

The role of MET in the pathophysiology of non-small cell lung cancer and in acquired resistance to epidermal growth factor receptor inhibitors is summarized. An update on progress in the clinical development of inhibitors of MET for treatment of non-small cell lung cancer is provided.
Keywords: Epidermal growth factor receptor tyrosine kinase inhibitor, Hepatocyte growth factor, MET, Non-small cell lung cancer, Tyrosine kinase inhibitor
Abstract
A better understanding of the pathophysiology and evolution of non-small cell lung cancer (NSCLC) has identified a number of molecular targets and spurred development of novel targeted therapeutic agents. The MET receptor tyrosine kinase and its ligand hepatocyte growth factor (HGF) are implicated in tumor cell proliferation, migration, invasion, and angiogenesis in a broad spectrum of human cancers, including NSCLC. Amplification of MET has been reported in approximately 5%–22% of lung tumors with acquired resistance to small-molecule inhibitors of the epidermal growth factor receptor (EGFR). Resistance to EGFR inhibitors is likely mediated through downstream activation of the phosphoinositide 3-kinase /AKT pathway. Simultaneous treatment of resistant tumors with a MET inhibitor plus an EGFR inhibitor can abrogate activation of downstream effectors of cell growth, proliferation, and survival, thereby overcoming acquired resistance to EGFR inhibitors. Development and preclinical testing of multiple agents targeting the HGF–MET pathway, including monoclonal antibodies targeting HGF or the MET receptor and small-molecule inhibitors of the MET tyrosine kinase, have confirmed the crucial role of this pathway in NSCLC. Several agents are now in phase III clinical development for the treatment of NSCLC. This review summarizes the role of MET in the pathophysiology of NSCLC and in acquired resistance to EGFR inhibitors and provides an update on progress in the clinical development of inhibitors of MET for treatment of NSCLC.
Implications for Practice:
Identification of the role of the HGF–MET pathway in cancer, and specifically in non-small cell lung cancer (NSCLC) has led to the development of pharmaceutical agents targeting this pathway. In particular, MET's role in secondary resistance to EGFR-directed therapies has led to the investigation of combining MET-directed agents with erlotinib in patients with metastatic NSCLC. This article reviews the early development of MET-directed therapies as well as currently ongoing Phase III studies. We await the results of these studies, which will determine whether targeting MET in combination with EGFR is a valid clinical option in patients whose cancers progress following treatment with EGFR inhibitors.
Introduction
Lung cancer is the leading cause of cancer-related death in the U.S., with an estimated 220,000 new cases diagnosed and 160,000 deaths annually [1]. Histologically, the majority of lung cancers (75%–85%) are classified as non-small cell lung cancer (NSCLC), of which adenocarcinoma (40%) and squamous cell carcinoma (30%–35%) are the two most common subtypes [2]. Standard first-line treatment of advanced NSCLC with platinum-based doublet chemotherapy is associated with a median survival duration of ∼10 months [3, 4], and second-line treatment with single-agent docetaxel or pemetrexed is associated with a median survival duration of ∼8 months [5]. Better understanding of the molecular pathophysiology and natural history of NSCLC has led to the development of targeted agents that promise to improve these outcomes.
The epidermal growth factor receptor (EGFR) regulates key cellular pathways involved in tumorigenesis and is frequently overexpressed in NSCLC. Agents blocking EGFR tyrosine kinase activity were the first targeted agents to demonstrate clinical benefit in patients with NSCLC who had failed standard first-line chemotherapy. In this setting, the EGFR tyrosine kinase inhibitor (TKI) erlotinib led to a significantly longer overall survival (OS) time than with placebo (6.7 months versus 4.7 months; p < .001) [6]. Subsequently, EGFR TKIs were demonstrated to have clinical benefit in the first-line setting in selected patients. A phase III, randomized study in previously untreated Asian patients with advanced adenocarcinoma who were nonsmokers or former light smokers reported a higher 12-month progression-free survival (PFS) rate among patients treated with gefitinib than among those treated with carboplatin plus paclitaxel (25% versus 7%) [7]. In that study, subgroup analysis demonstrated that gefitinib resulted in a significantly better PFS outcome in patients with tumors harboring activating EGFR mutations (hazard ratio [HR], 0.48; p < .001). However, in patients with tumors lacking EGFR mutations, the PFS interval was significantly longer for patients who received carboplatin plus paclitaxel (HR, 2.85; p < .001). Thus, EGFR mutation status was shown to be a strong predictor of clinical benefit derived from gefitinib in this patient population. Two additional randomized trials conducted in Japan in previously untreated patients with NSCLC also demonstrated a better PFS outcome in patients with EGFR mutations who received gefitinib than in those who received doublet chemotherapy (carboplatin plus paclitaxel or cisplatin plus docetaxel) [8, 9]. Likewise, a study conducted in China in patients with confirmed EGFR mutations demonstrated a significantly longer PFS time in those who received first-line erlotinib than in those who received gemcitabine plus carboplatin (13.1 months versus 4.6 months; p < .0001) [10]. However, the duration of response to EGFR TKIs is often short, and ultimately all patients develop resistance.
Resistance to EGFR TKIs occurs through both primary and secondary mechanisms [11, 12]. Primary resistance has been demonstrated in patients with KRAS mutations, which are mutually exclusive of EGFR mutations, and the presence of KRAS mutations has been shown to predict lack of response to EGFR TKIs for some tumors [13, 14]. Secondary (acquired) resistance can occur via secondary EGFR mutations or parallel activation of downstream signaling pathways. In approximately half of the patients with acquired resistance to EGFR TKIs, a methionine-for-threonine substitution at position 790 (T790M) in exon 20 leads to acquired resistance to EGFR inhibitors, and additional secondary mutations (T854A, D761Y) have recently been identified [11, 15, 17]. Resistance to EGFR TKIs has also been demonstrated in tumor cells harboring MET gene amplification [17]. Likewise, expression of the MET receptor ligand hepatocyte growth factor (HGF) has also been shown to confer resistance to EGFR-directed therapies [18–22]. These data suggest that activation of the HGF–MET pathway may be a potential mechanism of resistance to EGFR TKIs.
In the last two decades, preclinical studies have defined multiple cellular pathways that promote lung cancer tumorigenesis and progression and, currently, clinical studies are under way to determine how agents that target those pathways can be most effectively used to treat patients with NSCLC. The National Cancer Institute's Lung Cancer Mutation Consortium (LCMC) recently reported that 60% of patients with NSCLC had tumor-specific driver mutations that could be used to guide treatment with either the currently approved anti-EGFR agents or agents targeting other pathways, including the MET pathway [23]. This review summarizes the role of MET in NSCLC and in acquired resistance to EGFR inhibitors, and it provides an update on progress in the clinical development of inhibitors of MET for treatment of NSCLC.
Methods
To evaluate the role of MET in NSCLC, a systematic review of the published English-language literature was performed using PubMed. Keywords included “c-met inhibitor” and “non-small cell lung cancer.” Additional references were obtained from the reference sections of articles identified using these search terms. In addition, abstracts from annual meetings of the American Society of Clinical Oncology, European Society for Medical Oncology, and American Association for Cancer Research were searched to identify recent presentations related to MET inhibitors being investigated for the treatment of NSCLC. Publications and abstracts that did not include clinical trial or mouse xenograft model data were excluded. The discussion of MET inhibitors in clinical development for NSCLC was limited to agents that have progressed to phase II or phase III clinical trial status.
MET and MET Inhibitors for NSCLC
MET is a heterodimeric transmembrane receptor tyrosine kinase composed of an extracellular α-chain and a membrane-spanning β-chain linked via disulfide bonds (Fig. 1) [24]. MET contains several conserved protein domains, including sema, PSI (in plexins, semaphorins, integrins), 4 IPT repeats (in immunoglobulins, plexins, transcription factors), TM (transmembrane), JM (juxtamembrane), and TK (tyrosine kinase) domains. The sole identified ligand for MET is HGF, also known as scatter factor. Binding of HGF to MET triggers receptor dimerization and transphosphorylation, leading to conformational changes in MET that activate the TK domain. MET mediates activation of downstream signaling pathways, including phosphoinositide 3-kinase (PI3K)/AKT, Ras-Rac/Rho, mitogen-activated protein kinase, and phospholipase C, that stimulate morphogenic, proliferative, and antiapoptotic activities common to many growth factors, as well as stimulating pathways involved in cell detachment, motility, and invasiveness (Fig. 2) [24, 25]. The pattern of gene expression observed on activation of MET resembles the mesenchymal–epithelial transition [26].
Figure 1.

Structure and function of the MET receptor tyrosine kinase.
Figure 2.

MET signal transduction pathways.
Abbreviations: EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; GRB, growth factor receptor-bound protein; HGF, hepatocyte growth factor; MAPK, mitogen-activated protein kinase; MEK, MAPK–extracellular signal related kinase kinase; mTOR, mammalian target of rapamycin; PAK, p21-activated kinase; PI3K, phosphoinositide 3-kinase; PLC, phospholipase C; SHC, SRC homology 2 domain containing transforming protein; SHP, small heterodimer protein; SOS, son of sevenless; STAT, signal transducer and activator of transcription.
MET was originally isolated from a human osteosarcoma-derived cell line and has subsequently been shown to be expressed primarily on epithelial cells [24]. Dysregulation of MET expression can occur by multiple mechanisms, including overexpression, constitutive kinase activation, gene amplification, paracrine or autocrine activation via HGF, MET mutation, and epigenetic changes [24, 27–29]. Amplification and/or overexpression of MET and/or HGF have been reported in multiple tumor types and correlate with poor clinical prognosis in patients with NSCLC and other solid tumors [30, 31]. Consistent with the role of MET in cell motility and morphogenesis, metastatic lesions typically exhibit higher expression levels of MET than primary tumors [24]. Taken together, these data suggest that MET plays an important role in tumor metastasis.
The critical role of MET in the pathophysiology of NSCLC has been established based on animal models and human NSCLC cell lines that demonstrate dysregulation of MET expression and are sensitive to MET inhibitors. An analysis of human primary NSCLC tumor samples and NSCLC-derived cell lines found MET expression in 100% (n = 23) of primary tumors and 89% (n = 9) of NSCLC cell lines [2]. MET was also strongly expressed in 67% (n = 9) of adenocarcinomas, and expression of activated phospho-MET was observed preferentially along the invasive fronts of NSCLC tumor tissue. Activating mutations have been identified in the MET gene that resulted in MET autophosphorylation and downstream phosphorylation of PI3K, 3-phosphoinositide-dependent protein kinase 1, AKT, mammalian target of rapamycin, and S6K. In another study, MET amplification up to 2.5-fold greater than normal and constitutive MET phosphorylation were reported in two of nine NSCLC cell lines [32]. In both studies, selective inhibition of MET with either small interfering RNA or a selective MET TKI (SU11274) inhibited growth and viability of MET-expressing tumor cells and abrogated MET-mediated downstream signaling.
Tivantinib (ARQ 197), a selective small-molecule inhibitor of MET, effectively abrogated constitutive and HGF-induced MET phosphorylation in lung cancer cell lines and inhibited phosphorylation of AKT, extracellular signal–related kinase (ERK)-1/ERK-2, and signal transducer and activator of transcription 3 [33]. Tivantinib also inhibited proliferation and induced caspase-dependent apoptosis in cell lines with constitutive MET activity. Similar results were observed using RNA interference-mediated depletion of MET, confirming that cellular responses to tivantinib were based on selective inhibition of MET.
Murine models of human NSCLC have demonstrated the antitumor activity of MET inhibitors. In a study of a divalent humanized anti-MET antibody (h224G11), in vivo growth of NSCLC tumor xenografts was significantly inhibited in animals that received anti-MET antibody and near complete inhibition of tumor growth was observed in animals receiving an anti-MET antibody plus vinorelbine [34]. In another study, administration of the MET-specific TKI PHA665752 reduced NSCLC tumorigenicity in mouse xenografts by 75% and induced regression of established tumors [35]. Administration of PHA665752 inhibited MET phosphorylation in mouse NSCLC xenografts, inhibited angiogenesis by >85%, and caused an angiogenic switch resulting in decreased production of vascular endothelial growth factor (VEGF) and increased production of the angiogenesis inhibitor thrombospondin-1. Administration of PHA665752 also decreased the number of premalignant lung lesions and induced apoptosis in tumor cells and vascular endothelial cells within lung lesions in Kras(LA1) mice [36]. These studies have provided critical proof-of-concept data and support clinical testing of MET inhibitors for the treatment of NSCLC.
MET Amplification and Acquired Resistance to EGFR Inhibitors
Acquired resistance to EGFR TKIs is an inevitable consequence of treatment, and recent studies indicate that it can occur as a result of secondary EGFR mutations or parallel activation of downstream signaling pathways, including MET. Approximately 5%–22% of NSCLC patients with secondary resistance to EGFR TKIs had evidence of amplification of the MET oncogene [11, 17, 37, 38]. In another study, de novo focal amplification of the MET-containing region 7q31.1 to 7q33.3 was observed in HCC827 NSCLC cells after exposure to increasing concentrations of gefitinib; four of 18 tumor samples (22%) from gefitinib-resistant NSCLC patients demonstrated MET amplification [17]. Amplification of MET was also detected in nine of 43 (21%) lung adenocarcinoma tumor samples from patients with gefitinib or erlotinib resistance, compared with two of 62 (3%) tumor samples from patients who had not been treated with an EGFR inhibitor [11]. EGFR and MET also show significant overlap of expression in primary NSCLC samples [39].
The mechanism by which cells acquire resistance to EGFR inhibitors may involve parallel activation of human epidermal growth factor receptor (HER)-3/PI3K/AKT signaling by MET (Fig. 3) [40]. Gefitinib treatment of HCC927 cells harboring activating EGFR mutations was shown to induce apoptosis, and this was dependent on downregulation of HER-3/PI3K/AKT phosphorylation and signaling [17]. In gefitinib-resistant HCC827 cells, phosphorylation of HER-3 and AKT was maintained in the presence of gefitinib; however, treatment of these cells with gefitinib plus PHA665752 or MET-specific short hairpin RNA fully suppressed HER-3 and AKT phosphorylation and re-established sensitivity to gefitinib. Thus, amplification of MET appears to promote resistance to EGFR inhibitors by stimulating EGFR-independent phosphorylation of HER-3 and downstream activation of the PI3K/AKT pathway. In this model, inhibition of MET blocked activation of HER-3/PI3K/AKT and restored sensitivity to EGFR inhibitors. Another study also suggested that MET activation may be associated with resistance to EGFR inhibitors [41].
Figure 3.

Proposed mechanism for acquired resistance to EGFR inhibitors by MET. (A): In erlotinib-sensitive cells, HER-3 phosphorylation by EGFR and downstream activation of PI3K/AKT is inhibited. (B): MET amplification phosphorylates HER-3 and activates PI3K/AKT in erlotinib-resistant cells. (C): MET inhibition by tivantinib and EGFR by erlotinib prevents phosphorylation of HER-3 and downstream activation of PI3K/AKT.
Abbreviations: EGFR, epidermal growth factor receptor; HER human epidermal growth factor receptor; PI3K, phosphoinositide 3-kinase.
Adapted from Nat Med 2007;13:675–677, with permission from Macmillan Publishers Ltd. Copyright 2007.
Approximately 5%–22% of NSCLC patients with secondary resistance to EGFR TKIs had evidence of amplification of the MET oncogene. In another study, de novo focal amplification of the MET-containing region 7q31.1 to 7q33.3 was observed in HCC827 NSCLC cells after exposure to increasing concentrations of gefitinib; four of 18 tumor samples (22%) from gefitinib-resistant NSCLC patients demonstrated MET amplification.
Recent studies in animal models of NSCLC using inhibitors of MET and EGFR have furthered our understanding of the interplay between the MET and EGFR signaling pathways and the possible synergistic benefits of dual inhibition of these pathways (Table 1) [39, 42–47]. For example, in multiple NSCLC xenograft models, including erlotinib-resistant xenografts, the combination of MGCD265 (a small-molecule inhibitor of MET, VEGF receptor [VEGFR], Tie-2, and RON) with erlotinib demonstrated significantly greater antitumor activity than with either agent alone without significant added toxicity or drug–drug interactions [45]. In a study of HGF-Tg-severe combined immunodeficient (SCID) mice harboring established NCI-H596 tumors, administration of the anti-MET monoclonal antibody onartuzumab (MetMab) resulted in roughly 65% tumor inhibition, whereas erlotinib alone had minimal effects [39]. However, the combination of onartuzumab plus erlotinib inhibited tumor growth by roughly 90%.
Table 1.
Dual EGFR–MET inhibition studies in lung cancer xenograft models
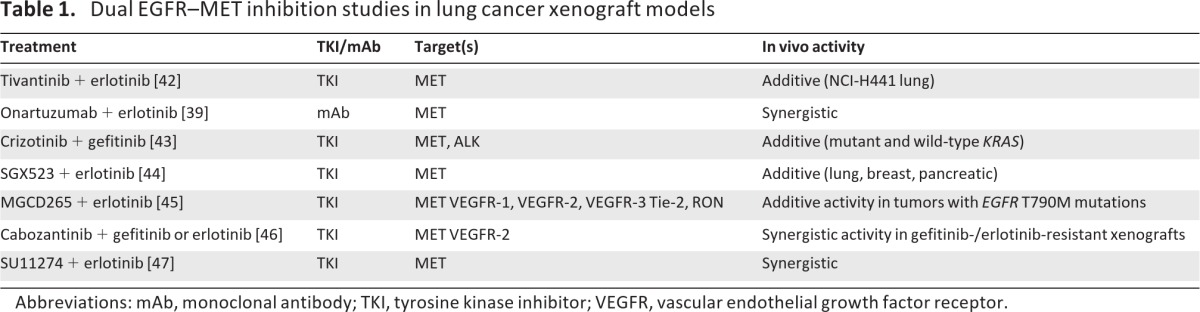
Abbreviations: mAb, monoclonal antibody; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
In studies with transgenic mice overexpressing human HGF that develop lung tumors when exposed to tobacco carcinogens, animals treated with anti-HGF antibody (L2G7) plus gefitinib developed fewer tumors than mice treated with either agent alone [43]. A higher rate of KRAS mutation was observed in lung tumors from mice treated with L2G7 alone than with combination treatment. However, mice treated with the MET/ALK inhibitor crizotinib exhibited less formation of both wild-type KRAS and mutant KRAS lung tumors. Likewise, combined treatment of mice with crizotinib plus gefitinib had an additive effect on the rate of lung tumor formation. In an HGF-overexpressing SCID mouse model, the MET-specific TKI SGX523 partially inhibited HGF-dependent growth of lung, breast, and pancreatic tumor xenografts. Simultaneous targeting of MET and EGFR pathways with SGX523 plus erlotinib demonstrated greater antitumor activity in lung, breast, and pancreatic xenografts than single-agent treatment in this model [44].
Dual inhibition of MET and EGFR has also been investigated in models of EGFR-resistant NSCLC. For example, the combination of cabozantinib (XL184, a MET, VEGFR-2, and RET inhibitor) and gefitinib inhibited proliferation, EGFR phosphorylation, and ErbB3 phosphorylation in gefitinib-resistant HCC827GR6 cells [46]. Likewise, in mice bearing gefitinib-/erlotinib-resistant HCC827GR6 NSCLC xenografts, neither cabozantinib nor erlotinib had an effect on AKT phosphorylation. However, the combination of cabozantinib and erlotinib significantly inhibited AKT phosphorylation. Combined administration of erlotinib and cabozantinib also resulted in regression of HCC827GR6 xenografts, whereas administration of either agent alone did not.
These studies suggest a role for MET in acquired resistance to EGFR inhibitors and demonstrate that combined inhibition of EGFR and MET can overcome resistance to EGFR inhibitors. Moreover, these studies suggest that combined treatment with EGFR and MET inhibitors may have greater antitumor activity than with either agent alone, and they establish a rationale for testing dual MET and EGFR inhibition for the treatment of patients with NSCLC.
Clinical Development of MET Inhibitors for NSCLC
The preclinical work described above provided rationale for clinical trials evaluating treatment of NSCLC patients with the combination of EGFR TKIs and MET inhibitors. Clinical development of dual MET–EGFR inhibition as second-line therapy for NSCLC has now progressed into phase III development. The agents that have been most extensively studied include cabozantinib, ficlatuzumab, onartuzumab, and tivantinib (Table 2) [48–52].
Table 2.
Early phase clinical trial results of anti-MET and anti-HGF agents in patients with NSCLC
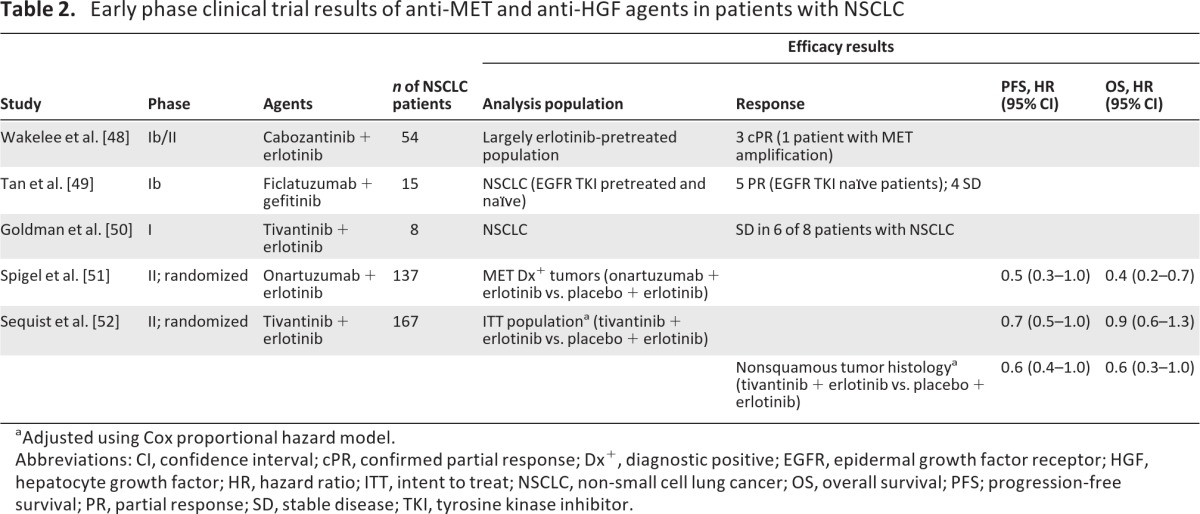
aAdjusted using Cox proportional hazard model.
Abbreviations: CI, confidence interval; cPR, confirmed partial response; Dx+, diagnostic positive; EGFR, epidermal growth factor receptor; HGF, hepatocyte growth factor; HR, hazard ratio; ITT, intent to treat; NSCLC, non-small cell lung cancer; OS, overall survival; PFS; progression-free survival; PR, partial response; SD, stable disease; TKI, tyrosine kinase inhibitor.
Cabozantinib was assessed in combination with erlotinib in a phase Ib/II trial in 54 patients with NSCLC, most of whom had received previous erlotinib treatment [48]. Of 36 evaluable patients, six patients (17%) achieved a best response of ≥30% reduction in tumor burden, including three patients who had previous erlotinib therapy. Additionally, three patients (8%) had a confirmed partial response (PR), including one patient with a MET-amplified tumor. Cabozantinib monotherapy is also being assessed in a phase II trial in pretreated patients with NSCLC (n = 59); preliminary results include best responses of a PR (n = 5), stable disease (SD) (n = 27), and progressive disease (PD) (n = 10) with a 12-week disease control rate of 42% [53].
In a phase Ib study, ficlatuzumab, an anti-HGF monoclonal antibody, was evaluated in combination with gefitinib in Asian patients with unresectable NSCLC (n = 15), most of whom had received previous EGFR TKI treatment (n = 10) [49]. Best responses among 12 patients in the recommended phase II dose cohort included a PR (n = 5), SD (n = 4), and PD (n = 3). All patients who had a best response of PR were EGFR TKI naïve, and the median duration of treatment was 12 weeks (range, 3.6–40 weeks). Based on these results, ficlatuzumab is being studied in combination with gefitinib versus gefitinib alone in a phase II study in Asian patients with NSCLC [54]. The study's primary endpoint is the objective response rate; secondary objectives include safety and tolerability, response duration, the PFS interval, the OS time, and an analysis of biomarkers.
The monoclonal antibody onartuzumab has been extensively studied in patients with previously treated NSCLC. A randomized phase II trial of onartuzumab or placebo in combination with erlotinib was conducted in 128 pretreated, EGFR TKI–naïve patients with NSCLC [51]. The intent-to-treat population did not demonstrate differences between treatment arms in the PFS (HR, 1.1; 95% CI, 0.7–1.6) and OS (HR, 0.8; 95% CI, 0.5–1.3) outcomes. In a prespecified subgroup analysis, however, the combination of onartuzumab plus erlotinib demonstrated a benefit over erlotinib alone in patients with MET-overexpressing tumors (defined as MET diagnostic [Met Dx]+ if >50% of tumor cells had staining 2+ or 3+ intensity for MET by immunohistochemistry [IHC]). Roughly half of the patients in this study (52%) were Met Dx+, which was associated with worse outcomes. In Met Dx+ patients, the PFS (HR, 0.53; 95% CI, 0.3–1.0) and OS (HR, 0.4; 95% CI, 0.2–0.7) outcomes were better with onartuzumab and erlotinib than with erlotinib plus placebo. In contrast, in Met Dx− patients, the PFS (HR, 1.8; 95% CI, 1.0–3.3) and OS (HR, 1.8; 95% CI, 0.8–4.0) outcomes were better in patients who received erlotinib plus placebo than in those who received onartuzumab plus erlotinib [51, 55]. Based on these results, a randomized phase III trial (ClinicalTrials.gov identifier, NCT01456325) comparing erlotinib plus onartuzumab with erlotinib plus placebo in patients with Met Dx+ NSCLC has been initiated.
The available data suggest that dual inhibition of MET and EGFR may overcome resistance and improve clinical outcomes. Phase I and phase II studies of different MET inhibitors have demonstrated the safety and efficacy of these novel agents in patients with advanced NSCLC.
Tivantinib is currently being investigated in a randomized, phase III trial in combination with erlotinib for the treatment of patients with nonsquamous NSCLC [56]. A phase I trial assessed the safety, pharmacokinetics, and preliminary antitumor activity of tivantinib in combination with erlotinib in patients with advanced solid tumors, including eight patients with NSCLC. Fifteen of 32 patients (47%) with advanced solid tumors had a PR (n = 1) or SD (n = 14), and six of eight patients (75%) with NSCLC achieved SD [50]. A recently reported randomized, placebo-controlled, phase II trial investigated erlotinib plus tivantinib in 173 previously treated, EGFR TKI–naïve patients [52]. The median PFS times were 3.8 months for erlotinib plus tivantinib and 2.3 months for erlotinib plus placebo (adjusted HR, 0.7; 95% CI, 0.5–1.0). Exploratory analyses revealed a benefit with tivantinib among patients with nonsquamous NSCLC, with superior PFS (adjusted HR, 0.6; 95% CI, 0.4–1.0) and OS (adjusted HR, 0.6; 95% CI, 0.3–1.0) outcomes. Among 50 evaluable patients in that trial, 27 (54%) had tumors that were MET+ by IHC [57]. Among 33 patients with nonsquamous tumors, 25 tumors (75%) were MET+, whereas only two of 17 squamous tumors (12%) were MET+. Among patients with MET+ tumors, treatment with tivantinib plus erlotinib was associated with better PFS (HR, 0.58) and OS (HR, 0.46) outcomes than with erlotinib plus placebo; there was no evidence of a worse outcome in patients with MET− tumors.
Based on these promising phase II results, the randomized phase III MET Inhibitor ARQ 197 Plus Erlotinib versus Erlotinib plus Placebo in NSCLC (MARQUEE) trial of dual tivantinib plus erlotinib therapy in patients with nonsquamous NSCLC has begun accruing patients [56]. Patients eligible for the MARQUEE trial must have stage IIIB or IV nonsquamous NSCLC and an Eastern Cooperative Oncology Group performance status score of 0 or 1, and must have received one or two previous lines of systemic anticancer therapy for advanced or metastatic disease, including one line of platinum-doublet therapy. This trial will also examine biomarker status, including KRAS, EGFR, and MET, and patients will be stratified according to EGFR and KRAS mutational status, number of previous therapies, sex, and smoking status.
Conclusion
Lung cancer remains the most common cancer in the world, and survival rates for patients with advanced or metastatic NSCLC are still low. However, targeted therapeutic approaches are improving clinical outcomes. In a recent study conducted by the LCMC, 60% of patients with NSCLC had tumor-specific driver mutations that could be used to guide treatment with agents targeting EGFR, MET, or other pathways. Current studies have also demonstrated the benefits of EGFR inhibitors in selected patients in both the first- and second-line settings. Moreover, recent studies suggest that MET amplification and activation may be involved in acquired resistance to EGFR inhibitors and may lead to downstream signaling that promotes cell survival, proliferation, and metastasis.
The available data suggest that dual inhibition of MET and EGFR may overcome resistance and improve clinical outcomes. Phase I and phase II studies of different MET inhibitors have demonstrated the safety and efficacy of these novel agents in patients with advanced NSCLC. Ongoing, randomized, phase III trials of onartuzumab and tivantinib in combination with erlotinib in selected NSCLC patients will provide important answers. Selection of patients for enrollment in these studies is based on either MET overexpression by IHC (onartuzumab trial) or nonsquamous histology (tivantinib trial). Both strategies should help select patients most likely to benefit from dual inhibition.
Acknowledgments
Financial support for medical editorial assistance was provided by Daiichi Sankyo, Inc., a member of the Daiichi Sankyo Group (Daiichi Sankyo Co., Ltd. is headquartered in Japan). We thank Bret A. Wing, Ph.D., ProEd Communications, Inc., for his medical editorial assistance with this manuscript.
Author Contributions
Conception/Design: Alan B. Sandler
Manuscript writing: Kyle W. Robinson, Alan B. Sandler
Final approval of manuscript: Alan B. Sandler
Disclosures
Alan B. Sandler: Aveo, Genentech/Roche, Daiichi-Sankyo, Pfizer, Exelixis (C/A); Genentech/Roche, Daiichi-Sankyo, Pfizer (H); Daiichi-Sankyo, Pfizer - funds paid to OHSU (RF); Other: Expert Testimony: Genentech/Roche, Pfizer.
The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Surveillance, Epidemiology, and End Results. Table 1.1. Estimated New Cancer Cases and Deaths for 2010. All Races, By Sex. [accessed September 4, 2012]. Available at http://seer.cancer.gov/csr/1975_2007/results_single/sect_01_table.01.pdf.
- 2.Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 3.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 4.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 5.Hanna N, Shepherd FA, Fossella FV, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol. 2004;22:1589–1597. doi: 10.1200/JCO.2004.08.163. [DOI] [PubMed] [Google Scholar]
- 6.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 9.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 10.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 11.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammerman PS, Jänne PA, Johnson BE. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2009;15:7502–7509. doi: 10.1158/1078-0432.CCR-09-0189. [DOI] [PubMed] [Google Scholar]
- 13.Jackman DM, Miller VA, Cioffredi LA, et al. Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: Results of an online tumor registry of clinical trials. Clin Cancer Res. 2009;15:5267–5273. doi: 10.1158/1078-0432.CCR-09-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raponi M, Winkler H, Dracopoli NC. KRAS mutations predict response to EGFR inhibitors. Curr Opin Pharmacol. 2008;8:413–418. doi: 10.1016/j.coph.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 16.Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–7525. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 18.Wang W, Li Q, Takeuchi S, et al. Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor-induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer. Clin Cancer Res. 2012;18:1663–1671. doi: 10.1158/1078-0432.CCR-11-1171. [DOI] [PubMed] [Google Scholar]
- 19.Yamada T, Matsumoto K, Wang W, et al. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin Cancer Res. 2010;16:174–183. doi: 10.1158/1078-0432.CCR-09-1204. [DOI] [PubMed] [Google Scholar]
- 20.Yamada T, Takeuchi S, Kita K, et al. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J Thorac Oncol. 2012;7:272–280. doi: 10.1097/JTO.0b013e3182398e69. [DOI] [PubMed] [Google Scholar]
- 21.Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 22.Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 23.Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI's Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29(suppl 18):CRA7506. [Google Scholar]
- 24.Cipriani NA, Abidoye OO, Vokes E, et al. MET as a target for treatment of chest tumors. Lung Cancer. 2009;63:169–179. doi: 10.1016/j.lungcan.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma PC, Maulik G, Christensen J, et al. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–325. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- 26.Boccaccio C, Comoglio PM. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 27.Furge KA, Kiewlich D, Le P, et al. Suppression of Ras-mediated tumorigenicity and metastasis through inhibition of the Met receptor tyrosine kinase. Proc Natl Acad Sci U S A. 2001;98:10722–10727. doi: 10.1073/pnas.191067898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moghul A, Lin L, Beedle A, et al. Modulation of c-MET proto-oncogene (HGF receptor) mRNA abundance by cytokines and hormones: Evidence for rapid decay of the 8 kb c-MET transcript. Oncogene. 1994;9:2045–2052. [PubMed] [Google Scholar]
- 29.Pennacchietti S, Michieli P, Galluzzo M, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 30.Okuda K, Sasaki H, Yukiue H, et al. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99:2280–2285. doi: 10.1111/j.1349-7006.2008.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lutterbach B, Zeng Q, Davis LJ, et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007;67:2081–2088. doi: 10.1158/0008-5472.CAN-06-3495. [DOI] [PubMed] [Google Scholar]
- 33.Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544–1553. doi: 10.1158/1535-7163.MCT-09-1173. [DOI] [PubMed] [Google Scholar]
- 34.Goetsch L, Lepecquet A-M, Geronimi F, et al. First bivalent fully antagonist anti-c-Met antibody targeting the c-Met receptor: II) in vivo activity. Proc Am Assoc Cancer Res. 2009;2009:2792. [Google Scholar]
- 35.Puri N, Khramtsov A, Ahmed S, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;67:3529–3534. doi: 10.1158/0008-5472.CAN-06-4416. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Wislez M, Fujimoto N, et al. A selective small molecule inhibitor of c-Met, PHA-665752, reverses lung premalignancy induced by mutant K-ras. Mol Cancer Ther. 2008;7:952–960. doi: 10.1158/1535-7163.MCT-07-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mark M, Zhang Y-W, Su Y, et al. Combination efficacy with MetMAb and erlotinib in a NSCLC tumor model highlight therapeutic opportunities for c-Met inhibitors in combination with EGFR inhibitors. Presented at the 99th American Association for Cancer Research Annual Meeting; April 12–16, 2008; San Diego, CA. [Google Scholar]
- 40.Arteaga CL. HER3 and mutant EGFR meet MET. Nat Med. 2007;13:675–677. doi: 10.1038/nm0607-675. [DOI] [PubMed] [Google Scholar]
- 41.Benedettini E, Sholl LM, Peyton M, et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am J Pathol. 2010;177:415–423. doi: 10.2353/ajpath.2010.090863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tokyo, Japan: Kyowa Hakko Kirin Co. Ltd.; Data on file. [Google Scholar]
- 43.Stabile LP, Rothstein ME, Keohavong P, et al. Targeting of both the c-Met and EGFR pathways results in additive inhibition of lung tumorigenesis in transgenic mice. Cancers (Basel) 2010;2:2153–2170. doi: 10.3390/cancers2042153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang YW, Staal B, Essenburg C, et al. MET kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res. 2010;70:6880–6890. doi: 10.1158/0008-5472.CAN-10-0898. [DOI] [PubMed] [Google Scholar]
- 45.Fournel M, Dupont I, Bonfils C, et al. Potent preclinical anti-tumor activity of MGCD265, an orally active Met/VEGFR multitargeted kinase inhibitor in phase II clinical development, in combination with an EGFR inhibitor. Proc Am Assoc Cancer Res. 2010;70(suppl 1):3612. [Google Scholar]
- 46.Janne PA, Wax M, Leach JW, et al. Targeting MET with XL184 to reverse EGFR tyrosine kinase inhibitor (TKI) resistance in NSCLC: Impact of preclinical studies on clinical trial design. Eur J Cancer Suppl. 2008;6:174. [Google Scholar]
- 47.Tang Z, Du R, Jiang S, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99:911–922. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakelee HA, Gettinger SN, Engelman JA, et al. A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib (E) in patients (pts) with non-small cell lung cancer (NSCLC) J Clin Oncol. 2010;28(suppl 15):3017. [Google Scholar]
- 49.Tan E, Park K, Lim WT, et al. Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol. 2011;29(suppl 15):7571. [Google Scholar]
- 50.Goldman J, Laux I, Chai F, et al. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118:5901–5911. doi: 10.1002/cncr.27575. [DOI] [PubMed] [Google Scholar]
- 51.Spigel DR, Ervin TJ, Ramlau R, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol. 2011;29(suppl 15):7505. [Google Scholar]
- 52.Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29:3307–3315. doi: 10.1200/JCO.2010.34.0570. [DOI] [PubMed] [Google Scholar]
- 53.Yasenchak C, Nackaerts K, Awada A, et al. Phase 2 results of XL184 in a cohort of patients (pts) with advanced non-small cell lung cancer (NSCLC) Eur J Cancer Suppl. 2010;8:126. [Google Scholar]
- 54.Mok T, Tan E, Park K, et al. Randomized phase II study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol. 2011;29(suppl 15):TPS213. [Google Scholar]
- 55.Yu W, Pandita A, Penuel E, et al. Exploratory biomarker analyses from OAM4558g: A placebo-controlled phase II study of erlotinib with or without MetMAb in patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29(## suppl):7529. [Google Scholar]
- 56.Sandler A, Schiller JH, Hirsh V, et al. A phase III, randomized, double-blind, placebo-controlled study of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated subjects with locally advanced or metastatic, nonsquamous, non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29(suppl 15):TPS217. doi: 10.1200/JCO.2014.60.7317. [DOI] [PubMed] [Google Scholar]
- 57.Rodig S, Sequist LV, von Pawel J, et al. An exploratory biomarker analysis evaluating the effect of the c-MET inhibitor tivantinib (ARQ 197) and erlotinib in NSCLC patients in a randomized, double-blinded phase 2 study. Presented at the Annual Meeting of the American Association for Cancer Research; March 31–April 4, 2012; Chicago, IL. [Google Scholar]