

A noncomparative, randomized, phase II study evaluating the antitumor efficacy of two doses of oral itraconazole was conducted in men with metastatic prostate cancer.
Keywords: Itraconazole, Prostate cancer, Angiogenesis, Hedgehog pathway
Abstract
Background.
The antifungal drug itraconazole inhibits angiogenesis and Hedgehog signaling and delays tumor growth in murine prostate cancer xenograft models. We conducted a noncomparative, randomized, phase II study evaluating the antitumor efficacy of two doses of oral itraconazole in men with metastatic prostate cancer.
Patients and Methods.
We randomly assigned 46 men with chemotherapy-naïve metastatic castration-resistant prostate cancer (CRPC) to receive low-dose (200 mg/day) or high-dose (600 mg/day) itraconazole until disease progression or unacceptable toxicity. The primary endpoint was the prostate-specific antigen (PSA) progression-free survival (PPFS) rate at 24 weeks; a 45% success rate in either arm was prespecified as constituting clinical significance. Secondary endpoints included the progression-free survival (PFS) rate and PSA response rate (Prostate Cancer Working Group criteria). Exploratory outcomes included circulating tumor cell (CTC) enumeration, serum androgen measurements, as well as pharmacokinetic and pharmacodynamic analyses.
Results.
The high-dose arm enrolled to completion (n = 29), but the low-dose arm closed early (n = 17) because of a prespecified futility rule. The PPFS rates at 24 weeks were 11.8% in the low-dose arm and 48.0% in the high-dose arm. The median PFS times were 11.9 weeks and 35.9 weeks, respectively. PSA response rates were 0% and 14.3%, respectively. In addition, itraconazole had favorable effects on CTC counts, and it suppressed Hedgehog signaling in skin biopsy samples. Itraconazole did not reduce serum testosterone or dehydroepiandrostenedione sulfate levels. Common toxicities included fatigue, nausea, anorexia, rash, and a syndrome of hypokalemia, hypertension, and edema.
Conclusion.
High-dose itraconazole (600 mg/day) has modest antitumor activity in men with metastatic CRPC that is not mediated by testosterone suppression.
Implications for Practice:
This study investigated two doses of an oral antifungal drug, itraconazole, to determine whether it has antitumor activity in men with metastatic castration-resistant prostate cancer. The results showed that while low-dose itraconazole (200 mg/day) did not have significant antitumor effects, high-dose itraconazole (600 mg/day) did have some activity in these patients. Moreover, the effects of itraconazole appeared to be associated with inhibition of Hedgehog signaling in skin biopsies, and were not caused by testosterone suppression. Therefore, itraconazole may be a non-hormonal treatment option for patients with castration-resistant prostate cancer who wish to prevent or delay the use of chemotherapy. While itraconazole is not as effective as other novel agents for advanced prostate cancer (e.g. abiraterone, enzalutamide), it is a generic drug that may be considered if the cost of these newer agents is prohibitive, or in parts of the world where abiraterone and enzalutamide may not be available.
Introduction
Although androgen-deprivation therapy is very effective initial therapy for men with advanced prostate cancer, all patients will eventually progress to a state known as castration-resistant prostate cancer (CRPC), which is invariably fatal. Until recently, life-prolonging therapies for patients with metastatic CRPC were limited, consisting only of docetaxel chemotherapy [1]. In the past 2 years, three additional modalities were added to our armamentarium for metastatic CRPC: the autologous immunotherapy product sipuleucel-T [2], the chemotherapy agent cabazitaxel [3], and the novel androgen-biosynthesis inhibitor abiraterone [4]. Moreover, two additional agents (the bone-targeting radiopharmaceutical radium-223 [5] and the androgen-signaling inhibitor enzalutamide [6]) were recently reported to extend survival in these patients. Despite these advances, none of these therapies are curative, and survival times for men with metastatic CRPC remain short (20–24 months) [7]. In this light, novel biological targets continue to be explored [8] in order to expand treatment options for men with CRPC.
Drug development is a lengthy and expensive process, taking, on average, 15 years and US$80 million to bring a single drug to market [9]. To increase the efficiency of this process, a drug library comprising >3,000 existing compounds has been created, enabling in vitro screening of old drugs for novel biological functions [10]. This drug library was recently screened for agents that may inhibit angiogenesis, a potentially important target of prostate cancer therapeutics [11]. An unexpected “hit” from this screen was the antifungal agent itraconazole, which was found to inhibit endothelial cell proliferation in vitro (unlike other azole antifungals) [12] and to impede endothelial cell migration and capillary tube formation [13]. Although its antiangiogenic target is uncertain, one study suggested that itraconazole inhibits mammalian target of rapamycin in endothelial cells by impairing cholesterol trafficking [14]. In vivo, itraconazole was found to inhibit neovascularization in a mouse Matrigel™ (BD Biosciences, San Diego, CA) model, to delay tumor growth in a castration-resistant xenograft mouse model (22Rv1), and to inhibit metastases in the AT6.3 prostate cancer mouse model [12]. Intriguingly, itraconazole was also discovered to potently inhibit Hedgehog (Hh) signaling, a developmental pathway regulating epithelial–mesenchymal interactions, cell survival, and angiogenesis [15]. To this end, in vitro studies showed that itraconazole inhibited proliferation of the Hh reporter cell line Shh-Light2 by antagonizing Smoothened [16]. Additionally, itraconazole induced tumor growth inhibition in a mouse medulloblastoma model (Ptch+/− p53−/−) with constitutive overactivation of Hh signaling. In this allograft model, itraconazole downmodulated intratumoral expression of GLI1, a Hh target gene [16].
Because itraconazole is already approved by the U.S. Food and Drug Administration (FDA) as an antifungal agent at oral doses in the range of 200–600 mg/day [17], we conducted a phase II study examining the antitumor efficacy of two doses of itraconazole (200 mg/day and 600 mg/day) in men with metastatic CRPC. This study was prompted by the encouraging clinical activity of other antiangiogenic agents in CRPC patients [18] and by other data suggesting that upregulation of Hh pathway components may drive CRPC [19]. In addition, the cost of generic itraconazole is only a fraction of that of other novel therapies for CRPC, such as abiraterone and enzalutamide.
Patients and Methods
Patients
Our target population was men with metastatic CRPC who had not received cytotoxic chemotherapy. Patients were required to have histologically confirmed prostate adenocarcinoma, progressive disease despite “castration levels” of serum testosterone (<50 ng/dL), and radiographically visible distant metastases on computed tomography (CT) or technetium-99 bone scans. Patients had to have three or more rising serum prostate-specific antigen (PSA) values taken 4 weeks apart with the last value being ≥2.0 ng/mL, in accordance with Prostate Cancer Working Group (PCWG) guidelines [20]. Other eligibility criteria included age >18 years, an Eastern Cooperative Oncology Group (ECOG) performance status score ≤2, a life expectancy >6 months, and adequate kidney, liver, and bone marrow function.
Patients were excluded if they had received an oral antiandrogen within 6 weeks, had ever received chemotherapy for metastatic CRPC, took systemic corticosteroids, had a malabsorption syndrome, took drugs metabolized by cytochrome P450 (CYP)3A4, had a prior malignancy within 3 years, had major infectious, pulmonary, or cardiac illnesses, had symptomatic congestive heart failure, or had a corrected QT interval >450 msec on electrocardiography. Prior ketoconazole treatment was permitted.
The review boards at all institutions approved the study, which was conducted according to good clinical practice guidelines. All patients provided written informed-consent.
Study Design
This was a noncomparative, open-label, randomized, phase II study conducted at four institutions of the Prostate Cancer Clinical Trials Consortium [21]. Patients were randomized (1:1) to receive low-dose (200 mg/day) or high-dose (600 mg/day) itraconazole. These doses were chosen because itraconazole is already FDA approved as an antifungal agent at doses in the range of 200–600 mg/day and because data from animal models suggested that, although 200 mg might be sufficient to inhibit angiogenesis, doses ≥600 mg might be required to suppress Hh signaling.
Itraconazole was supplied as generic 100-mg capsules (Sandoz, Princeton, NJ). Patients assigned to the low-dose arm received two 100-mg capsules once daily; patients in the high-dose arm received three 100-mg capsules twice daily. Because itraconazole absorption depends on gastric acidity, patients were instructed to take itraconazole capsules with a carbonated beverage and together with food or within 30 minutes after a meal. Patients were not permitted to take concurrent antacids, histamine blockers, or proton pump inhibitors. Treatment continued either until unmanageable drug-related toxicity or until clinical or radiographic progression. Importantly, treatment was not discontinued for PSA elevations [20].
Assessments
Clinical evaluations included a physical examination, vital sign measurements, assessment of ECOG score, review of concomitant medications, laboratory evaluations (chemical and hematologic studies), and review of adverse events and were performed every 4 weeks. Efficacy assessments included serum PSA measurement every 4 weeks and CT (chest, abdomen, and pelvis) and whole-body technetium-99 bone scan evaluations every 12 weeks.
Outcome Measures
The primary endpoint was freedom from PSA progression (the PSA progression-free survival [PPFS] rate) at 24 weeks after randomization. PSA progression was defined as a ≥25% increase in PSA from nadir (and by ≥2 ng/mL), requiring confirmation ≥4 weeks later (PCWG criteria) [20]. Although the PPFS rate is not a validated surrogate of clinical benefit, this endpoint was chosen in order to screen for preliminary evidence of clinical activity in the setting of a small phase II trial. A key secondary endpoint, which might be considered more clinically meaningful, was freedom from progression (the progression-free survival [PFS] rate) at 24 weeks. Progression was defined [20] as clinical progression (worsening disease-related symptoms or new cancer-related complications), radiographic progression (on CT scan, ≥20% enlargement in the sum diameter of soft-tissue target lesions according to the Response Evaluation in Solid Tumors [RECIST], version 1.0 [22]; on bone scan, two or more new confirmed bone lesions), or death, whichever occurred first.
Secondary endpoints included the median PPFS duration, PSA response rate (≥50% PSA decline from baseline, maintained for ≥4 weeks), best PSA response (maximal percentage PSA decrease from baseline), median PFS time, and objective response rate in measurable soft-tissue lesions (partial response, ≥30% decrease in the sum diameter of target lesions; progressive disease, ≥20% increase in the sum diameter of target lesions or one or more new lesion; stable disease, change in the sum diameter of target lesions that do not meet the above parameters; RECIST, version 1.0 [22]). A final secondary endpoint was safety; adverse events were graded using the National Cancer Institute Common Toxicity Criteria for Adverse Events, version 3.0.
Circulating Tumor Cell Analysis
Blood samples (7.5 mL) for circulating tumor cell (CTC) enumeration were collected at baseline and after 4 weeks and 12 weeks on study and were analyzed using the CellSearch® system (Veridex, Raritan, NJ), as previously described [23]. Results were expressed as numbers of CTCs per 7.5 mL blood.
Pharmacokinetics and Pharmacodynamics
Pharmacokinetics
Plasma samples were collected at baseline and prior to itraconazole administration (minimum concentration [Cmin]) and at 4 weeks and 12 weeks on study. Itraconazole and 4-hydroxyitraconazole concentrations were assessed using a validated liquid chromatography–mass spectrometry assay, over the range of 2–2,000 ng/mL.
Analysis of Adrenal Axis
To examine whether or not itraconazole suppressed adrenal cortical function, several adrenal-axis hormones were evaluated at baseline and after 4 weeks and 12 weeks on study: testosterone, dehydroepiandrostenedione sulfate (DHEA-S), cortisol, aldosterone, and adrenocorticotropic hormone (ACTH). Serum testosterone and serum aldosterone were measured using a liquid chromatography–mass spectrometry assay. Using this method, the lower limit of detection of testosterone is 1 ng/dL. Serum DHEA-S and plasma ACTH levels were measured using a chemiluminescence immunoassay. Serum cortisol was measured using an enzyme immunoassay.
Vascular Endothelial Growth Factor Levels
To evaluate antiangiogenic effects in an exploratory analysis, plasma was collected for vascular endothelial growth factor (VEGF) measurement at baseline and after 4 weeks and 12 weeks on study. Total VEGF concentrations were measured using the Quantikine® enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN).
Hh Pathway Analysis
Because Hh signaling is present in skin and hair follicles, we examined GLI1 mRNA expression (a marker of Hh pathway activation) using 3-mm skin punch biopsies from hair-containing skin obtained at baseline and after 4 weeks and 12 weeks on study. RNA was extracted from skin biopsy specimens, and GLI1 expression levels were assessed by real-time reverse-transcription polymerase chain reaction (SABiosciences-Qiagen, Frederick, MD), as previously described [24].
Statistical Analysis
Based on prior studies [25], we estimated that up to 20% of patients with metastatic CRPC who had not received prior chemotherapy would be free from PSA progression (as defined above) after 24 weeks on study. We hypothesized that itraconazole (at either dose level) would prevent PSA progression at 24 weeks in ∼45% of men (i.e., we considered a 25% absolute improvement >20% to be clinically meaningful). Twenty-nine patients per arm would grant 83% power to detect an improvement in the 24-week PPFS rate (the primary endpoint) from 20% (historical controls) to 45% using a two-sided α of 0.05. A 45% 24-week PPFS rate in each arm was predefined to constitute a success (indicating worthiness for further study). To monitor for treatment futility, both arms had prespecified early-stopping rules that were applied after nine (one third of the total) and 15 (one half of the total) patients were evaluable for the primary endpoint. In each arm, if there were fewer than two of nine men who achieved the primary endpoint or if there were fewer than four of 15 men who achieved the primary endpoint, then that arm would close for futility. These stopping rules were consistent with observing an upper bound of a one-sided exact 90% confidence interval (CI) that excluded our hypothesized success rate of 45%.
The study was not powered to allow inferential statistics comparing treatment arms. Kaplan–Meier analysis was used to estimate time-to-event endpoints and 95% CIs. Patient baseline characteristics were compared between arms using Fisher's exact test, Student's t-test, or the Wilcoxon rank-sum test (p-values are merely descriptive because all differences are a result of chance variation induced by randomization). Pharmacodynamic and pharmacokinetic endpoints were reported as trends over time using descriptive statistics; associations between these exploratory measures and clinical outcomes were sought using Pearson's correlation coefficient (r).
Results
Patients
The high-dose arm was enrolled to completion (29 patients) whereas the low-dose arm closed early because of futility after 17 men were enrolled (in this arm, there were two successes in the first nine patients and enrollment continued until 15 were evaluable for the primary endpoint; at that time, two additional patients were enrolled but no more achieved the primary endpoint) (Fig. 1). Baseline patient characteristics appeared generally balanced (Table 1); there was a trend toward lower baseline PSA levels in the low-dose arm and a trend toward more bone-only metastases in the high-dose arm. One third of patients in both arms had received prior ketoconazole. The median treatment durations were 11.9 weeks in the low-dose arm and 23.6 weeks in the high-dose arm.
Figure 1.

Consort diagram.
Table 1.
Baseline demographic and clinical characteristics
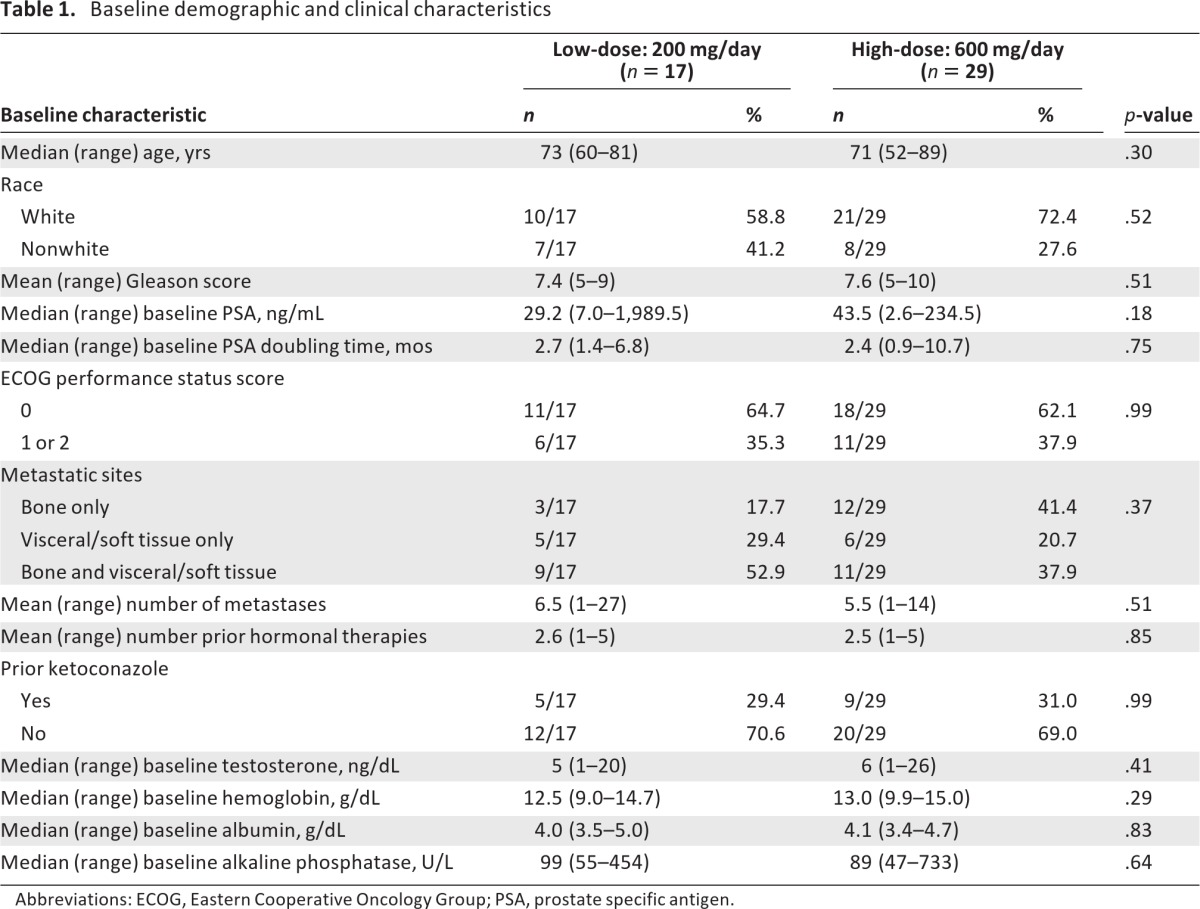
Abbreviations: ECOG, Eastern Cooperative Oncology Group; PSA, prostate specific antigen.
Primary Endpoint
All 17 patients in the low-dose arm and 25 of 29 patients in the high-dose arm (four men came off study before 24 weeks because of toxicity) were evaluable for the primary endpoint. In the low-dose arm, the 24-week PPFS rate estimate was 11.8% (two of 17 men; 95% CI, 1.5%–36.4%), failing to achieve the primary endpoint. Conversely, the high-dose arm met the primary endpoint, demonstrating a 24-week PPFS rate estimate of 48.0% (12 of 25 men; 95% CI, 27.8%–68.7%).
Secondary Endpoints
The median PPFS times were 11.9 weeks (95% CI, 5.6–20.0 weeks) and 17.0 weeks (95% CI, 12.4–32.0 weeks) in the low-dose and high-dose arms, respectively (Fig. 2A). The 24-week PFS rate estimates were 18.8% (95% CI, 6.8%–52.0%) and 61.6% (95% CI, 46.1%–84.6%) in the two arms, respectively. The median PFS times were 11.9 weeks (95% CI, 11.9–28.1 weeks) and 35.9 weeks (95% CI, 21.6–47.4 weeks) (Fig. 2B). PSA response rates (≥50% PSA decline) were 0% (95% CI, 0%–19.5%) and 14.3% (95% CI, 4.0%–32.7%) (Fig. 2C), respectively. Among those with measurable disease at baseline, 7.7% (95% CI, 1.8%–33.9%) and 11.1% (95% CI, 3.4%–33.1%) of patients in the two arms achieved a partial objective response, respectively (Fig. 2D). Finally, the median PSA doubling time (PSADT) estimates were longer in both study arms after treatment initiation, although this change was only statistically significant in the high-dose arm (baseline median PSADT, 2.4 months; on-study median PSADT, 7.7 months; difference, +5.3 months; p < .01) and not in the low-dose arm (baseline median PSADT, 2.7 months; on-study median PSADT, 5.8 months; difference, +3.1 months; p = .07).
Figure 2.

Clinical effects of itraconazole. (A): Kaplan–Meier curves of PPFS in men receiving low-dose and high-dose itraconazole. (B): Kaplan–Meier curves of PFS in each treatment arm. (C): Waterfall plots showing best PSA responses among men receiving low-dose and high-dose itraconazole. The asterisk denotes a clipped PSA value. Prior treatment with ketoconazole is indicated by the hashed bars. (D): Waterfall plots showing best objective responses in measurable lesions according to Response Evaluation Criteria in Solid Tumors, version 1.0. Prior treatment with ketoconazole is indicated by the hashed bars (and daggers).
Abbreviations: CI, confidence interval; PFS, progression-free survival; PPFS, PSA progression-free survival; PSA, prostate-specific antigen.
Safety
Adverse events were generally more frequent in the high-dose than in the low-dose arm (Table 2). Common toxicities in both arms included fatigue, pain, nausea and constipation. Also, a constellation of adverse events comprising hypertension, hypokalemia, and edema was of special interest, suggesting a syndrome of secondary mineralocorticoid excess (see adrenal-axis evaluations below). Manifestations of this syndrome were more frequent in the high-dose arm.
Table 2.
Adverse events
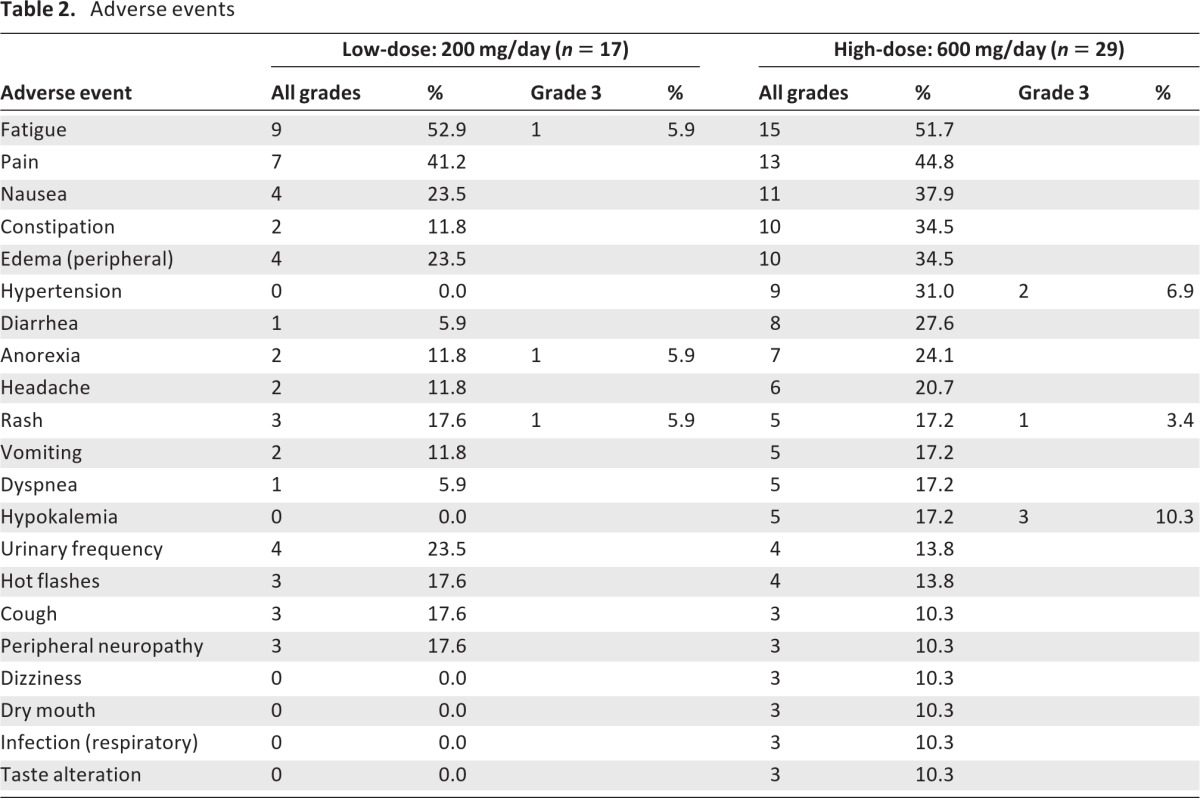
Grade 3 adverse events in the low-dose arm included fatigue (5.9%), anorexia (5.9%), and rash (5.9%). Grade 3 toxicities in the high-dose arm included hypokalemia (10.3%), hypertension (6.9%), and rash (3.4%). There were no grade 4 toxicities. The percentages of patients who came off study as a result of toxicities were 5.9% in the low-dose arm (one patient developed a rash) and 13.8% in the high-dose arm (one patient developed fatigue, one patient developed anorexia, one patient developed a rash, and one patient developed temporal arteritis [not drug related]).
CTC Enumeration
Fifteen patients in the low-dose arm (88.2%) and 25 patients in the high-dose arm (86.2%) had paired baseline and post-treatment blood samples collected for CTC enumeration. Thirty-two men had favorable baseline CTC counts (<5 CTCs per 7.5 mL blood); 96.9% of them retained favorable CTC counts for 12 weeks. Eight men had unfavorable baseline CTC counts (≥5 CTCs per 7.5 mL blood); five (62.5%) of them converted to favorable CTC counts post-treatment. Data from those patients converting from unfavorable to favorable CTC counts are shown here: 28→3, 15→1, 7→0, 6→0, and 6→0 CTCs per 7.5 mL blood.
Pharmacokinetics and Pharmacodynamics
Pharmacokinetics
Sixteen patients in the low-dose arm (94.1%) and 26 patients in the high-dose arm (89.7%) had paired baseline and post-treatment plasma samples for pharmacokinetic analyses. The mean plasma itraconazole trough concentration (Cmin) values were 370.0 ng/mL (range, 86.9–653.1 ng/mL) and 1,517.0 ng/mL (range, 673.8–2,360.2 ng/mL) in low- and high-dose arms, respectively. The mean plasma 4-hydroxyitraconazole Cmin values were 723.5 ng/mL (range, 289.2–1,157.8 ng/mL) and 2,630.8 ng/mL (range, 1,036.0–4,225.6 ng/mL), respectively. There were significant correlations between a higher itraconazole Cmin level and both a longer PPFS duration (r = 0.56; p = .003) and a greater PSA decline (r = 0.39; p = .03) (supplemental online Fig. 1). Similar statistically significant correlations were observed with 4-hydroxyitraconazole (data not shown).
Adrenal Axis Analysis
Neither low-dose nor high-dose itraconazole caused suppression of serum testosterone or DHEA-S levels. Unexpectedly, low-dose and high-dose itraconazole appeared to slightly increase serum testosterone (Fig. 3A) and DHEA-S (Fig. 3B) levels, respectively. Additionally, high-dose (but not low-dose) itraconazole potently suppressed serum aldosterone (Fig. 3C) while raising plasma ACTH (Fig. 3D). There were no effects with either itraconazole dose on serum cortisol at 4 weeks or 12 weeks (data not shown).
Figure 3.

Endocrine effects of itraconazole. (A): Effect of low- and high-dose itraconazole on serum testosterone concentrations (data are shown as medians and interquartile ranges). (B): Effect of low- and high-dose itraconazole on serum DHEA-S concentrations. (C): Effect of low- and high-dose itraconazole on serum aldosterone concentrations. (D): Effect of low- and high-dose itraconazole on plasma ACTH concentrations.
Abbreviations: ACTH, adrenocorticotropic hormone; DHEA-S, dehydroepiandrostenedione-sulfate.
VEGF Analysis
Low-dose itraconazole was not associated with a change in plasma VEGF level at either 4 weeks (p = .59) or 12 weeks (p = .11). Likewise, high-dose itraconazole was not associated with a VEGF level change at either 4 weeks (p = .72) or 12 weeks (p = .76).
Hh pathway analysis
Fifteen patients in the low-dose arm (88.2%) and 25 patients in the high-dose arm (86.2%) had paired baseline and post-treatment skin punch biopsy samples collected for GLI1 expression analysis. GLI1 was downmodulated in 33% and 68% of patients in the low- and high-dose arms, respectively (Fig. 4A). The percentage of patients who achieved a twofold or greater downmodulation in GLI1 with itraconazole was 28% (11 of 40), compared with 68% of patients receiving vismodegib (a potent Hh pathway antagonist) in prior studies [26]. The median PPFS time was longer in men who achieved GLI1 downmodulation (p = .028) (Fig. 4B) and there was also a trend toward a longer PFS interval in men with GLI1 downmodulation (p = .128) (Fig. 4C). Finally, there was a significant correlation between a stronger GLI1 downmodulation and a greater PSA decline (r = 0.38; p = .01) (Fig. 4D). Interestingly, all five patients who achieved favorable CTC conversions also had downmodulation of GLI1.
Figure 4.

GLI1 modulation by itraconazole. (A): Waterfall plots showing GLI1 modulation in skin punch biopsies, depicted as fold change in GLI expression post-treatment compared with baseline values. (B): Kaplan–Meier curves depicting PPFS according to GLI1 modulation status. (C): Kaplan–Meier curves depicting PFS according to GLI1 modulation status. (D): Scatterplot showing the association between GLI1 modulation and PSA change.
Abbreviations: PFS, progression-free survival; PPFS, PSA progression-free survival; PSA, prostate-specific antigen.
Figure 4.

Continued.
Discussion
This phase II study is the first to examine itraconazole as an antineoplastic agent in human cancer. We demonstrate that, in men with metastatic chemotherapy-untreated CRPC, low-dose itraconazole (200 mg/day) lacks significant antitumor efficacy, whereas high-dose itraconazole (600 mg/day) may have modest clinical activity, as suggested by longer PPFS and PFS times than in historical data [25]. Importantly, the PFS duration observed here (35.9 weeks) is comparable with PFS time estimates (range, 30–40 weeks) of other FDA-approved and experimental agents in this patient population (mitoxantrone, docetaxel, tasquinimod, and cabozantinib) [1, 18, 27], although the PFS time is not a surrogate of clinical benefit. Notably, itraconazole's activity does not appear to be mediated by testosterone suppression (although a comprehensive analysis of the androgen axis was not conducted), and it may possibly be associated with downmodulation of Hh signaling. Alternatively, itraconazole may have beneficial off-target effects on other unknown targets.
Another azole antifungal, ketoconazole, has been used off label for many years as a therapy for CRPC. Ketoconazole functions by suppressing extragonadal androgen synthesis [28] (nonselectively inhibiting multiple CYP enzymes), but carries significant toxicity without evidence that it extends the survival duration [29]. However, the selective CYP17 inhibitor abiraterone was shown to improve survival outcomes in men with docetaxel-pretreated metastatic CRPC, [4], resulting in its FDA approval. Here, we demonstrate that itraconazole does not suppress circulating testosterone or DHEA-S levels (although androstenedione and dihydrotestosterone levels were not measured), suggesting an alternative or additional antitumor mechanism. Moreover, itraconazole appeared to have activity in both ketoconazole-pretreated and ketoconazole-naïve patients.
Tumor angiogenesis and Hh signaling are both involved in prostate cancer growth, progression, and metastasis [15, 30]. Although blocking each pathway separately has failed to yield new prostate cancer therapeutics [26, 31], inhibition of both pathways simultaneously with itraconazole represents a rational approach. In this study, we did not observe modulation of circulating VEGF levels, but that does not necessarily mean that itraconazole lacks antiangiogenic effects in man. Our ability to interrogate angiogenesis was limited by the lack of tumor biopsy samples and because we evaluated only one of many circulating angiogenic factors (although none have consistently been associated with clinical benefit from antiangiogenic therapies). Additionally, although we observed GLI1 downmodulation in skin biopsy samples, we did not interrogate Hh signaling in tumors themselves; therefore, we provide only indirect evidence that Hh pathway suppression is a potential mechanism of action of itraconazole. Finally, the association between GLI1 downmodulation and itraconazole's clinical activity may not be causal, and it may simply reflect a pharmacodynamic effect that is not linked to drug efficacy. Nevertheless, the results of this study provide the impetus to examine other more potent Hh pathway inhibitors (e.g., vismodegib, LDE225) in men with CRPC.
Of particular interest was the occurrence of a syndrome of hypokalemia, hypertension, and edema in a dose-dependent manner. Although these manifestations are usually related to hyperaldosteronism [32], aldosterone levels were potently suppressed in our patients. This raises the possibility of a syndrome of secondary mineralocorticoid excess (with elevated aldosterone precursors), as has been reported in abiraterone-treated patients [4, 33]. To this end, we discovered raised levels of corticosterone and deoxycorticosterone in a patient who developed all three features of this syndrome. However, unlike abiraterone (and ketoconazole), itraconazole did not suppress cortisol production and does not require glucocorticoid supplementation. Indeed, the combination of itraconazole and corticosteroids is contraindicated and can induce Cushing's syndrome by impairing corticosteroid metabolism by CYP3A4 [34]. Finally, the slight rises observed in serum testosterone and DHEA-S levels may have resulted from elevation of upstream ACTH, although these increases in androgen levels were modest.
In conclusion, this study suggests that high-dose itraconazole (600 mg/day) may have modest antitumor activity in men with metastatic CRPC that could potentially be associated with Hh pathway suppression, although an androgen-mediated effect cannot be excluded. Ongoing trials are now assessing the impact of itraconazole as an antineoplastic agent in patients with lung cancer, breast cancer, and basal cell carcinoma. Future studies in prostate cancer patients will compare itraconazole with placebo in men with nonmetastatic CRPC, aiming to extend the metastasis-free survival duration in this population. In addition, clinical trials using more potent Hh antagonists (e.g., vismodegib, LDE225) in men with CRPC are also being planned.
See www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgment
This study was conducted within the Department of Defense/Prostate Cancer Foundation Prostate Cancer Clinical Trials Consortium (DOD/PCF-PCCTC). We thank the patients who volunteered to participate in this study, and their families. We are grateful to all the study staff members at each site, the research nurses, the pharmacy staff, and the lead research nurse, Rana Sullivan.
The study was supported by the Commonwealth Foundation for Cancer Research (M.A.C.), the David H. Koch Charitable Foundation (M.A.C.), a Conquer Cancer Foundation 2009 Young Investigator Award (E.S.A.), DOD grants W81XWH-09–1-0149 (M.A.C.) and PC051385 (M.A.C.), and the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973 and UL1 RR025005). This publication was made possible by Grant Number UL1RR025005 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Author Contributions
Conception/Design: Emmanuel S. Antonarakis, Amanda L. Blackford, Michelle A. Rudek, Michael A. Carducci
Provision of study material or patients: Emmanuel S. Antonarakis, Elizabeth I. Heath, David C. Smith, Dana Rathkopf, Daniel C. Danila, Michael A. Carducci
Collection and/or assembly of data: Emmanuel S. Antonarakis, Elizabeth I. Heath, David C. Smith, Dana Rathkopf, Amanda L. Blackford, Daniel C. Danila, Serina King, Anja Frost, A. Seun Ajiboye, Ming Zhao, Janet Mendonca, Sushant A. Kachhap, Michelle A. Rudek, Michael A. Carducci
Data analysis and interpretation: Emmanuel S. Antonarakis, Amanda L. Blackford, Daniel C. Danila, Anja Frost, A. Seun Ajiboye, Ming Zhao, Janet Mendonca, Sushant A. Kachhap, Michelle A. Rudek, Michael A. Carducci
Manuscript writing: Emmanuel S. Antonarakis, Elizabeth I. Heath, David C. Smith, Dana Rathkopf, Amanda L. Blackford, Daniel C. Danila, Serina King, Anja Frost, A. Seun Ajiboye, Ming Zhao, Janet Mendonca, Sushant A. Kachhap, Michelle A. Rudek, Michael A. Carducci
Final approval of manuscript: Emmanuel S. Antonarakis, Elizabeth I. Heath, David C. Smith, Dana Rathkopf, Amanda L. Blackford, Daniel C. Danila, Serina King, Anja Frost, A. Seun Ajiboye, Ming Zhao, Janet Mendonca, Sushant A. Kachhap, Michelle A. Rudek, Michael A. Carducci
Disclosures
The authors indicated no financial relationships.
References
- 1.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 2.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 4.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson S, Franzén L, Parker C, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: A randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8:587–594. doi: 10.1016/S1470-2045(07)70147-X. [DOI] [PubMed] [Google Scholar]
- 6.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 7.Antonarakis ES, Eisenberger MA. Expanding treatment options for metastatic prostate cancer. N Engl J Med. 2011;364:2055–2058. doi: 10.1056/NEJMe1102758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: New estimates of drug development costs. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 10.Chong CR, Sullivan DJ., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 11.Kluetz PG, Figg WD, Dahut WL. Angiogenesis inhibitors in the treatment of prostate cancer. Expert Opin Pharmacother. 2010;11:233–247. doi: 10.1517/14656560903451716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong CR, Xu J, Lu J, et al. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2:263–270. doi: 10.1021/cb600362d. [DOI] [PubMed] [Google Scholar]
- 13.Aftab BT, Dobromilskaya I, Liu JO, et al. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011;71:6764–6772. doi: 10.1158/0008-5472.CAN-11-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Dang Y, Ren YR, et al. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci U S A. 2010;107:4764–4769. doi: 10.1073/pnas.0910872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signaling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 16.Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen-Ortho, Inc. Itraconazole prescribing information, 2001. [Accessed December 12, 2012]. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020083s040s041s044lbl.pdf.
- 18.Pili R, Häggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29:4022–4028. doi: 10.1200/JCO.2011.35.6295. [DOI] [PubMed] [Google Scholar]
- 19.Tzelepi V, Karlou M, Wen S, et al. Expression of Hedgehog pathway components in prostate carcinoma microenvironment: Shifting the balance towards autocrine signaling. Histopathology. 2011;58:1037–1047. doi: 10.1111/j.1365-2559.2011.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris MJ, Basch EM, Wilding G, et al. Department of Defense prostate cancer clinical trials consortium: A new instrument for prostate cancer clinical research. Clin Genitourin Cancer. 2009;7:51–57. doi: 10.3816/CGC.2009.n.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 23.Shaffer DR, Leversha MA, Danila DC, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:2023–2029. doi: 10.1158/1078-0432.CCR-06-2701. [DOI] [PubMed] [Google Scholar]
- 24.Seifert AW, Zheng Z, Ormerod BK, et al. Sonic hedgehog controls growth of external genitalia by regulating cell cycle kinetics. Nat Commun. 2010;1:1–9. doi: 10.1038/ncomms1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carducci MA, Saad F, Abrahamsson PA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer. 2007;110:1959–1966. doi: 10.1002/cncr.22996. [DOI] [PubMed] [Google Scholar]
- 26.LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: Results of a phase II randomized discontinuation trial. J Clin Oncol. 2012 doi: 10.1200/JCO.2012.45.0494. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trump DL, Havlin KH, Messing EM, et al. High-dose ketoconazole in advanced hormone-refractory prostate cancer: Endocrinologic and clinical effects. J Clin Oncol. 1989;7:1093–1098. doi: 10.1200/JCO.1989.7.8.1093. [DOI] [PubMed] [Google Scholar]
- 29.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: A phase III trial (CALGB 9583) J Clin Oncol. 2004;22:1025–1033. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 30.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu EM, Jain M, Aragon-Ching JB. Angiogenesis inhibitors in prostate cancer therapy. Discov Med. 2010;10:521–530. [PubMed] [Google Scholar]
- 32.Tomaschitz A, Pilz S, Ritz E, et al. Aldosterone and arterial hypertension. Nat Rev Endocrinol. 2010;6:83–93. doi: 10.1038/nrendo.2009.263. [DOI] [PubMed] [Google Scholar]
- 33.Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 34.Bolland MJ, Bagg W, Thomas MG, et al. Cushing's syndrome due to interaction between corticosteroids and itraconazole. Ann Pharmacother. 2004;38:46–49. doi: 10.1345/aph.1D222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.