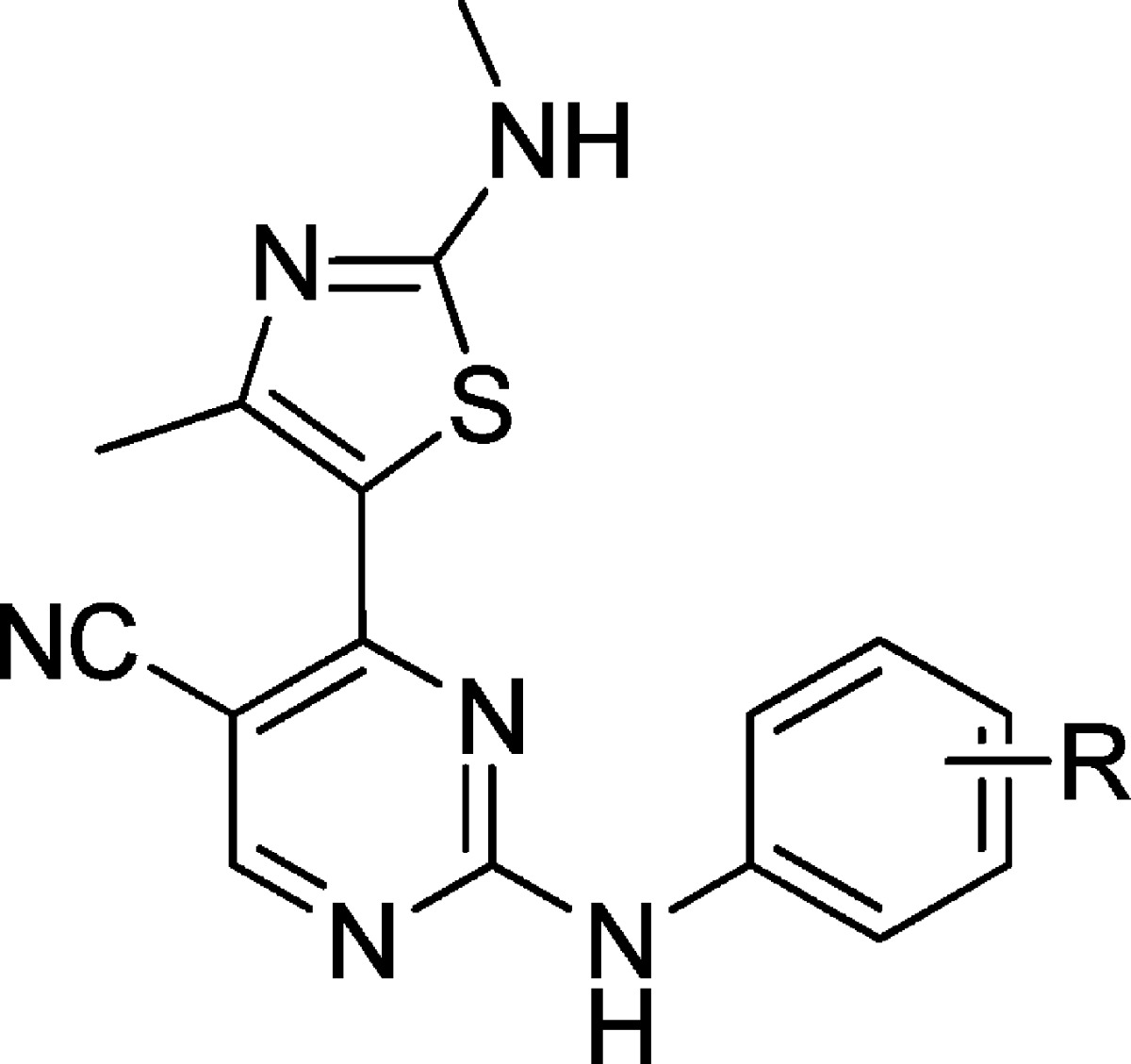
Table 2. Polar Interactions Identified for the 2-Amino-4-heteroarylpyrimidine Substituents within the CDK9 and CDK2 Active Sites.
| polar interactions |
|||
|---|---|---|---|
| inhibitor | R | CDK9 | CDK2 |
| 12t | m-acetylhomopiperazin | ||
| 3 | m-OH | Glu107 | water, Asp86 |
| 4 | m-SO2-morph, p-Me | water, Asp86, Lys89 | |
| 6 | m-CO-morph | no data | |
| 12u | m-homopiperazin | ||
| 12c | m-SO2NH2 | Glu107 | Asp86,a Lys89,b His84b |
Interactions observed for 12c in the inward conformation.
Interactions observed for 12c in the outward conformation.
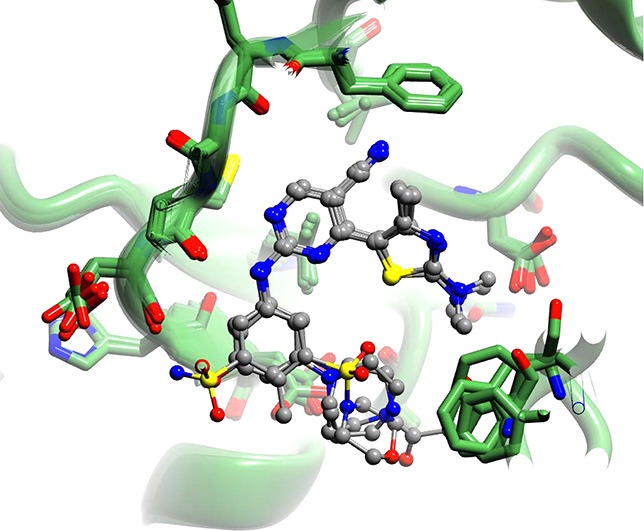
The core of each of the inhibitors adopts a similar pose within the active sites of CDK9/cyclin T and CDK2/cyclin A (Figure 2). The thiazole, pyrimidine, and aniline moieties occupy similar positions within the ATP binding sites, and each of the inhibitors forms similar hydrogen bonds to the hinge regions of each kinase (Figure 2, and as detailed in ref (22)). The pyrimidine ring is sandwiched between the hydrophobic side chains of Ala46 (Ala31) (CDK9 (CDK2) numbering throughout) and Leu156 (Leu134). At the back of the ATP binding site, the C5-carbonitrile contacts the gatekeeper residue Phe103 (Phe80), forming a lone pair−π interaction (mean distance from the nitrile nitrogen to atoms of the phenyl ring is 3.7 Å).
Figure 2.

The binding mode for the 2-amino-4-heteroarylpyrimidine class of inhibitors is similar in the ATP-binding sites of CDK9/cyclin T and CDK2/cyclin A. (A, B) A summary of the common interactions made by 2-amino-4-heteroarylpyrimidine inhibitors bound to CDK9/cyclin T (A) or CDK2/cyclin A (B) is illustrated using structural formulas. (C, D) Superimposed backbone conformations for 2-amino-4-heteroarylpyrimidine complexes of CDK9/cyclin T (green carbons, panel C) or CDK2/cyclin A (blue carbons, panel D) are illustrated in ribbon representation, with the inhibitor (gray carbon atoms) and contacting amino acids shown in cylinder representation. Hydrogen bonds are shown as dotted lines. (E, F) Binding of compound 6 to CDK2 in two different conformations, colored as for panel D, and with the corresponding refined 2mFo – DFc electron density contoured at 1σ.
In the majority of these structures the thiazole moiety has less well-defined electron density bound to CDK9/cyclin T or CDK2/cyclin A than do the other heterocyclic moieties of the inhibitor, suggesting that it may adopt multiple alternative conformations. In CDK9 the thiazole ring within the ATP-binding site causes the side chain of Lys48 to become defined by electron density, presumably by preferentially stabilizing a unique conformation. By contrast, the equivalent residue in apo-CDK9/cyclin T (PDB code 3BLH) is mobile and lacks electron density. In the major CDK9/cyclin T-bound conformation, the methylamino group substituted on the thiazole hydrogen bonds with Asp167 (Figure 2). In CDK2/cyclin A the electron density indicates two main conformations for the thiazole moiety. The first resembles the conformation adopted by the thiazole group when bound to CDK9/cyclin T and is stabilized by the formation of favorable interactions between the methylamino group and Asp145. The second conformation is approximately coplanar with the pyrimidine and hence stabilized by conjugation of the aromatic systems. Toward the front of the ATP binding pocket, the aniline ring is contacted from above by Ile25 (Ile11) with which it forms favorable van der Waals interactions.
Within the CDK2/cyclin A and CDK9/cyclin T crystal structures, meta-substituents of the aniline group are observed in two distinct locations. The first has the substituents positioned toward the thiazole ring, close to the location of the ribose moiety of ATP in other kinase structures. We term this the “inward” conformation. By contrast, the second orientation corresponds to a rotation of 180° around the nitrogen–phenyl bond so that the substituents are positioned away from the thiazole and are therefore directed toward hinge residues. We term this the “outward” conformation (Figure 2 and Supporting Information Figure S2).
4-(Thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitriles Form Few Polar Contacts with CDK9 and CDK2
Analysis of the structures shows that there are few specific hydrogen bonds or ionic interactions between the CDKs and the substituents that decorate the 2-amino-4-heteroarylpyrimidine scaffold (Table 2). This is particularly apparent for the CDK9/cyclin T complexes in which only the peptide carbonyl of Glu107 hydrogen-bonds with the sulfonamide and hydroxyl groups of 12c and 3, respectively (Figure 2C). In the CDK2/cyclin A complexes, 3, 4, and both orientations of the sulfonamide of 12c form polar interactions with CDK2. The hydroxyl group of 3 hydrogen-bonds with Asp86. The SO2 of 4 forms interactions with a water molecule, the Lys89 side chain, and the peptide bond of Asp86, and the sulfonamide of 12c is positioned for favorable interaction with Asp86 in an inward conformation or with Lys89 and His84 in the outward conformation (Figure 2D).
The homopiperazine moieties of 12u and 12t appear not to form polar contacts with either CDK9 or CDK2 (Table 2). 12u and 12t are both selective for CDK9/cyclin T over CDK2/cyclin A, with 12t displaying ∼20-fold selectivity and 12u displaying ∼80-fold selectivity. These selectivities arise without apparent formation of any polar interactions and must therefore be derived from other contacts between kinase and inhibitor. We deduce that most of the CDK9/cyclin T specificity demonstrated by this pair of inhibitors results from a capacity of CDK9/cyclin T to bind to them in a way that optimizes apolar intermolecular contacts, without introducing strain in the inhibitor. This capacity may, in turn, be determined by the malleability of the CDK9 ATP binding site, which allows it to accommodate the inhibitors in their most favored conformations.
Larger Inhibitors Can Be Accommodated in the CDK9 ATP Binding Site
Consistent with the hypothesis that relatively large inhibitors might selectively bind to CDK9 because of the comparative malleability of its ATP binding site, 12t, 4, 6, and 12u, which all have a bulkier substituent on the aniline ring, demonstrate appreciable selectivity for CDK9 over CDK2 when compared to other analogues shown in Table 1. Our crystal structures identify different conformations for the respective m-homopiperazine and m-acetylhomopiperazine groups of 12u and 12t when bound to CDK9/cyclin T and CDK2/cyclin A (Figure 3). The m-acetylhomopiperazine and m-homopiperazine moieties of 12u and 12t are both accommodated by CDK9 in the inward conformation. This preference differs from that observed for CDK2, where the rings are either disordered or predominantly adopt an outward conformation (Table 2). We propose that the inward conformation observed in the CDK9/cyclin T complexes is the preferred conformation for the inhibitors and that its exclusion from the CDK2/cyclin A complexes explains, in part, the selectivity of the inhibitors for CDK9/cyclin T over CDK2/cyclin A.
Figure 3.

CDK9/cyclin T has a more flexible ATP-binding site than CDK2/cyclin A. (A, B) Compound 12u, as bound to CDK9/cyclin T (green carbons, panel A) or CDK2/cyclin A (blue carbons, panel B), is shown in ball and stick representation, with carbons colored gray. Final refined 2mFo – DFc electron density is shown in cyan, contoured at 1σ. (C, D) Corresponding illustration of the binding of compound 12t to CDK9/cyclin T (C) or CDK2/cyclin A (D). Within CDK9/cyclin T, 12t and 12u interact in an inward conformation, whereas in the context of CDK2/cyclin A, 12t and 12u bind either partially (12t) or predominantly (12u) in an outward conformation.
The crystal structures of 4 and 6 show that both compounds bind in an inward conformation to CDK2/cyclin A and that 4 adopts an inward conformation when bound to CDK9/cyclin T (no data are available for 6 bound to CDK9/cyclin T). Both inhibitors show a modest selectivity for CDK9/cyclin T over CDK2/cyclin A (6.0-fold and 3.5-fold, respectively), consistent with the hypothesis that selectivity is limited where both CDK2/cyclin A and CDK9/cyclin T are able to accommodate the inhibitor in an identical conformation. The additional hydrogen bonds made by the sulfonamide moiety of 4 to Asp86 and Lys89 of CDK2 may help stabilize the compound in the inward conformation and thereby mitigate other binding penalties associated with this pose. Compounds 2 and 5 show excellent potency against CDK2 and significant selectivity over CDK9. The introduction of polar groups, including hydroxyl and sulfonamide, at the para position of equivalent aryl groups in other series of CDK inhibitors has had similarly dramatic effects on their potency and selectivity for CDK2.27 These effects have been ascribed to the formation of optimal and concerted stacking and hydrogen-bonding interactions with the main chain of residues beyond the hinge and with the side chain of Asp86, respectively.
Inhibitor Binding Can Induce Subdomain Movements in CDK9 and CDK2
Inhibitor binding to CDK9/cyclin T can provoke unique tertiary structural changes around the ATP-binding site.28,29 The inhibitors included in this study induce similar structural changes in CDK9, confirming the enzyme’s structural malleability. By contrast, the binding of most inhibitors to CDK2/cyclin A results in no significant structural changes within the active site.
There are two main differences observed between the apo- and inhibitor-bound states of CDK9. First, the glycine-rich loop folds down to enclose or shield the inhibitor from solvent (Figure 4). Concurrently the β3/αC loop adopts an altered conformation in which it partially occupies the site vacated by the movement of the glycine-rich loop. In this conformation Leu51 adopts the structural role played by Phe30 in the apo structure. In both the 12u- and 12t-bound structures there is a further slight movement of the glycine-rich loop so that it can make an additional contact with Asp127 in the CDK9 C-terminal lobe. Weak electron density and high B-factors indicate that the glycine-rich loop has conformational flexibility in its downward position. In comparison it remains in a more open conformation in the majority of CDK2/cyclin A/inhibitor structures.
Figure 4.

Inhibitor-induced conformational changes. (A) The protein conformations in novel CDK9/cyclin T/inhibitor structures (green ribbon representation) are compared with the conformation of CDK9/cyclin T in the absence of inhibitor (PDB code 3BLQ, orange ribbon representation). (B) The protein conformations in representative novel CDK2/cyclin A/inhibitor structures (blue ribbon representations) are compared with that of CDK2/cyclin A in the absence of inhibitor (PDB code 1QMZ, yellow ribbon representation). (C) Detail of the conformational change induced by the binding of 12u (cylinder representation) on CDK9/cyclin T (green ribbon representation), relative to CDK9/cyclin T in the absence of inhibitor (PDB code 3BLQ, orange ribbon representation). (D) Detail of the conformational change induced by the binding of 3 and 12t (cylinder representation) on CDK2/cyclin A (blue ribbon representation), relative to CDK2/cyclin A in the absence of inhibitor (PDB code 1QMZ, yellow ribbon representation). Kinase complexes were superimposed on their kinase C-terminal lobes (i.e., CDK2 residues 101–286 and CDK9 residues 123–316).
Second, the N- and C-terminal lobes of CDK9 adopt different relative orientations in the presence and absence of inhibitor (Figure 4). This difference corresponds to a rotation that is particularly apparent on inspection of the cyclin subunit after superimposing CDK9 subunits on their C-terminal kinase lobes. A similar reconfiguration has been reported previously in the CDK9/cyclin T/HIV-Tat (PDB code 3MIA) structure and in certain other CDK9/cyclin T/inhibitor structures.28,30
Discussion
The strong correlation between ΔTm and IC50 values for 2-amino-4-heteroarylpyrimidine inhibitors confirms that DSF both cross-validates CDK9 inhibition assays and offers a potential surrogate for IC50 determination in assessing inhibitor binding. Analysis of the 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine series by this technique confirms that they predominantly stabilize CDK9/cyclin T in a manner independent of the C-terminal tail. In addition, the data confirm that inhibitors 12t, 3, and 6, are potent CDK9/cyclin T inhibitors that demonstrate varying degrees of CDK isotype specificity. Biophysical techniques such as DSF (e.g., ref (24)) and calorimetry31are playing an increasing role in drug discovery, and this work provides a further example of how such techniques can provide an orthogonal measure of protein–ligand interactions.
Deregulation of the P-TEFb pathway has been identified in several cancers,32 and a number of CDK9 inhibitors are in clinical trials. 4-(Thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitriles down-regulate the antiapoptotic protein Mcl-1 and show selective cytotoxic activity against patient-derived primary CLL cancer cells.22 We have determined that members of this inhibitor class bind within the ATP-binding site and stabilize CDK9/cyclin T, employing a number of interactions that do not depend on the presence of the CDK9 tail sequence. Furthermore we have shown that these inhibitors adopt a conserved binding pose and that, within this series, different aniline substituents bind in subtly different ways.
Bis-hetaryl scaffolds have shown up as privileged chemotypes among CDK inhibitors (reviewed in ref (33)), but the molecules described here stand out for their CDK9 inhibition. The compounds included in this study are potent CDK9 inhibitors and show a range of potencies toward CDK2. Where they display appreciable selectivity for CDK9, the major determinant appears to be their ability to simultaneously make favorable hinge region contacts and van der Waals contacts, as opposed to selective electrostatic or hydrogen bonds. Within the series, differences in specificity can be rationalized by considering the identity and size of the aniline substituent. Bulky substituted anilines can be accommodated within the ATP binding site of CDK9, but when bound to CDK2, they often extend out of the binding site into the solvent exposed channels. This binding mode is unique to this set of compounds as other smaller CDK9 inhibitors such as flavopiridol, 8, and 4-arylazo-3,5-diamino-1H-pyrazole (11, CAN508)35 achieve potency by probing alternative pockets within the ATP-binding site of CDK9.28,29,36
Molecular dynamics simulations (e.g., ref (34)) conducted on CDKs and CDK/cyclin complex have illustrated that parts of the ATP-binding site, and most notably the glycine-rich lid, are highly dynamic in nature, presumably associated with a demand for flexibility during the kinase catalytic cycle. The studies presented here provide further, experimentally supported insight into the nature of plastic deformations that the CDK glycine-rich loop may undergo and identify a markedly higher degree of plasticity in the CDK9 than the CDK2 structure.
This study is the largest to date to provide structural details of the interactions of a series of inhibitors with CDK9/cyclin T and a comparison of their binding to CDK2/cyclin A. Taken together, these results suggest that the malleable CDK9 active site, unlike that of CDK2, can accommodate large and flexible compounds providing a structural explanation for the observed selectivity of such compounds for CDK9/cyclin T over CDK2/cyclin A. This property, which provides a rationale for the impressive selectivity of 12u and 12t toward CDK9/cyclin T over CDK2/cyclin A, will help to focus chemical synthesis in future inhibitor development.
Experimental Section
Protein Preparation
CDK9/cyclin T (residues 1–259) (termed CDK9/cyclin T) and CDK9 (residues 1–330)/cyclin T (residues 1–259, Q77R, E96G, F241L) (termed CDK9330/cyclin T) were expressed and purified as reported in refs (23) and (28), respectively. Expression and purification of CDK2/cyclin A (residues 174–432) were undertaken in a similar manner to that in ref (37).
Crystallization
CDK9330/cyclin T crystals were obtained at 4 °C using 10–16% PEG 1000, 100 mM Na/K phosphate, pH 6.2, 500 mM NaCl, and 4 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) as the precipitant solution. Crystals were soaked in the precipitant solution containing 1 mM inhibitor before being buffer exchanged into a cryoprotectant solution containing the precipitant solution supplemented with 15% glycerol and 1 mM inhibitor. Crystals were subsequently flash-cooled in liquid nitrogen.
For cocrystallization, CDK2/cyclin A was first concentrated to 11.9 mg/mL and then incubated in the presence of 0.2 mM inhibitors for 20 min on ice. The solution was filtered through a microfiltration unit (Amicon Ultrafree-MC) and subsequently mixed in equal volumes with mother liquor composed of 1–1.25 M ammonium sulfate, 0.5–0.9 M KCl, 100 mM HEPES, pH 7.0, and 5 mM dithiothreitol (DTT). Crystals were cryoprotected in 7 M sodium formate before flash cooling in liquid nitrogen.
Data Collection and Processing
Data were collected at the Diamond Light Source on beamlines I-02, I-03, I-04 or at the European Synchrotron Radiation Facility on beamline ID14-EH4. MOSFLM38 or XDS39 was used to index and integrate the data. Data were scaled using SCALA.40 Initial models were produced by molecular replacement using PHASER41 or rigid body refinement in phenix.refine.42 Models were iteratively improved by alternate rounds of refinement using either REFMAC43 or phenix.refine and rebuilding in COOT.44 External restraints to apo-CDK9/cyclin T were identified in PROSMART40 and applied to CDK9/cyclin T structures refined in REFMAC. Inhibitor restraints were produced using phenix.elbow builder and confirmed by visual inspection of the resulting PDB and cif files.
Thermal Denaturation
An amount of 0.95 μg of protein was incubated with 20 μM inhibitor, 5× SYPRO Orange (Molecular Probes) in 20 mM HEPES, pH 7.0, 500 mM NaCl, 10% glycerol, 3 mM DTT. The total reaction volume was 15 μL. The temperature was raised in 1 °C steps starting at 25 °C and terminating at 80 °C. The fluorescence emitted at 570 nm was measured using a Stratagene mx3005P RT-PCR 305 machine. The temperature dependence of the fluorescence during the protein denaturation process was approximated by the equation
where ΔuG is the difference in unfolding free energy between the folded and unfolded state, R is the gas constant, and yF and yU are the fluorescence intensity of the probe in the presence of completely folded and unfolded protein, respectively.45 The baselines of the denatured and native state were approximated by a linear fit. The observed temperature shifts, ΔTm, for each inhibitor were recorded as the difference between the transition midpoints of sample and reference wells within the same plate and determined by nonlinear least-squares fit.
Chemistry
1H NMR and 13C NMR spectra were obtained using a Bruker 400 Ultrashield spectrometer at 400 and 100 MHz, respectively. These were analyzed using the Bruker TOPSPIN 2.1 program. Chemical shifts are reported in parts per million relative to an internal tetramethylsilane standard. Coupling constants (J) are reported to the nearest 0.1 Hz. The following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. High resolution mass spectra were obtained using a Waters 2795 single quadrupole mass spectrometer/micromass LCT platform. Purity for final compounds was greater than 95% and was measured using Waters high performance liquid chromatograph (Waters 2487 dual wavelength absorbance detector) with a Phenomenex Gemini-NX 5u C18 110A 250 mm × 4.60 mm column, UV detector at 254 nm, using system A (10% MeOH containing 0.1% TFA for 4 min, followed by linear gradient of 10–100% MeOH over 6 min at a flow rate of 1 mL/min), system B (10% MeCN containing 0.1% TFA for 2 min, followed by linear gradient of 10–100% over 10 min at a flow rate of 1 mL/min), and system C (10% MeCN containing 0.1% TFA for 4 min, followed by linear gradient of 10–100% over 10 min at a flow rate of 1 mL/min). Melting points were determined with an Electrothermal melting point apparatus.
Preparation of compounds 12a, 12c, 12t, 12u, and 27a has been described in ref (22). The following compounds were prepared by the same methods.
4-(4-Methyl-2-(methylamino)thiazol-5-yl)-2-((4-(morpholinosulfonyl)phenyl)amino)pyrimidine-5-carbonitrile (1)
1 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 1-(4-(morpholinosulfonyl)phenyl)guanidine. Yellow solid (13% yield); mp 250–251 °C. Anal. RP-HPLC: tR 12.37 min (method A), 12.00 (method B), purity 96%. 1H NMR (DMSO-d6): δ 2.46 (s, 3H, CH3), 2.86 (apparent t, 4H, J = 4.8 Hz, 2 × CH2), 2.91 (d, 3H, J = 4.8 Hz, CH3), 3.64 (apparent t, 4H, J = 4.8 Hz, 2 × CH2), 7.71 (dt, 2H, J = 8.8, 2.0 Hz, 2 × Ph-H), 8.03 (dt, 2H, J = 8.8, 2.0 Hz, 2 × Ph-H), 8.32 (q, 1H, J = 4.8 Hz, NH), 8.88 (s, 1H, Py-H), 10.69 (s, 1H, NH). 13C NMR (DMSO-d6): δ 20.09, 31.33, 46.39, 65.74, 95.30, 114.13, 117.96, 120.20, 127.96, 129.16, 144.12, 156.48, 159.13, 161.45, 164.21, 170.84. HR-MS (ESI+): m/z [M + H]+ calcd for C20H22N7O3S2, 472.1226, found, 472.1217.
4-((5-Cyano-4-(4-methyl-2-(methylamino)thiazol-5-yl)pyrimidin-2-yl)amino)benzenesulfonamide (2)
2 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 4-guanidinobenzenesulfonamide. Yellow solid (67% yield); mp 254–255 °C. Anal. RP-HPLC: tR 11.40 min (method A), 11.27 (method B), purity 98%. 1H NMR (DMSO-d6): δ 2.45 (s, 3H, CH3), 2.91 (d, 3H, J = 4.8 Hz, CH3), 7.71 (d, 2H, J = 8.8 Hz, 2 × Ph-H), 8.03 (d, 2H, J = 8.8 Hz, 2 × Ph-H), 8.31 (q, 1H, J = 4.4 Hz, NH), 8.86 (s, 1H, Py-H), 10.57 (s, 1H, NH). 13C NMR (DMSO-d6): δ 20.11, 31.33, 94.98, 114.28, 118.04, 120.05, 126.94, 138.36, 142.55, 156.25, 159.17, 161.45, 164.16, 170.79. HR-MS (ESI+): m/z [M + H]+ calcd for C16H16N7O2S2, 402.0807, found, 402.0809.
2-((3-Hydroxyphenyl)amino)-4-(4-methyl-2-(methylamino)thiazol-5-yl)pyrimidine-5-carbonitrile (3)
3 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 1-(3-hydroxyphenyl)guanidine. Yellow solid (18% yield); mp 242–243 °C. Anal. RP-HPLC: tR 11.75 min (method A), 11.50 (method B), purity 99%. 1H NMR (DMSO-d6): δ 2.43 (s, 3H, CH3), 2.89 (d, 3H, J = 4.4 Hz, CH3), 6.44–6.51 (m, 1H, Ph-H), 7.10 (t, 1H, J = 8.0 Hz, Ph-H), 8.03 (d, 1H, J = 8.4 Hz, Ph-H), 7.22 (s, 1H, Ph-H), 8.24 (q, 1H, J = 4.8 Hz, NH), 8.77 (s, 1H, Py-H), 9.39 (s, 1H, OH), 10.11 (s, 1H, NH). 13C NMR (DMSO-d6): δ 19.92, 31.29, 93.93, 108.19, 110.88, 111.89, 118.31, 129.60, 140.36, 155.80, 157.99, 159.41, 161.46, 163.90, 170.57. HR-MS (ESI+): m/z [M + H]+ calcd for C16H15N6OS, 339.1028, found, 339.1078.
4-(4-Methyl-2-(methylamino)thiazol-5-yl)-2-((4-methyl-3-(morpholinosulfonyl)phenyl)amino)pyrimidine-5-carbonitrile (4)
4 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 1-(4-methyl-3-(morpholinosulfonyl)phenyl)guanidine. Yellow solid (35% yield); mp 244–246 °C. Anal. RP-HPLC: tR 12.35 min (method A), 12.15 (method B), purity 98%. 1H NMR (DMSO-d6): δ 2.43 (s, 3H, CH3), 2.53 (s, 3H, CH3), 2.89 (d, 3H, J = 4.4 Hz, CH3), 3.06 (apparent t, 4H, J = 4.8 Hz, CH2 × 2), 3.63 (apparent t, 4H, J = 4.8 Hz, CH2 × 2), 7.43 (d, 1H, J = 8.4 Hz, Ph-H), 7.95 (dd, 1H, J = 8.4, 2.0 Hz, Ph-H), 8.18 (d, 1H, J = 2.0 Hz, Ph-H), 8.28 (q, 1H, J = 4.8 Hz, NH), 8.82 (s, 1H, Py-H), 10.46 (bs, 1H, NH). 13C NMR (DMSO-d6): δ 19.96, 20.21, 31.36, 45.77, 66.04, 118.11, 121.45, 125.00, 131.82, 133.67, 135.28, 137.79, 159.26, 161.55, 164.12, 170.82. HR-MS (ESI+): m/z [M + H]+ calcd for C21H24N7O3S2, 486.1383, found 486.1421.
2-((4-Hydroxyphenyl)amino)-4-(4-methyl-2-(methylamino)thiazol-5-yl)pyrimidine-5-carbonitrile (5)
5 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 1-(4-hydroxyphenyl)guanidine. Yellow solid (18% yield); mp 251–252 °C. Anal. RP-HPLC: tR 11.59 min (method A), 11.35 (method B), purity 95%. 1H NMR (DMSO-d6): δ 2.35 (s, 3H, CH3), 2.88 (d, 3H, J = 4.8 Hz, CH3), 6.73 (dt, 1H, J = 9.2, 3.0 Hz, Ph-H), 7.42 (d, 1H, J = 8.8 Hz, Ph-H), 8.20 (q, 1H, J = 4.8 Hz, NH), 8.69 (s, 1H, Py-H), 9.28 (s, 1H, OH), 10.11 (s, 1H, NH). 13C NMR (DMSO-d6): δ 19.93, 31.26, 92.80, 115.46, 118.53, 123.43, 130.59, 154.22, 155.62, 159.59, 161.44, 163.83, 170.44. HR-MS (ESI+): m/z [M + H]+ calcd for C16H15N6OS, 339.1028, found, 339.1089.
4-(4-Methyl-2-(methylamino)thiazol-5-yl)-2-((3-(morpholine-4-carbonyl)phenyl)amino)pyrimidine-5-carbonitrile (6)
6 was obtained from 3-(dimethylamino)-2-(4-methyl-2-(methylamino)thiazole-5-carbonyl)acrylonitrile and 1-(3-(morpholine-4-carbonyl)phenyl)guanidine. Yellow solid (26% yield); mp 131–132 °C. Anal. RP-HPLC: tR min 12.87 (method A), 12.98 (method B), purity 98%. 1H NMR (DMSO-d6): δ 2.43 (s, 3H, CH3), 2.89 (d, 3H, J = 4.8 Hz, CH3), 3.61 (br s, 8H, 4 × CH2), 7.10 (dt, 1H, J = 7.6, 1.2 Hz, Ph-H), 7.41 (t, 1H, J = 8.0 Hz, Ph-H), 7.72–7.80 (m, 1H, Ph-H), 7.85 (s, 1H, Ph-H), 8.28 (q, 1H, J = 4.8 Hz, NH), 8.81 (s, 1H, Py-H), 10.37 (s, 1H, NH). 13C NMR (DMSO-d6): δ 19.95, 31.34, 42.59, 48.14, 66.58, 94.44, 114.17, 118.17, 119.30, 121.82, 122.01, 129.23, 136.33, 139.50, 156.00, 159.36, 161.49, 164.07, 169.34, 170.70. HR-MS (ESI+): m/z [M + H]+ calcd for C16H15N6OS, 436.1556, found, 436.1616.
N-Methyl-5-(2-((4-methyl-3-(morpholinosulfonyl)phenyl)amino)pyrimidin-4-yl)-4-phenylthiazol-2-amine (7)
7 was obtained from tert-butyl (5-(3-(dimethylamino)acryloyl)-4-phenylthiazol-2-yl)(methyl)carbamate and 1-(4-methyl-3-(morpholinosulfonyl)phenyl)guanidine. Yellow solid (38% yield); mp 98–99 °C. Anal. RP-HPLC: tR 11.34 min (method A), 11.78 (method B), purity 99%. 1H NMR (DMSO-d6): δ 2.91 (d, 3H, J = 4.8 Hz, CH3), 3.07 (apparent t, 4H, J = 4.8 Hz, 2 × CH2), 3.64 (apparent t, 4H, J = 4.8 Hz, 2 × CH2), 6.27 (d, 1H, J = 5.2 Hz, Py-H), 7.36 (d, 1H, J = 8.8 Hz, Ph-H), 7.41–7.60 (m, 5H, 5 × Ph-H), 7.97 (dd, 1H, J = 8.0, 2.0 Hz, Ph-H), 8.10 (d, 1H, J = 5.2 Hz, Py-H), 8.20 (q, 1H, J = 4.8 Hz, NH), 8.26 (d, 1H, J = 2.4 Hz, Ph-H), 9.78 (s, 1H, NH). 13C NMR (DMSO-d6): δ 20.16, 31.45, 45.82, 66.07, 107.61, 119.48, 119.68, 123.30, 129.10, 129.33, 129.38, 129.61, 133.50, 135.13, 136.51, 139.41, 154.53, 157.41, 159.34, 159.77, 170.44. HR-MS (ESI+): m/z [M + H]+ calcd for C25H27N6O3S2, 523.1586, found, 523.1571.
Acknowledgments
We thank the staff at the Diamond Light Source on beamlines I-02, I-03, I-04 and at the European Synchrotron Radiation Facility on beamline ID14-EH4 for providing excellent facilities and E. Lowe for data collection management. This study was supported by Cancer Research UK gGrants C21568/A8988 and C21568/A12474 and by MRC Grant G0901526. A.J.H. was supported by a Wellcome Trust studentship (Grant 083113/Z/07/A).
Glossary
Abbreviations Used
- CDK
cyclin-dependent kinase
- CLL
chronic lymphocytic leukemia
- DRB
5,6-dichlorobenzimidazole 1-β-d-ribofuranoside
- DSF
differential scanning fluorimetry
- DTT
dithiothreitol
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- NMR
nuclear magnetic resonance
- PEG
polyethylene glycol
- Pol II
RNA polymerase II
- P-TEFb
positive transcription elongation factor b
- SAR
structure–activity relationship
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
Supporting Information Available
Two figures showing thermal denaturation profiles and inhibitor binding and one table listing crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Northern Institute for Cancer Research, Medical School, Newcastle University, Newcastle upon Tyne NE2 4HH, U.K.
Author Contributions
A.J.H. undertook all crystallographic experiments. A.J.H. and S.B. performed the DSF studies and interpreted the results. H.S., S.S., and S.H. prepared and characterized the compounds. S.W. designed the compound library and helped in interpreting the results. P.M.F. and C.P. assisted in compound selection for the study. A.J.H., M.E.N., and S.W. wrote the paper with the assistance of J.A.E., P.M.F., and S.B. All authors contributed to the design of the study. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Accession Codes
The structures of the CDK2/cyclin A and CDK9330/cyclin T inhibitor complexes have been deposited in the PDB with the following accession codes: 4BCF, 4BCH, 4BCI, 4BCJ, 4BCM, 4BCN, 4BCK, 4BCO, 4BCQ.
Supplementary Material
References
- Wang S.; Fischer P. M. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [DOI] [PubMed] [Google Scholar]
- Peng J.; Zhu Y.; Milton J. T.; Price D. H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao S. H.; Fujinaga K.; Marion J. E.; Taube R.; Sausville E. A.; Senderowicz A. M.; Peterlin B. M.; Price D. H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [DOI] [PubMed] [Google Scholar]
- Marshall N. F.; Price D. H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [DOI] [PubMed] [Google Scholar]
- Lam L. T.; Pickeral O. K.; Peng A. C.; Rosenwald A.; Hurt E. M.; Giltnane J. M.; Averett L. M.; Zhao H.; Davis R. E.; Sathyamoorthy M.; Wahl L. M.; Harris E. D.; Mikovits J. A.; Monks A. P.; Hollingshead M. G.; Sausville E. A.; Staudt L. M. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001, 2, 0041.1–0041.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojo I.; Zhang B.; Fenton R. G. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 2002, 8, 3527–3538. [PubMed] [Google Scholar]
- Raje N.; Kumar S.; Hideshima T.; Roccaro A.; Ishitsuka K.; Yasui H.; Shiraishi N.; Chauhan D.; Munshi N. C.; Green S. R.; Anderson K. C. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005, 106, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. S.; Kimball S. D.; Misra R. N.; Rawlins D. B.; Hunt J. T.; Xiao H. Y.; Lu S.; Qian L.; Han W. C.; Shan W.; Mitt T.; Cai Z. W.; Poss M. A.; Zhu H.; Sack J. S.; Tokarski J. S.; Chang C. Y.; Pavletich N.; Kamath A.; Humphreys W. G.; Marathe P.; Bursuker I.; Kellar K. A.; Roongta U.; Batorsky R.; Mulheron J. G.; Bol D.; Fairchild C. R.; Lee F. Y.; Webster K. R. Discovery of aminothiazole inhibitors of cyclin-dependent kinase 2: synthesis, X-ray crystallographic analysis, and biological activities. J. Med. Chem. 2002, 45, 3905–3927. [DOI] [PubMed] [Google Scholar]
- Joshi K. S.; Rathos M. J.; Joshi R. D.; Sivakumar M.; Mascarenhas M.; Kamble S.; Lal B.; Sharma S. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol. Cancer Ther. 2007, 6, 918–925. [DOI] [PubMed] [Google Scholar]
- Chao S. H.; Price D. H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793–31799. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Li T.; Price D. H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd J. C.; Lin T. S.; Dalton J. T.; Wu D.; Phelps M. A.; Fischer B.; Moran M.; Blum K. A.; Rovin B.; Brooker-McEldowney M.; Broering S.; Schaaf L. J.; Johnson A. J.; Lucas D. M.; Heerema N. A.; Lozanski G.; Young D. C.; Suarez J. R.; Colevas A. D.; Grever M. R. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 2007, 109, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps M. A.; Lin T. S.; Johnson A. J.; Hurh E.; Rozewski D. M.; Farley K. L.; Wu D.; Blum K. A.; Fischer B.; Mitchell S. M.; Moran M. E.; Brooker-McEldowney M.; Heerema N. A.; Jarjoura D.; Schaaf L. J.; Byrd J. C.; Grever M. R.; Dalton J. T. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. S.; Ruppert A. S.; Johnson A. J.; Fischer B.; Heerema N. A.; Andritsos L. A.; Blum K. A.; Flynn J. M.; Jones J. A.; Hu W.; Moran M. E.; Mitchell S. M.; Smith L. L.; Wagner A. J.; Raymond C. A.; Schaaf L. J.; Phelps M. A.; Villalona-Calero M. A.; Grever M. R.; Byrd J. C. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Wood G.; Meades C.; Griffiths G.; Midgley C.; McNae I.; McInnes C.; Anderson S.; Jackson W.; Mezna M.; Yuill R.; Walkinshaw M.; Fischer P. M. Synthesis and biological activity of 2-anilino-4-(1H-pyrrol-3-yl) pyrimidine CDK inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4237–4240. [DOI] [PubMed] [Google Scholar]
- Wu S. Y.; McNae I.; Kontopidis G.; McClue S. J.; McInnes C.; Stewart K. J.; Wang S.; Zheleva D. I.; Marriage H.; Lane D. P.; Taylor P.; Fischer P. M.; Walkinshaw M. D. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: structural basis for ligand-induced disordering of the activation loop. Structure 2003, 11, 399–410. [DOI] [PubMed] [Google Scholar]
- Wang S.; Griffiths G.; Midgley C. A.; Barnett A. L.; Cooper M.; Grabarek J.; Ingram L.; Jackson W.; Kontopidis G.; McClue S. J.; McInnes C.; McLachlan J.; Meades C.; Mezna M.; Stuart I.; Thomas M. P.; Zheleva D. I.; Lane D. P.; Jackson R. C.; Glover D. M.; Blake D. G.; Fischer P. M. Discovery and characterization of 2-anilino-4- (thiazol-5-yl)pyrimidine transcriptional CDK inhibitors as anticancer agents. Chem. Biol. 2010, 17, 1111–1121. [DOI] [PubMed] [Google Scholar]
- McIntyre N. A.; McInnes C.; Griffiths G.; Barnett A. L.; Kontopidis G.; Slawin A. M.; Jackson W.; Thomas M.; Zheleva D. I.; Wang S.; Blake D. G.; Westwood N. J.; Fischer P. M. Design, synthesis, and evaluation of 2-methyl- and 2-amino-N-aryl-4,5-dihydrothiazolo[4,5-h]quinazolin-8-amines as ring-constrained 2-anilino-4-(thiazol-5-yl)pyrimidine cyclin-dependent kinase inhibitors. J. Med. Chem. 2010, 53, 2136–2145. [DOI] [PubMed] [Google Scholar]
- Liu X.; Shi S.; Lam F.; Pepper C.; Fischer P. M.; Wang S. CDKI-71, a novel CDK9 inhibitor, is preferentially cytotoxic to cancer cells when compared with flavopiridol. Int. J. Cancer 2011, 130, 1216–1226. [DOI] [PubMed] [Google Scholar]
- Shao H.; Shi S.; Huang S.; Hole A. J.; Abbas A. Y.; Baumli S.; Liu X.; Lam F.; Foely D.; Fischer P. M.; Noble M. E. M.; Endicott J. A.; Pepper C.; Wang S.. J. Med. Chem., manuscript submitted. DOI: 10.1021/jm301475f. [DOI] [Google Scholar]
- Baumli S.; Hole A. J.; Wang L. Z.; Noble M. E.; Endicott J. A. The CDK9 tail determines the reaction pathway of positive transcription elongation factor b. Structure 2012, 20, 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. [DOI] [PubMed] [Google Scholar]
- Sehgal P. B.; Derman E.; Molloy G. R.; Tamm I.; Darnell J. E. 5,6-Dichloro-1-beta-d-ribofuranosylbenzimidazole inhibits initiation of nuclear heterogeneous RNA chains in HeLa cells. Science 1976, 194, 431–433. [DOI] [PubMed] [Google Scholar]
- Bullock A. N.; Debreczeni J. E.; Fedorov O. Y.; Nelson A.; Marsden B. D.; Knapp S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. J. Med. Chem. 2005, 48, 7604–7614. [DOI] [PubMed] [Google Scholar]
- Davies T. G.; Bentley J.; Arris C. E.; Boyle F. T.; Curtin N. J.; Endicott J. A.; Gibson A. E.; Golding B. T.; Griffin R. J.; Hardcastle I. R.; Jewsbury P.; Johnson L. N.; Mesguiche V.; Newell D. R.; Noble M. E.; Tucker J. A.; Wang L.; Whitfield H. J. Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat. Struct. Biol. 2002, 9, 745–749. [DOI] [PubMed] [Google Scholar]
- Baumli S.; Lolli G.; Lowe E. D.; Troiani S.; Rusconi L.; Bullock A. N.; Debreczeni J. E.; Knapp S.; Johnson L. N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S.; Endicott J. A.; Johnson L. N. Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem. Biol. 2010, 17, 931–936. [DOI] [PubMed] [Google Scholar]
- Tahirov T. H.; Babayeva N. D.; Varzavand K.; Cooper J. J.; Sedore S. C.; Price D. H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett N. C.; Chaires J. B. Thermodynamic studies for drug design and screening. Expert Opin. Drug Discovery 2012, 7, 299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano G.; Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008, 7, 3664–3668. [DOI] [PubMed] [Google Scholar]
- Cicenas J.; Valius M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett C. P.; Noble M. E. Molecular motions of human cyclin-dependent kinase 2. J. Biol. Chem. 2005, 280, 13993–14005. [DOI] [PubMed] [Google Scholar]
- Krystof V.; Cankar P.; Frysova I.; Slouka J.; Kontopidis G.; Dzubak P.; Hajduch M.; Srovnal J.; de Azevedo W. F. Jr.; Orsag M.; Paprskarova M.; Rolcik J.; Latr A.; Fischer P. M.; Strnad M. 4-Aylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J. Med. Chem. 2006, 49, 6500–6509. [DOI] [PubMed] [Google Scholar]
- Baumli S.; Hole A. J.; Noble M. E.; Endicott J. A. The CDK9 C-helix exhibits conformational plasticity that may explain the selectivity of CAN508. ACS Chem. Biol. 2012, 7, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N. R.; Noble M. E.; Endicott J. A.; Johnson L. N. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W. Recent changes to the MOSFLM package for processing film and image plate data. Jnt CCP4/ESF-EACBM Newsl. Protein Crystallogr. 1992, 26, 27–33. [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative computational project, number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994, D50, 760–763 [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulis D.; Kranz J. K.; Salemme F. R.; Todd M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005, 44, 5258–5266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.