

Abstract
The adenosine receptors (ARs) provide an example of how to accurately predict ligand recognition, even prior to the availability of a crystallographic structure. Homology modeling has been used to gain structural insight, in conjunction with site-directed mutagenesis and structure activity relationships of small molecular ligands. Recent X-ray structures greatly improved the accuracy of knowledge of AR ligand recogntion and furthermore characterized conformational changes induced by receptor activation. Now homology modeling extends these structural insights to related GPCRs and suggests new ligand structures. This strategy is also being applied to the eight subtypes of P2Y receptors for extracellular nucleotides, which lack X-ray structures and are best modeled by homology to the CXCR4 (peptide) receptor. Neoceoptors, as studied for three of the four AR subtypes, create a molecular complementarity between a mutant receptor and a chemically tailored agonist ligand to selectively enhance affinity, implying direct physical contact and thus validating docking hypotheses.
Keywords: Purines, pyrimidines, G protein-coupled receptors, mutagenesis, molecular modeling, neoceptor, G protein-coupled receptors
1. INTRODUCTION
Extracellular nucleosides and nucleotides act as signaling molecules through four subtypes of adenosine receptors (ARs) and eight subtypes of P2Y receptors (P2YRs) (Table 1). The ARs are a well-developed field of medicinal chemistry, with each of the subtypes having highly selective agonists and antagonists that have generated new therapeutic concepts for a wide variety of diseases (Gessi et al., 2011). The P2YRs are activated by various adenine and uracil mono- and dinucleotides (Abbracchio et al., 2006), but, except for the P2Y12R, are less well developed clinically or preclinically than the ARs.
Table 1.
Representative molecular models of ARs and P2Y receptors.
| Family | Subtype | G protein | Native agonist (human, pEC50) | Modeling template |
|---|---|---|---|---|
| ARs | A1, | Gi, Go | adenosine (6.51) | bacteriorhodopsin, rhodopsin, agonist-bound A2AAR (IJzerman et al., 1992; Palaniappan et al., 2007; Ivanov et al., 2007b; Tosh et al., 2011) |
| A2A | Gs, Golf | adenosine (6.14) | bacteriorhodopsin, rhodopsin, agonist-bound A2AAR (IJzerman et al., 1992; Kim et al., 2005; Ivanov et al., 2007b; Jacobson et al., 2005; Kim et al., 2005; Deflorian et al., 2012) | |
| A2B | Gs, Gq | adenosine (4.59) | rhodopsin (Ivanov et al., 2007a, 2007b, 2008) | |
| A3 | Gi | adenosine (6.53), inosine (6.60) |
rhodopsin, antagonist-bound and agonist-bound A2AAR, (Jacobson et al. 2001; Gao et al., 2006; Ivanov et al., 2007b; Dal Ben et al, 2010; Tosh et al., 2011; Cheong et al., 2012) | |
| P2Y1-like | P2Y1 | Gq | ADP (5.09) | bacteriorhodopsin, rhodopsin (van Rhee et al., 1995; Moro et al., 1998; Costanzi et al., 2004) |
| P2Y2 | Gq, Gi | UTP (8.10), ATP (7.07) |
bacteriorhodopsin, rhodopsin, CXCR4 receptor (Erb et al., 1995; Ivanov et al., 2007a; Hillmann et al., 2009; Maruoka et al., 2011) | |
| P2Y4 | Gq, Gi | UTP (5.60)a | rhodopsin, CXCR4 receptor (Costanzi et al., 2004; Maruoka et al., 2011) | |
| P2Y6 | Gq | UDP (6.52) | rhodopsin (Costanzi et al., 2004; Besada et al., 2006) | |
| P2Y11 | Gq, Gs | ATP (4.77) | rhodopsin (Zylberg et al., 2007) | |
| P2Y12-like | P2Y12 | Gi | ADP (7.22) | rhodopsin, antagonist-bound A2AAR, CXCR4 receptor (Costanzi et al., 2004; Deflorian and Jacobson, 2011) |
| P2Y13 | Gi | ADP (7.94) | rhodopsin (Costanzi et al., 2004) | |
| P2Y14 | Gi | UDP-glucose (6.45), UDP-galactose(6.17), UDP (6.80) |
rhodopsin, antagonist-bound A2AAR (Costanzi et al., 2004; Ivanov et al., 2008) |
ATP is a competitive antagonist at the hP2Y4R and agonist at the rat or mouse P2Y4R.
Prior to the determination of the first X-ray crystallographic structure of an AR, i.e. the A2A subtype, the only means of probing the structures of these G protein-coupled receptors (GPCRs) was through homology modeling. Now, modeling is used in conjunction with the rapidly increasing knowledge of GPCR structure (Yarnitzky et al., 2010; Katritch and Abagyan, 2011). As family A (rhodopsin-like) GPCRs, both the ARs and P2YRs consist of a single polypeptide chain that spans the plasma membrane seven times with α-helical transmembrane domains (TMs, numbered from TM1 to TM7) connected by three extracellular loops (ELs, numbered from EL1 to EL3) and three intracellular loops (ILs, numbered from IL1 to IL3).
The exact three-dimensional structures of most GPCRs have long been elusive. Rhodopsin, a light-activated receptor found in retinal rod cells, led the way as a prototypical GPCR. Its structure, which was successfully determined by X-ray crystallography in 2000 (Palczewski et al., 2000), offered the first high-resolution glimpse into the three-dimensional topology of the members of this superfamily of transmembrane proteins. More recently, starting in 2007, a number of breakthroughs in GPCR crystallography yielded the solution of the structures of several other receptors, among which was the A2AAR (Jaakola et al., 2008). Importantly, the few GPCRs solved crystallographically, besides being directly applicable to the computer-aided discovery of their ligands (Topiol and Sabio, 2010), also provide a platform for the construction of three-dimensional models of other members of the superfamily by homology modeling. Founded on the observation that evolutionarily related proteins – or homologous proteins – share a great deal of structural similarity, this technique allows the construction of a molecular model of a protein on the basis of the known structure of one of its homologues. Thus, in the absence of experimental structures, homology modeling, in tandem with molecular docking, has been employed for several decades to generate hypothetical models of GPCRs in complex with their ligands. Comparisons of models and experimental structures of receptor ligand complexes, even in the context of blind assessments, demonstrated that the construction of accurate models of GPCRs in complex with their ligands is a concrete possibility (Costanzi, 2008; Katritch and Abagyan, 2011). Not surprisingly, the models are particularly accurate when: a) the target receptor shares a significant sequence similarity with one of the crystallized homologues; and/or b) the proposed interactions of the target receptor with its ligands are well characterized experimentally. In contrast, for the receptors that are more distantly related to the available templates and have less-characterized interactions with their ligands, the structural predictions still remain very challenging.
2. RECEPTORS FOR EXTRACELLULAR NUCLEOSIDES AND THEIR MODELED STRUCTURES
2.1. X-ray crystallography of A2AAR
X-ray crystallographic structures of both agonist- and antagonist-bound forms of the A2AAR have provided unprecedented three-dimensional detail concerning molecular recognition in the binding site. A high resolution X-ray structure of the inactive human (h) A2AAR in complex with the antagonist 4-(2-[7-amino-2-(2-furyl)[1,2,4] triazolo[2,3-a][1,3,5]triazin-5-yl-amino]ethyl)phenol (ZM241385) was reported (Jaakola et al., 2008). In conjunction with the recent reports of the crystal structure of the A2AAR in agonist-bound conformations, it is now possible to describe the conformational changes that take place upon agonist binding, which lead to receptor activation. Some changes resemble movements in activated structures of other receptors or opsin, and others are unique, such as an outward tipping movement of TM7 on the exofacial side (Xu et al., 2011). The first agonist-bound A2AAR was crystallized and its structure determined as a consequence of the bulky and exended substituents of the agonist 2-(3-[1-(pyridin-2-yl)piperidin-4-yl]ureido)ethyl-6-N-(2,2-diphenylethyl)-5′-N-ethylcarboxamidoadenosine-2-carboxamide (UK432097). These multiple projections from the main pharmacophore filled remaining spaces in the receptor binding site to stabilize the complex. Indeed the melting temperature (Tm) of this complex was 65°C, which was 6° higher than the previous complex with ZM241,385. Complexes of the A2AAR with the native agonist adenosine and the non-selective adenosine-5′-N-ethyluronamide (NECA) were also reported (Lebon et al., 2011), but they required stabilizing mutations. These structures are very similar to the UK complex in the characteristic movements in the TM region upon agonist binding. Three agonist-bound A2AAR structures have been expeditiously applied to analysis of other nucleosides binding to the same receptor (Deflorian et al., 2012).
2.2. Molecular modeling of ARs
The X-ray structures follow upon a two-decade long progression of knowledge of the AR binding site(s) and other structural features based on molecular modeling, mutagenesis and analysis of structure activity relationships (SARs) of ligands. The early modification of both adenosine agonists and alkylxanthine antagonists empirically located regions in these two ligand classes that were amenable to chemically functionalized chain extension without losing the ability to bind to the ARs (Jacobson, 2009). This observation suggested, long before the structure or even polypeptide composition of the ARs was known, that the putative binding sites had regions that were accessible to the external environment and therefore less sterically demanding. That is the functionalized chains protruded beyond the conformationally and sterically restricted binding region of the core pharmacophore. Points on the adenosine scaffold that displayed this characteristic and could be extended chemically, without a limiting length, included the N6 position for the A1AR and the C2 position for the A2AAR. In the xanthine series, such modification of the C8 position was relatively insensitive in receptor binding, thus serving as a suitable site for derivatization of AR antagonist functionalized congeners.
This orientation of adenosine and the xanthine analogues in the putative AR binding site was effectively visualized in the first AR molecular modeling in 1991, based on the crude template of bacteriorhodopsin (IJzerman et al., 1992). The first indication of separate binding regions on the first ARs cloned, i.e. the canine A1 and A2AARs, featured a hydrophilic region that was situated more distal from the exofacial side than a putative hydrophobic binding region for the nucleobase of agonists or the purine ring system of xanthine antagonists. Thus, in the proposed overlay of adenosine and potent functionalized antagonist 8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl]oxy]phenyl]-l,3-dipropylxanthine (xanthine amine congener, XAC), the ribose moiety of the agonist projects deeper into the binding site than the adenine moiety, which is superimposed on the purine ring of XAC (Figure 1A). This allows the terminal amino group of XAC, a site for generalized coupling to much larger moieties without losing AR affinity, to extend into the extracellular medium. This mode of binding was roughly predictive of the recently determined X-ray structures of adenosine and XAC (Doré et al., 2011) bound to a thermostabilized hA2AAR (Figure 1B). The terminal amino chain of XAC proved to be very flexible and not anchored to the receptor in a specific conformation. It occupied a groove formed between Tyr9 (1.35, using the universal GPCR residue identifier, Ballesteros et al. 1995) and Tyr271 (7.36), two residues that can adopt two different rotameric states depending on the ligand. Thus, the objective in the design of XAC as a functionalized congener, in which the distal amino group escapes the steric constraints of the pharmacophore binding site, was finally explained structurally.
Figure 1.
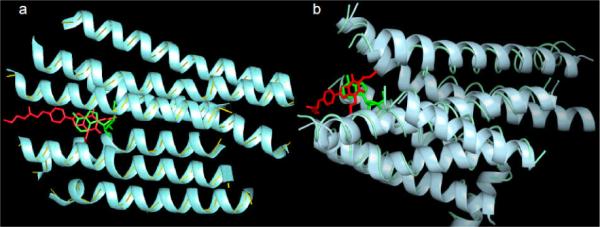
A comparison of the first AR modeling results (A) using early modeling techniques (IJzerman et al., 1992) with the new X-ray structures (B) (Doré et al., 2011). The concept of the amino group of XAC being accessible to the extracellular medium has been validated. B. XAC (atoms colored in red) in the recently released crystal structure 3REY (represented as ribbon colored in cyan) and ADO (atoms colored in green) in the crystal structure 2YDO (represented as thin tube colored in light blue).
The A2AAR structures can reliably serve as modeling templates, with some adjustment, for other ARs, due to the relatively high sequence identity between ARs (average 47% between human subtypes). The presence of conserved residues in the vicinity of the putative ligand binding site were probed through site-directed mutagesis (Kim et al., 1995). Residues in TMs 3, 5, 6, and 7 and in the C-terminal half of EL2 were also implicated in ligand binding with respect to other AR subtypes. First detected through chemical modification and mutagenesis of ARs, two His residues, occurring at position 6.52 in three subtypes and at 7.43 in four subtypes, were found to be important in ligand recognition. His at position 7.43 was proposed to be in proximity of the ribose moiety of adenosine agonists. Also, the hydrophilic side chains of Ser and Thr at positions 7.42 and 3.36 were suggested to be in this region, consistent with the selective loss of agonist potency upon mutation to Ala. Furthermore, a conserved Asn at position 6.55 was essential for recognition of both agonists and antagonists. The A3AR is the most divergent from the other subtypes with respect to residues in the ligand binding region. For example, instead of His at position 6.52, it features a Ser residue. The predictions concerning these key residues were confirmed and explained with great clarity in the X-ray structures of the A2AAR and are currently being extended to the other AR subtypes through the use of homology modeling (Tosh et al., 2011).
A flowchart depicting current modeling and docking approaches for closely related GPCR structures is shown in Figure 2. Structure-based drug design strategies have been used to elucidate specific ligand recognition determinants and ultimately lead to the design of new small molecules for a specific target. The structure of the target receptor protein is the first requirement in structure-based drug design. In the absence of a high resolution structure of a given receptor, computational techniques like homology modeling can be used to build a 3D model. Briefly, the best structural template is chosen from the Protein Data Bamk (PDB), and the sequence of the target receptor is aligned to the template structure using highly conserved residues and the known shared structural features to guide the automated or semi-automated alignment. The sequence alignment and the template structures are the input for the homology modeling. Energy minimization or molecular dynamics can be used to further refine and optimize the resulting 3D models. The ligand-receptor interactions can be identified by means of structure-based approaches, e.g. molecular docking. The putative ligand-receptor complexes from the docking can clarify structural elements for molecular recognition and lead to a further optimization of the compounds and the design of new derivatives. The binding site of a given GPCR can be mapped for each class of small molecule ligands (Zhukov et al., 2011). Also, the virtual searching of chemically diverse databases for novel chemotypes that bind to a given GPCR structure has been productive (Carlsson et al., 2011; van der Horst et al., 2011).
Figure 2.
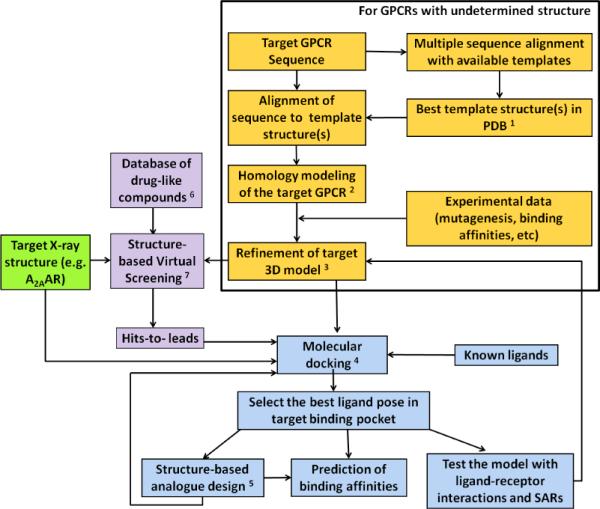
Flowchart of structure-based drug design approaches.
1. The selection of a template is based on sequence identities, conserved key residues, binding site similarities, consideration of disulfide bridges, and shared structural features.
2. DSModeler, Prime, ICM, and MOE are commercial software packages widely used for homology modeling with the structure-based alignment and the template structure as inputs and root main square deviation (RMSD) of Cα, side chain, and heavy atoms as criteria for homology modeling success.
3. Ligand-supported homology modeling, energy minimization or/and molecular dynamics are used to relax unfavorable contacts.
4. Automated docking in rigid binding site, induced-fit docking, or docking by Monte Carlo simulations may be used. Among the available docking software packages are: AutoDock, DOCK, Glide, GOLD, ICM, and OEDocking. SAR information can help to identify the binding pocket in the receptor.
5. Modification of the lead compounds on the basis of the property of the binding site to attain binding and specificity for the target protein. Approaches include: fragment addition, fragment replacement, connection of fragments with new scaffolds, and other techniques.
6. Database preparation with several physical and chemical filters, e.g. number of rotatable bonds in the compounds, polar surface area, and various versions of Lipinski's rule-of-five based on lipophilicity, hydrophobicity, and molecular weight.
7. Structure-based virtual screening (SBVS) with high-throughput docking of compound database to the 3D structure of the target. Glide, Gold, ICM, AutoDock are common docking tools for SBVS. The post-docking analysis for the selection of the hits to be tested includes shape complementarities, cluster analysis, consensus scoring, geometric analysis, and visualization of binding modes.
3. RECEPTORS FOR EXTRACELLULAR NUCLEOTIDES AND THEIR MODELED STRUCTURES
ATP, UTP, and other nucleotides are released from intracellular sources in response to stress, (such as hypoxia, ischemia, or mechanical stress) as a result of tissue damage, through vesicular release as a cotransmitter, or through pannexin hemichannels. Upon release, these nucleotides can activate P2X channels or P2YRs, which tend to mobilize a response to the challenge, such as intensifying the immune response. Over time, through the action of nucleotidases on adenine nucleotides, adenosine is produced, or it may also originate from cellular release.
The P2YRs belong to a branch of class A GPCRs chiefly composed of receptors for nucleotides, lipids, metabolites of the Krebs cycle, and protease-activated receptors (Costanzi et al., 2004). Second messenger coupling and phylogenetic analyses based on sequence comparisons revealed that the P2YRs can be subdivided into two distinct groups: the P2Y1-like subfamily, comprising the P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11 subtypes, and the P2Y12-like subfamily, comprising the P2Y12, P2Y13 and P2Y14 subtypes (Table 1). The missing numbers are due to earlier erroneous attribution of certain receptors, which in fact are not activated by nucleotides, to the P2Y family. The sequence identity is significantly high between members of the same subfamily, while it is relatively low across the two subfamilies (from 14% for P2Y1R/P2Y14R to 50% for P2Y2R/P2Y4R). Notably, the classification based on sequence alignment coincides with a pharmacological division based on the coupling of the receptors to different G proteins: the members of P2Y1-like subfamily activate the phospholipase C (PLC) signaling pathway via Gq, while those of the P2Y12-like subfamily inhibit the adenylyl cyclase pathway via Gi.
Experimentally elucidated structures are not yet available for any of the P2YRs. In the absence of such structures, in the last decade we have constructed models of the members of this family of receptors based on the various available templates. The earlier models were all based on rhodopsin, which, at the time, was the only crystallographically solved GPCR (Ivanov et al., 2006). We incorporated into the model experimentally derived hypotheses on the structures of the P2YRs. These included the presence of a disulfide bridge that putatively connects the third transmembrane domain (TM3) with the second extracellular loop in virtually all GPCRs and a second disulfide bridge between the N-terminal domain with EL3 that was proposed, on the basis of mutagenesis studies conducted at the P2Y1 and P2Y2Rs, as a constraint to help characterize the extracellular region. Our models also incorporated a salt bridge connecting EL2 of P2Y1R with the extracellular end of TM6, which was supported by experimental data. Specifically, data from mutagenesis studies revealed that the residues involved in formation of these extracellular bridges are fundamental to receptor function.
On the basis of these rhodopsin-based in silico structures, through molecular docking experiments, we generated models of the complexes of the P2YRs with both natural and synthetic nucleotides ligands, including agonists and antagonists (Table 1). Experimentally derived data were amply used to guide the molecular docking. Most notably, modeling-guided mutagenesis data revealed two distinct sets of cationic residues in the P2Y1R-like and the P2Y12R-like subfamilies to be implicated in the recognition of the phosphate moieties of the nucleotides. Three cationic residues were identified at positions 3.29 of TM3, 6.55 of TM6, and 7.39 of TM7 for the members of the P2Y1R-like subfamily and at positions EL2.52 of EL2, 6.55 of TM6, and 7.43 of TM7 for the P2Y12R-like subfamily. Our molecular models of the P2YRs in complex with their ligands were constructed by anchoring the phosphates of the nucleotides to these key cationic residues (Costanzi et al., 2004).
The puckering conformation of the sugar moiety of the ligands adopted in our models was also experimentally supported. Specifically, the ribose conformation necessary for the recognition of nucleotides by each of the P2Y1R-like subtypes was inferred through the synthesis of rigid methanocarba analogs of the ribonucleotides, constrained in two isomeric forms as either the northern (N) or the southern (S) conformation by the fusion of a cyclopentane and a cyclopropane ring (Costanzi et al., 2005), and most of these subtypes preferred to bind with their ligands in the (N) conformation. The only the exception was the P2Y6R, which recognized the sugar moiety of its ligands only in the (S) conformation. Notably, this eccentric characteristic of the P2Y6R was strikingly anticipated by our models based on Monte Carlo searches followed by molecular dynamics simulations.
More recently, the structure of the CXCR4 chemokine receptor was solved crystallographically (Wu et al., 2010). Among the currently crystallized GPCRs, this receptor is the most suitable template for the modeling of the P2YRs. As we previously illustrated, CXCR4 is a member of a branch of peptide-activated GPCRs that appears to be phylogenetically the closest to the P2Y branch (Deflorian and Jacobson, 2011). Among the crystallographically solved GPCRs, this receptor shares the highest sequence similarity with P2YRs (about 25% calculated relatively to the entire portion of the CXCR4 receptor that was solved crystallographically, with the exclusion of the C-terminal domain) (Maruoka et al., 2011). Moreover, the CXCR4 receptor contains the abovementioned second disulfide bridge connecting the N-terminus (through a Cys located two positions upstream when compared to the P2Ys) with a Cys situated between EL3 and TM7 (at position 7.25, as in the P2YRs). Before the CXCR4 structure was solved, none of the structurally characterized GPCRs featured a similar disulfide bridge.
For these reasons, we built new models of the members of the P2Y family based on this new peptide receptor template. Different conformations of the EL2 domain are immediately apparent when comparing the models of a P2YR based on either rhodopsin or CXCR4 (Figure 3). Specifically, the rhodopsin-based models feature a loop that occludes like a plug the extracellular opening of the interhelical binding cavity. On the contrary, the CXCR4-based models feature a more solvent-exposed EL2 domain that leaves the interhelical binding cavity more open toward the extracellular milieu. Moreveer, the choice of receptor template for modeling of the P2Y4R had major effect on the position of the pharmacophore, in either of two ligands: UTP or a selective agonist, MRS4062. Such a difference is probably attributable to the peculiarity of the biology of rhodopsin when compared to the CXCR4 receptor and the great majority of GPCRs. Unlike most GPCRs, whose ligands diffuse from the extracellular space into the receptor, rhodopsin features a ligand already covalently bound within the interhelical cavity.
Figure 3.
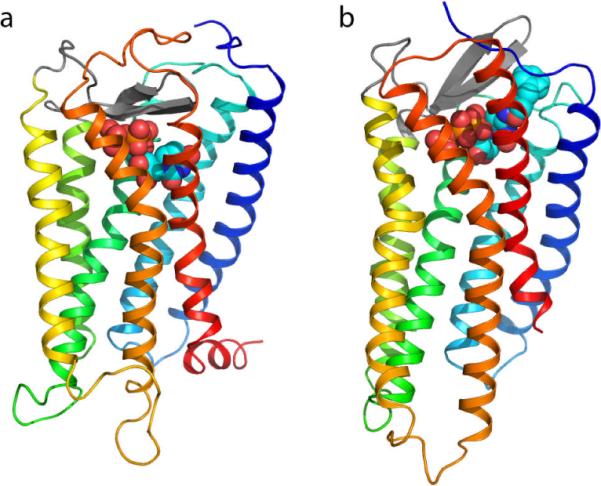
Side-by-side view of two P2Y4R models (Maruoka et al., 2011): an older rhodopsin-based model is shown in panel a, while a more recent CXCR4-based model is shown in panel b. The two models can be readily recognized by the distinctive structure of their second extracellular loop (EL2) domains – the beta-hairpins represented with thick gray antiparallel arrows. Specifically, in the rhodopsin-based model EL2 lays low over the extracellular opening of the helical bundle, while in the CXCR4-based model it projects towards the extracellular space. As a result, the docked ligand is pushed more deeply toward the center of the receptor in the first than in the latter. The structure of the receptors is schematically represented as a ribbon with a color gradient going from blue at the N-terminus to red at the C terminus (TM1: dark blue; TM2: cyan; TM3: green; TM4: yellow/green; TM5: yellow; TM6: orange, TM7: red). The ligands – UTP in the rhodopsin-based model, an N4-substituted derivative of CTP (MRS4062, N4-(3-phenylpropoxy)-cytidine-5'-triphosphate) in the CXCR4-based model – are represented as van der Waals spheres (orange, P; red, O).
Notably, the new CXCR4 receptor-based P2YR models explain ligand SAR more effectively than those based on rhodopsin, suggesting that they may be closer to the actual structure of the receptors. For instance, our new models of the pyrimidine-nucleotide binding P2Y2 and P2Y4Rs, unlike their rhodopsin-based counterparts, were in agreement with the activity and selectivity profile of compounds bearing relatively large substituents attached to the nucleobase (Maruoka et al., 2011). Moreover, a model of the P2Y12R also based on the CXCR4 receptor was revealed to be in excellent agreement with the SAR of both agonists and antagonists (Deflorian and Jacobson, 2011).
4. NEOCEPTORS: REENGINEERING GPCRS FOR RECOGNITION OF MODIFIED AGONISTS
The reengineering of enzymes, such as kinases, to create an orthogonal correspondence between a mutated protein and a modified inhibitor or ligand has provided a means of probing the binding site and the mechanistic pathways involved in the action of a given protein. The neoceptor approach as applied to GPCRs is similar in that the binding site is reengineered for activation by a chemically modified agonist. These GPCRs engineered for activation by strategically modified agonist analogues were termed neoceptors (Jacobson et al., 2001), because the ligand recognition profile of such mutant receptors was entirely different from the agonist SAR of the parent, native receptor. Molecular modeling, based on a large body of data for a given GPCR, was used to predict how to simultaneously mutate a residue(s) in the receptor binding site and to make complementary structural changes in agonists. This could be based on new energetically favorable H-bonding groups or electrostatic interactions, such as introducing oppositely charged groups at predicted points of contact in the receptor and ligand. The neoceptor approach is similar but not identical to strategies of RASSLs and DREADs (Conklin et al., 2008). Because structural correspondence of the protein and its ligands is taken into account from the outset, detecting a selective enhancement in such a simultaneously and orthogonally modified pair is a means of testing docking hypotheses. Examples are provided for three subtypes of ARs (Jacobson et al., 2007), which confirmed elements of the recognition of the ribose moiety of nucleoside agonists, predicted using mutagenesis, modeling, and analysis of SAR.
4.1. A3AR neoceptors
The first example of a neoceptor was demonstrated for the A3AR (Jacobson et al., 2001). The A3AR is of interest in the treatment of cardiac ischemia, inflammation, and neurodegenerative diseases. This receptor was mutated at the site of a conserved His residue in TM7, that had been proposed to coordinate the ribose moiety, specfically in the region of the 2' and 3' hydroxyl groups. The ribose moiety was predicted to bind between hydrophilic residues of TM3 and TM7. A negatively charged side chain in the H272E (7.43) and H272D mutant receptors expressed in COS-7 cells enhanced the binding affinity of 3'-amino-3'-deoxyadenosine, which would be mostly positively charged at physiological pH. Thus, an enhanced affinity in the complex predicted by modeling to form a novel salt bridge could be explained by direct proximity of the oppositely charged groups. Although the ratio of enhancement was only 7-fold, the fact that this nucleoside was considerably less potent at the wild-type (WT) A3AR was indicative of a contact in this region, either directly or possibly through a water molecule. Also, a standard AR agonist, NECA, was 19-fold less potent at the H272E A3AR than at the WT receptor. Substituting the other ribose hydroxyl groups with an amine, either at the 2' or 5' position, did not result in an enhancement of affinity. Thus, orthogonality of the modified pair of receptor and agonist in comparison to the native species was demonstrated to be site-specific. Curiously, introduction in the 3'-amino-3'-deoxyadenosine series of a substituted N6-benzyl group, known to favor selectivity at the A3AR, reduced the enhancement ratio, possibly because this recognition element functioned well in both mutant and WT receptors, and the novel electrostatic pair would have a relatively less dominant role in determining the affinity.
In this context it is to be mentioned that the modeling predicted a nearly 5 Å gap between the ribose 2' or 3'-amino groups (in place of hydroxyl) and the newly introduced carboxylate group of the mutant receptor. At the time it was puzzling why the distance was so large, and in fact would be too large for a direct H-bond. In hindsight, it could be explained by analogy to the rearrangement of TM7 in the agonist-bound A2AAR. TM7 moves closer to the ribose moiety of AR agonists in order to form a direct H-bond, and this movement is though to be characteristic of the activation of the A2AAR. The modeling in the first A3AR neoceptor study in 2001 would not have taken that movement into account, because it was based on homology to the inactive rhodopsin X-ray structure.
A subsequent refinement of the A3AR neoceptors involved the same mutant receptors, i.e. having a negatively charged side chain in place of His272 (7.43), but matched with additional modification of the ribose moiety of the agonist. Instead of simple replacement of the 3'-OH group with amino, this hydroxyl was replaced with more highly H-bonding and neutral sustituents, such as a primary urea group. Alternately, in the series of N6-substituted benzyl analogues of adenosine, an 3'-aminomethylene group was introduced in place of the 3'-hydroxyl, which allowed the positive charge to project further toward TM7, thereby providing a 20-fold affinity enhancement in the H272E receptor. This increased affinity at the mutant receptor was dependent on the N6 substitution, which likely shifted slightly the orientation of the entire molecule, because an N6-methyl analogue was not enhanced. Also, the enhancment was absent in molecules bearing the potency enhancing 5'-methyluronamide group. N6-(3-iodobenzyl)-3'-ureidoadenosine (MRS3481) was the most enhanced in affinity at this mutant A3AR in comparison to all other modifications examined. An azido, guanidino, or uronamido replacement of a ribose hydroxyl did not enhance affinity at the mutant A3AR. The affinity enhancements for the ureido analogue and a corresponding 3'-acetamidomethyl analogue were >100-fold and >20-fold, respectively. The ureido adenosine derivative activated PLC via the H272E mutant A3AR with an EC50 of 180 nM and was inactive at the WT mutant A3AR. Adenosine is known to be cardioprotective via activation of an A3AR, so we tested the ability of the matched pair of the MRS3481 and the H272E neoceptor to induce protection against hypoxia. This tailored nucleoside concentration-dependently activated phospholipase D in chick primary cardiomyocytes (associated with cardioprotection) and protected the cells against hypoxia, only when transfected with the mutant (H272E), but not the WT hA3AR. Therefore, the same signaling pathways associated with the WT A3AR were preserved in the neoceptor. Thus, the neoceptor approach could be applied to a known protective function of adenosine, suggesting the possible application of this stratefgy either to mechanistic studies or to pharmacological gene therapy. Neoceptor pairs should be useful for probing signaling pathways and could potentially be applied to diseases following organ-targeted delivery of the neoceptor gene.
4.2. A1AR neoceptors
By analogy to the A3AR, mutation in the hA1AR of the corresponding H278 (7.43) to either negatively charged (Asp, Glu) or smaller uncharged residues (Ala, Leu) resulted in enhanced affinity of 2'- and 3'-ureido adenosine analogues (Palaniappan et al., 2007). In this case, the N6 group selected was cyclopentyl, which is known to be associated with high affinity and selectivity at the A1AR. For example, the affinity of N6-cyclopentyl-3'-ureido-3'-deoxyadenosine was enhanced by >100-fold at the mutant H278E A1AR, while the potency of adenosine and other 3'-OH adenosine analogues was decreased at this mutant receptor. Replacement of His278 with Ala or Leu produced a similar enhancement of the more sterically bulky 3'-derivatives of the nucleoside agonists, suggesting that the gain resulted from steric rather than electrostatic factors. Mutations of another hydrophilic residue in the putative region for ribose binding, Thr277, did not enhance the affinity of 2'- and 3'-ureidoadenosine derivatives. The positional selectivity of the enhancement in H278 mutant A1ARs was also indicated by the failure of introducing H-bonding groups placed on the N6 or 5' substituents to enhance potency.
4.3. A2AAR neoceptors
However, at the reengineered hA2AAR (Jacobson et al., 2005), mutation of the conserved His278 (7.43) to Asp failed to enhance nucleoside affinity of 3'-modified analogues, suggesting a model of binding of the agonist notidentical to that of the A3AR. The principle of orthogonal affinity enhancement surrounded changes at the 5' position. Strategically mutated hA2AAR neoceptors, e.g., with anionic residues in TMs 3, 5, and 7 intended for pairing with positively charged amine-modified nucleosides, were were tested for activation by nucleosides modified at the 5', 2, and N6-positions. A 5'-(2-aminoethyl)uronamide of adenosine but not a 5'-(2-hydroxyethyl)uronamide displayed enhanced binding affinity at a T88D (3.36) mutant A2AAR, suggesting a critical role of extended positively charged primary amine in direct interaction with the side chain of position 3.36. The combination of an aminoethyluronamide modification with the N6-(2-methylbenzyl) group enhanced affinity at the A2AAR Q89D (3.37) and N181D (5.42) mutant receptors, but not with T88D. Amino groups placed near the adenine C2- or N6-position only slightly affected the binding to mutant receptors; thus, the enhancing interaction was site specific. The binding affinity of a 5'-hydrazide derivative was 670-fold enhanced at mutant Q89D-A2AAR compared to the WT A2AAR, and functional potency in stimulation of cAMP formation was similarly increased. Thus, matched pairs of A2AAR-derived neoceptors and tailored agonists were shown to be pharmacologically orthogonal with respect to the native species.
4.4. Generality of neoceptor approach
Thus, the neoceoptor approach has been applied to three AR subtypes to create a molecular complementarity, based on a salt bridge or increased H-bonding, between a mutant receptor and a chemically tailored agonist ligand. The observed selective enhancement of affinity implied direct physical contact, thereby validating the homology molecular modeling and docking hypotheses. This strategy can potentially be applied to other GPCRs for which no X-ray structures are yet available, such as P2YRs for extracellular nucleotides. In general, the multicomponent GPCR modeling process is an effective means of probing molecular recognition, which can lead to progress in ligand design and understanding receptor signaling. Iterative cycles of mutagenesis, SAR analysis, and molecular modeling, based on increasingly predictive structural templates, are needed for a given GPCR.
ACKNOWLEDGMENTS
The support of the NIDDK, Intramural Research Program is acknowledged.
REFERENCES
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII. Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacological Reviews. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Stuart CS, editor. Methods in Neurosciences. Vol. 25. Academic Press; San Diego, CA: 1995. pp. 366–428. [Google Scholar]
- Besada P, Shin DH, Costanzi S, Ko H, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. Structure-activity relationships of uridine 5'-diphosphate analogues at the human P2Y6 receptor. J. Med. Chem. 2006;49:5532–5543. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J, Yoo L, Gao ZG, Irwin J, Shoichet B, Jacobson KA. Structure-based discovery of adenosine A2A receptor ligands. J. Med. Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong SL, Federico S, Venkatesan G, Mandel AL, Shao YM, Moro S, Spalluto G, Pastorin G. The A3 adenosine receptor as multifaceted therapeutic target: pharmacology, medicinal chemistry, and in silico approaches. Med. Res. Rev. 2012 doi: 10.1002/med.20254. DOI: 10.1002/med.20254. [DOI] [PubMed] [Google Scholar]
- Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, Nissenson RA, Wess J, Bockaert J, Roth BL. Engineering GPCR signaling pathways with RASSLs. Nat. Methods. 2008;5:673–678. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: Structural comparison based on sequence analysis, mutagenesis, and homology modeling. J. Med. Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y6 receptor: Molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J. Med. Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S. On the applicability of GPCR homology models to computer-aided drug discovery: a comparison between in silico and crystal structures of the β2-adrenergic receptor. J. Med. Chem. 2008;51:2907–2914. doi: 10.1021/jm800044k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Ben D, Lambertucci C, Lammi C, Marucci G, Thomas A, Volpini R, Cristalli G. Molecular modeling study on potent and selective adenosine A3 receptor agonists. Bioorg. Med. Chem. 2010;18:7923–7930. doi: 10.1016/j.bmc.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Deflorian F, Jacobson KA. Comparison of three GPCR structural templates for modeling of the P2Y12 nucleotide receptor. J. Comput. Aided Mol. Des. 2011;25:329–338. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorian F, Kumar TS, Phan K, Gao ZG, Xu F, Wu H, Katritch V, Stevens RC, Jacobson KA. Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of the agonist-bound A2A adenosine receptor. J. Med. Chem. 2012;55:538–552. doi: 10.1021/jm201461q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tean B, Hurrell E, Bennett K, Congreve M, Magnani F, Tatte CG, Weir M, Marshall FH. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb L, Garrad R, Wang Y, Quinn T, Turner JT, Weisman GA. Site-directed mutagenesis of P2U purinoceptors. Positively charged amino acids in transmembrane helices 6 and 7 affect agonist potency and specificity. J. Biol. Chem. 1995;270:4185–4188. doi: 10.1074/jbc.270.9.4185. [DOI] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Fazzi D, Stefanelli A, Varani K, Borea PA. Adenosine receptor targeting in health and disease. Expert Opin. Investig. Drugs. 2011;20:1591–1609. doi: 10.1517/13543784.2011.627853. [DOI] [PubMed] [Google Scholar]
- Hillmann P, Ko GY, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje HD, Müller CE. Key Determinants of Nucleotide-Activated G Protein-Coupled P2Y2 Receptor Function Revealed by Chemical and Pharmacological Experiments, Mutagenesis and Homology Modeling. J. Med. Chem. 2009;52:2762–2775. doi: 10.1021/jm801442p. [DOI] [PubMed] [Google Scholar]
- IJzerman AP, van Galen, P JM, Jacobson KA. Molecular modeling of adenosine receptors. I. The ligand binding site on the A1 receptor. Drug Des. Discov. 1992;9:49–67. [PMC free article] [PubMed] [Google Scholar]
- Ivanov AA, Costanzi S, Jacobson KA. Defining the nucleotide binding sites of P2Y receptors using rhodopsin-based homology modeling. J. Comput. Aided Mol. Des. 2006;20:417–426. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- Ivanov AA, Fricks I, Harden TK, Jacobson KA. Molecular dynamics simulation of the P2Y14 receptor. Ligand docking and identification of a putative binding site of the distal hexose moiety. Bioorg. Med. Chem. Lett. 2007a;17:761–766. doi: 10.1016/j.bmcl.2006.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AA, Palyulin VA, Zefirov NS. Computer aided comparative analysis of the binding modes of the adenosine receptor agonists for all known subtypes of adenosine receptors. J. Mol. Graph. Model. 2007b;25:740–754. doi: 10.1016/j.jmgm.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Ivanov AA, Wang B, Klutz AM, Chen VL, Gao ZG, Jacobson KA. Probing distal regions of the A2B adenosine receptor by quantitative structure - activity relationship modeling of known and novel agonists. J. Med. Chem. 2008;51:2088–2099. doi: 10.1021/jm701442d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V, Griffith M, Hanson M, Cherezov V, Chien E, Lane J, IJzerman A, Stevens R. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, Van Rompaey P, Van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J. Med. Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chemistry and Biology. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG, Liang BT. Neoceptors: Reengineering GPCRs to recognize tailored ligands. Trends Pharmacol. Sci. 2007;28:111–116. doi: 10.1016/j.tips.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA. Functionalized congener approach to the design of ligands for G protein–coupled receptors (GPCRs) Bioconjugate Chem. 2009;20:1816–1835. doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Duong HT, Sonina T, Lim SK, Van Rompaey P, Van Calenbergh S, Mamedova L, Kim HO, Kim MJ, Kim AY, Liang BT, Jeong LS, Jacobson KA. Orthogonal activation of the reengineered A3 adenosine receptor (neoceptor) using tailored nucleoside agonists. J. Med. Chem. 2006;49:2689–2702. doi: 10.1021/jm050968b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Abagyan R. GPCR agonist binding revealed by modeling and crystallography. Trends in Pharm. Sciences. 2011;32:637–43. doi: 10.1016/j.tips.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wess J, van Rhee, A M, Shöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011 doi: 10.1038/nature10136. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka H, Jayasekara MPS, Barrett MO, Franklin DE, de Castro S, Kim N, Costanzi S, Harden TK, Jacobson KA. Pyrimidine nucleotides with 4-alkyloxyimino and terminal tetraphosphate δ-ester modifications as selective agonists of the P2Y4 receptor. J. Med. Chem. 2011;54:4018–4033. doi: 10.1021/jm101591j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. Human P2Y1 receptor: Molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J. Med. Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan KK, Gao ZG, Ivanov AA, Greaves R, Adachi H, Besada P, Kim HO, Kim AY, Choe SA, Jeong LS, Jacobson KA. Probing the binding site of the A1 adenosine receptor reengineered for orthogonal recognition by tailored nucleosides. Biochemistry. 2007;46:7437–7448. doi: 10.1021/bi7001828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Topiol S, Sabio M. X-ray structure breakthroughs in the GPCR transmembrane region. Biochem. Pharmacol. 2009;78:11–20. doi: 10.1016/j.bcp.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Tosh DK, Phan K, Deflorian F, Wei Q, Gao ZG, Jacobson KA. Truncated (N)-methanocarba nucleosides as A1 adenosine receptor agonists and partial agonists: Overcoming lack of a recognition element. ACS Med. Chem. Lett. 2011;2:626–631. doi: 10.1021/ml200114q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst E, van der Pijl R, Mulder-Krieger T, Bender A, IJzerman AP. Substructure-based virtual screening for adenosine A2A receptor ligands. ChemMedChem. 2011;6:2302–2311. doi: 10.1002/cmdc.201100369. [DOI] [PubMed] [Google Scholar]
- van Rhee AM, Fischer B, van Galen PJM, Jacobson KA. Modelling the P2Y purinoceptor using rhodopsin as template. Drug. Design Discov. 1995;13:133–154. [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnitzky T, Levit A, Niv MY. Homology modeling of G-protein-coupled receptors with X-ray structures on the rise. Curr Opin. Drug Discov. Devel. 2010;13:317–325. [PubMed] [Google Scholar]
- Zhukov A, Andrews SP, Errey JC, Robertson N, Tehan B, Mason JS, Marshall FH, Weir M, Congreve M. Biophysical mapping of the adenosine A2A receptor. J. Med. Chem. 2011;54:4312–4323. doi: 10.1021/jm2003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberg J, Ecke D, Fischer B, Reiser G. Structure and ligand-binding site characteristics of the human P2Y11 nucleotide receptor deduced from computational modelling and mutational analysis. Biochem. J. 2007;405:277–286. doi: 10.1042/BJ20061728. [DOI] [PMC free article] [PubMed] [Google Scholar]