

This article reviews the molecular signaling of antiangiogenic and HER2-directed therapies that may underpin cardiac toxicity and the hypothesized cardioprotective properties of aerobic exercise.
Keywords: Exercise, Cardiotoxicity, Molecular therapeutics, Solid malignancies
Abstract
Molecularly targeted therapeutics (MTT) are the future of cancer systemic therapy. They have already moved from palliative therapy for advanced solid malignancies into the setting of curative-intent treatment for early-stage disease. Cardiotoxicity is a frequent and potentially serious adverse complication of some targeted therapies, leading to a broad range of potentially life-threatening complications, therapy discontinuation, and poor quality of life. Low-cost pleiotropic interventions are therefore urgently required to effectively prevent and/or treat MTT-induced cardiotoxicity. Aerobic exercise therapy has the unique capacity to modulate, without toxicity, multiple gene expression pathways in several organ systems, including a plethora of cardiac-specific molecular and cell-signaling pathways implicated in MTT-induced cardiac toxicity. In this review, we examine the molecular signaling of antiangiogenic and HER2-directed therapies that may underpin cardiac toxicity and the hypothesized molecular mechanisms underlying the cardioprotective properties of aerobic exercise. It is hoped that this knowledge can be used to maximize the benefits of small molecule inhibitors, while minimizing cardiac damage in patients with solid malignancies.
Implications for Practice:
Cardiotoxicity, a frequent and devastating adverse complication of some molecularly targeted therapies (MTTs), can lead to potentially life-threatening cardiovascular complications, therapy discontinuation, and poor quality of life. In non-cancer patients with left ventricular dysfunction and heart failure, aerobic exercise is one of the mainstay clinical interventions for the prevention and treatment of cardiovascular disease. However, few studies have investigated the efficacy of aerobic exercise in the prevention and/or treatment of MTT-induced cardiac injury. This topic is of particular importance because cardiac function is a strong predictor of cardiovascular and all-cause mortality, quality of life, and fatigue, and maybe even cancer-specific mortality. Here, we provide a comprehensive overview of cardiac molecular and cell-signaling pathways specific to MTT-induced cardiac toxicity. This review also outlines many pertinent aerobic exercise-induced molecular signaling pathways that may uniquely prevent and/or treat MTT cardiac injury. Overall, information presented in this review provides critical information for basic scientists, clinicians, and exercise oncology researchers who are investigating the application of exercise in cancer control.
Introduction
The emergence of molecularly targeted therapeutics (MTTs) has revolutionized the management of solid malignancies. Antiangiogenic and human epidermal growth factor receptor 2 (HER2)-directed MTTs are approved by the U.S. Food and Drug Administration (FDA) for the treatment of several solid malignancies, either as monotherapy or in combination with standard chemotherapy [1, 2]. The biologic selectivities of these drugs were expected to substantially reduce off-target toxicity, although it is now apparent that MTTs cause adverse cardiovascular consequences, such as hypertension and progressive left ventricular (LV) dysfunction, ultimately leading to symptomatic heart failure.
Several excellent reviews have described the biologic and molecular mechanisms underlying MTT-induced cardiotoxicity and risk for cardiotoxicity [1–8]; however, comparably little attention has been focused on strategies to prevent and/or mitigate anticipated injury. MTTs target multiple cellular pathways including highly coordinated myocardial molecular signaling. Pleiotropic interventions will therefore be required to effectively prevent and/or treat MTT-induced cardiotoxicity. Aerobic exercise therapy has the unique capacity to modulate, without toxicity, multiple gene expression pathways in several organ systems, including a plethora of cardiac-specific molecular and cell-signaling pathways implicated in MTT-induced cardiac toxicity. Here we review molecular signaling of antiangiogenic and HER2-directed therapies that may underpin cardiac toxicity and the hypothesized cardioprotective properties of aerobic exercise.
The Biology of Tyrosine Kinases
Receptor tyrosine kinases (RTKs) are enzymes that act as critical mediators of normal cellular signal transduction and regulate diverse cellular processes including cell cycle progression, metabolism, transcription, and apoptosis (reviewed extensively elsewhere [9, 10]). All RTKs are embedded in plasma membranes and consist of an extracellular ligand-binding domain and an intracellular kinase domain. RTKs are not only key regulators of normal cellular processes, but they also are central to malignant transformation and tumor proliferation when constitutively activated via gene amplification, overexpression, or mutations [11]. Strategies for the prevention or interception of deregulated RTK signaling include the development of selective agents that target either the extracellular ligand-binding domain or the intracellular tyrosine kinase binding region [2, 4]. Monoclonal antibodies (mAbs) are designed to inhibit kinase activation by binding to the extracellular portion of RTKs or by binding to growth factor ligands that activate RTKs. Mechanistically, anti-RTK mAbs block the ligand-receptor interaction, thus inhibiting activation of the tyrosine kinase domain, and/or induce downregulation of receptor expression [12]. In contrast, small-molecule tyrosine kinase inhibitors (TKIs) bind to the intracellular portion of RTKs, thereby inhibiting the phosphorylation of downstream substrates.
Mechanisms of HER2-Directed Therapy Cardiac Injury
Overexpression and/or gene amplification of the RTK HER2 (also known as ErbB2) is present in approximately 20% of women with breast cancer [13], as well as approximately 10% and 5% of patients with non-small cell lung cancer, [14] and gastric cancer, respectively [15]. Randomized trials demonstrate that HER2-directed agents cause significant improvements in disease-free survival and overall survival among women with early [16, 17] and metastatic [18] HER2-positive breast cancer. However, trastuzumab (the first FDA-approved HER2-directed mAb) and pertuzumab (a newer mAb in phase III testing) are associated with cardiac toxicity (Table 1).
Table 1.
Incidence of cardiotoxicity in HER2-directed and angiogenesis inhibitor clinical trials
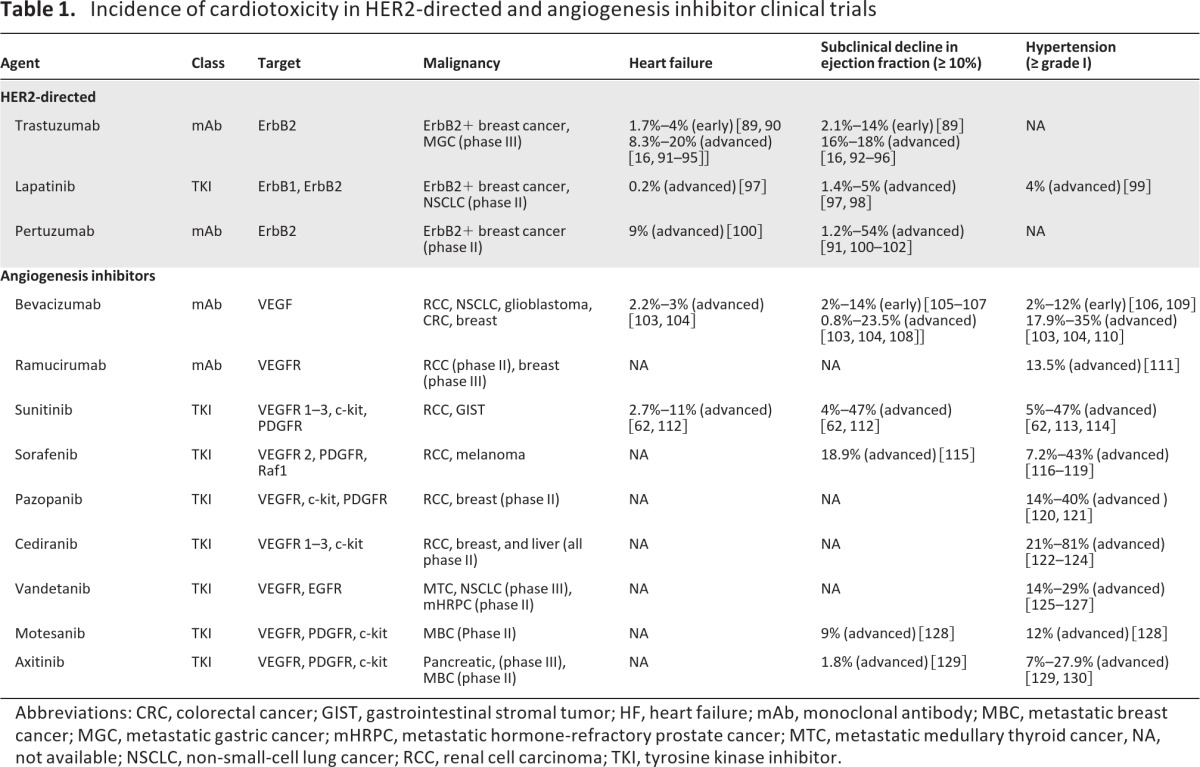
Abbreviations: CRC, colorectal cancer; GIST, gastrointestinal stromal tumor; HF, heart failure; mAb, monoclonal antibody; MBC, metastatic breast cancer; MGC, metastatic gastric cancer; mHRPC, metastatic hormone-refractory prostate cancer; MTC, metastatic medullary thyroid cancer, NA, not available; NSCLC, non-small-cell lung cancer; RCC, renal cell carcinoma; TKI, tyrosine kinase inhibitor.
HER2-Directed Therapy Inhibition of Cardiac Molecular Signaling
We focus here on the putative role of ErbB2 and transforming growth factor β (TGFβ) signaling as major pathways mediating anti-ErbB2 cardiotoxicity (Fig. 1).
Figure 1.

Mechanisms underlying HER2-directed therapy cardiotoxicity. Inhibition of ErbB receptors with HER2-directed therapies impacts numerous signaling pathways resulting in suppression of myofilament protein synthesis via the PI3K-Akt pathway (pathway A), suppression of protein hypertrophy via the MAPK pathway (pathway B), suppression of cell survival via Src/Fak pathway (pathway C), suppression of myofilament protein synthesis and upregulation of protein degradation via TGF-β1 and C/EBPβ signaling (pathway D), and alterations in cardiac energy metabolism via downregulation of AMPK (pathway E).
Abbreviations: AMPK, AMP-activated protein kinase; C/EBPβ, CCAAT/enhancer binding protein; Fak, focal adhesion kinase; MAPK, mitogen-activated protein kinase; Nrg1, Neuregulin-1β; PI3K, phosphatidylinositol 3-kinase; Smad, small mother against decapentaplegic; TGFβ, transforming growth factor β.
ErbB2
ErbB2 belongs to the family of human epidermal growth factor (EGF) receptors comprised of the EGF receptor (ErbB1), ErbB2, ErbB3, and ErbB4 [19]. Hyperactivation of the ErbB2/ErbB3/PI3K complex in tumor cells leads to upregulation of the phosphatidylinositol 3-kinase (PI3K)-Akt pathways resulting in cellular proliferation [20], increased mitogen-activated protein kinase (MAPK) activity causing protein hypertrophy [21], and reduced cytostasis via CCAAT/enhancer-binding protein (C/EBPβ) [22]. The antiproliferative activity of HER2-directed therapy is therefore based on disruption of the ErbB2-ErbB3 complex in tumor cells, thus reducing intracellular Akt activity [19, 23] and MAPK signaling [24]. Conversely, HER2-directed therapy dramatically alters cardiomyocyte function and/or survival via inhibition of Nrg1/ErbB/Akt signaling.
The essential role of ErbB2 signaling for cardiomyocyte proliferation was first revealed through germline deletion of ErbB2 receptors in mice, which proved lethal in mid gestation with failure of proper ventricle formation [25]. Transgenic mice with cardiac-specific deletion of ErbB2 after cardiac development survive embryogenesis [25] but develop progressive cardiomyopathy in adulthood [16]. In the healthy heart, neuregulin-1β (Nrg1) is released by endocardial and myocardial endothelial microvascular cells and binds to ErbB4 on cardiomyocytes leading to activation of the ErbB2/ErbB4/PI3K/Akt complex. HER2-directed agents inhibit Nrg1 release [26] and significantly decrease both total and phosphorylated Akt in neonatal rat cardiomyocytes [27], thus potentially limiting cell growth, glucose uptake, and protein regulation [28–30] and ultimately triggering the progression of heart failure [31].
TGFβ
During carcinogenesis, TGFβ signaling initially prevents malignant progression via the small mother against decapentaplegic (Smad) pathway [32]. Eventually, malignant cells overexpressing ErbB2 circumvent the growth-suppressive effects of TGFβ by downregulating TGFβ receptors or altering downstream pathways [22]. In the heart, TGFβ regulates ventricular remodeling (cardiomyocyte hypertrophy) via activation of the Smad/3 and 4 complexes and matrix metalloproteinases [33]. Inhibition of ErbB2 signaling with trastuzumab also activates TGFβ and C/EBPβ signaling in breast cancer cells [22]. Whether trastuzumab induces TGFβ and C/EBPβ signaling in cardiomyocytes is unknown; however, increased cardiac TGFβ and C/EBPβ expression results in pathological remodeling [34].
Aerobic Exercise-Induced Cardioprotection from HER2-Directed Agents
Figure 2 outlines the modulation of ErbB and TGFβ signaling through aerobic exercise. The cardioprotective properties of increased Nrg1/ErbB signaling are well described [26, 29]. For example, drug-induced enhancement of myocardial Nrg1/ErbB signaling significantly improved both cardiac performance and survival in four different rodent models of LV failure [31] and induced differentiated cardiomyocytes to proliferate [35]. In vitro studies in isolated cardiac endothelial cells (the main source of Nrg1 in the heart) show that mechanical strain increases endothelial Nrg1 synthesis and release [36], whereas Nrg1 release is directly inhibited by angiotensin II and adrenergic agonists [37]. Aerobic exercise increases endocardial mechanical strain [38] with a concomitant reduction in angiotensin II [39]. Interestingly, Lebrasseur et al. [40] found that resistance or aerobic exercise increased proteolytic processing of mature Nrg1 transmembrane protein to soluble Nrg1 in rat skeletal muscle. Thus, we contend that increased Nrg1 synthesis in the ventricle in response to exercise-induced mechanical stress will evoke suppression of neurohormonal factors, leading to cardioprotection.
Figure 2.

Mechanisms underlying modulation of HER2-directed therapy cardiotoxicity through aerobic exercise. Aerobic exercise induces cardioprotection via upregulation of Nrg1 synthesis and release, thus activating pathways A–C; inhibition of TGF-β1 and C/EBPβ signaling and upregulation of GATA4 (pathway D); and activation of AMPK expression (pathway E).
Abbreviations: AMPK, AMP-activated protein kinase; C/EBPβ, CCAAT/enhancer binding protein; Fak, focal adhesion kinase; MAPK, mitogen-activated protein kinase; Nrg1, Neuregulin-1β; PI3K, phosphatidylinositol 3-kinase; Smad, small mother against decapentaplegic; TGFβ, transforming growth factor β.
Aerobic exercise may also modulate a number of myocardial intracellular processes, thus overcoming HER2-inhibitor receptor blockade. McMullen et al. [41, 42] elegantly demonstrated that exercise increases myocardial Akt, with subsequent attenuation of pathologic LV remodeling, fibrosis, and protein degradation. Whether similar processes occur during administration of targeted cancer therapeutics has not been investigated; however, exercise training increases myocardial PI3K activity [43] with an effective reduction in infarct size and cardiomyocyte apoptosis in rats subjected to myocardial ischemia reperfusion [44], as well as improves lifespan in mice with dilated cardiomyopathy [45]. These investigations provide evidence for the protective effects of PI3K/Akt signaling in settings of targeted therapy-induced cardiac stress.
Finally, aerobic exercise inhibits TGFβ and C/EBPβ signaling to effectively prevent and/or attenuate pathological cardiac hypertrophy in various mouse models. For instance, aerobic exercise attenuates isoprenaline-induced increases in myocardial levels of TGFβ and C/EBPβ mRNA, with parallel inhibition of pathological myocardial hypertrophy [34]. Exercise-induced reduction of TGFβ and C/EBPβ expression, in turn, increases both cardiomyocyte size and cell division resulting in physiological hypertrophy [34]. Interestingly, exercise or experimental downregulation of C/EBPβ expression has also been shown to upregulate GATA4, a regulator of cardiomyocyte proliferation during myocardial regeneration in zebrafish [46]. These data indicate that aerobic exercise inhibits TGF-β1 and C/EBPβ expression and upregulates GATA4, ultimately leading to modulation of protein degradation and upregulation of protein synthesis.
In the only human study examining MTTs and exercise to date, our group found that 16 weeks of supervised aerobic training failed to attenuate trastuzumab-induced LV dilation and reduced ejection fraction in patients with HER2-positive operable breast cancer [47]. This observation is in contrast to work by us and others demonstrating that aerobic training can reverse LV remodeling in patients with stable heart failure [48, 49]. These discordant findings were likely due to the fact that participants in the trastuzumab trial attended an insufficient number of training sessions and did not receive a high enough training stimulus to achieve beneficial adaptations.
Mechanisms of Angiogenesis Inhibition Directed Therapy-Induced Cardiac Injury
Angiogenesis, the formation of new capillary blood vessels, is predominantly regulated via vascular endothelial growth factor (VEGF) and is fundamental for both physiologic and pathologic processes (reviewed extensively in references [50, 51]). VEGF inhibition, alone or in combination with conventional chemotherapy, is now approved by the FDA as first-line therapy for a broad range of advanced solid malignancies [50]. The incidence and magnitude of cardiac injury with multitargeted TKIs is particularly high, whereas treatment with monoclonal mAbs appears to cause comparably less injury (Table 1).
Antiangiogenic Therapy Inhibition of Cardiac and Vascular Molecular Signaling
Putative pathways underlying the cardiotoxic properties of monoclonal and multitargeted agents are illustrated in Figure 3.
Figure 3.

Mechanisms underlying anti-angiogenic therapy cardiotoxicity. Inhibition of VEGF signaling with tyrosine kinase inhibitor-directed therapies impacts numerous signaling pathways resulting in inhibition of angiogenesis, and protein synthesis and degradation via the PI3K-Akt-NO pathway; and inhibition of cell proliferation and differentiation via the MAPK-ERK pathway. Monoclonal inhibitors include bevacizumab and ramucirumab; multikinase inhibitors include sunitinib, axitinib, pazopanib, motesanib, vadetanib, and sorafenib.
Abbreviations: MAPK, mitogen-activated protein kinase; NO, nitric oxide; PI3K, phosphatidylinositol 3-kinase; VEGF, vascular endothelial growth factor.
Monoclonal Inhibition
Circulating VEGF binds to its receptors platelet derived growth factor receptor (PDGFR), VEGFR1 (also known as Flt-1), and VEGFR2 (also known as Flk-1 or KDR). Monoclonal therapeutic inhibition of the VEGF pathway is achieved via antibodies targeting a VEGF ligand (e.g., bevacizumab binding to VEGF), decoy receptors for VEGF (e.g., aflibercept), or antibodies targeting the extracellular domain of VEGFRs (e.g., ramucirumab binding to VEGFR2), thus limiting endothelial cell sprouting, migration, proliferation, and tube formation [52]. Of importance, suppression of VEGF/VEGFR signaling causes pathological alterations in cardiac and vascular tissues [53, 54]. For instance, global VEGF knockout in rodents causes embryonic lethality [55], whereas postnatal murine downregulation of myocardial VEGF expression initiates a cascade of events leading to progressive diastolic and systolic LV dysfunction [43]. Cardiac-specific VEGF knockout mice display abnormal cardiac muscle capillarity number [56] and classic features of cardiomyopathy (i.e., reduced cardiac output, fractional shortening, decreased dP/dt) [57]. Physiologically, cardiomyocyte binding of VEGF activates Akt, initiating signaling pathways regulating nitric oxide (NO) synthase, angiogenesis, and progenitor cell differentiation into cardiomyocytes [58–60]. Anti-VEGF agents likely inhibit vascular NO release, thus promoting vasoconstriction, increased peripheral resistance, and increased blood pressure, [53], as well as limiting endothelial progenitor cell (EPC) [59] and cardiac progenitor cell (CPC) [61] development and ultimately restraining cardiomyocyte differentiation. Unfortunately, limited clinical data and mechanistic information are available to substantiate these potential pathways.
Multitargeted Inhibition
Multitargeted TKIs may lead to a greater incidence and magnitude of cardiotoxicity due to binding of multiple proteins and/or downstream pathways (Table 1). For instance, Chu et al. [62] demonstrated that sunitinib, which targets VEGFR1–3, c-kit, and PDGFR, leads to profound structural and functional abnormalities in cardiomyocyte mitochondria, leading to a decrease in ATP production. In addition, blockade of VEGFR2 with TKIs decreases endothelial nitric oxide synthase (eNOS) activation and NO release, promoting vasoconstriction and increased blood pressure [63]. Of significance, the combined cardiac and vascular injury may enhance the severity of cardiac injury. Izumiya et al. [64] demonstrated that VEGF inhibition contributed to progression from compensatory cardiac hypertrophy to LV failure under hypertensive conditions. Chronic hypertension ultimately leads to a compensatory increase in myocardial muscle mass to maintain normal cardiac output [65]. In both hypertensive and pre-hypertensive states, there is slow but steady hypertrophy of the LV [66], leading to a decreased ability to relax initially during exercise and subsequently at rest [67]. Whether concentric remodeling occurs in patients receiving antiangiogenic therapies has not been investigated.
Aerobic Exercise-Induced Cardioprotection from Angiogenesis Inhibition
Aerobic exercise-induced increase in vascular and myocardial VEGF/VEGFR signaling may prevent/treat antiangiogenesis-induced cardiotoxicity. These conceptual pathways are illustrated in Figure 4. Upregulation of myocardial VEGF using naked DNA gene therapy enhances capillary density and decreases endothelial cell and cardiomyocyte apoptosis, leading to improvements in cardiac function in a diabetic rat model [43]. Aerobic exercise augments the aging-induced [68] and infarct-induced [69] decrease in VEGF mRNA and protein expression in murine cardiac tissue. Increased VEGF expression occurs via upstream peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) through the transcription factor estrogen-related receptor-α and is independent of hypoxia-inducible factor 1α-induced VEGF expression [70]. Aerobic exercise rapidly upregulates PGC-1α mRNA in skeletal muscle, with a concomitant increase in mitochondrial content leading to resistance to fatigue and a higher number of oxidative fibers [71]. Thus, because PGC-1α is obligatory for the exercise-induced increase in VEGF expression, it is evident that PGC-1α has a particularly prominent role in regulating training-induced VEGF expression.
Figure 4.

Mechanisms underlying anti-angiogenic therapy cardiotoxicity through aerobic exercise. Aerobic exercise induces cardioprotection via upregulation of VEGF expression; a nitric oxide-dependant increase in endothelial progenitor cells; and activation of STAT3 resulting in erythropoietin secretion and binding to cardiac progenitor cells, causing differentiation into endothelial cells.
Abbreviations: CPC, cardiac progenitor cells; MAPK, mitogen-activated protein kinase; NO, nitric oxide; PI3K, phosphatidylinositol 3-kinase; EC, endothelial cell; EPO, erythropoietin; VEGF, vascular endothelial growth factor.
An additional putative cardioprotective mechanism is exercise-induced augmentation in the production and mobilization of CPCs and EPCs via acute increases in interleukin-6 (IL-6), NO, and VEGF-dependent mechanisms [72–74]. Aerobic exercise leads to an NO-dependent increase in circulating EPCs in mice and humans [72], thus contributing to neovascularization and vascular repair, leading to improved endothelial function and myocardium recovery after ischemia [75–78]. IL-6 also plays multiple functions in angiogenesis and vascular remodeling and may stimulate EPC proliferation, migration, and tube formation following exercise [73, 79].
Aerobic exercise rapidly upregulates PGC-1α mRNA in skeletal muscle, with a concomitant increase in mitochondrial content leading to resistance to fatigue and a higher number of oxidative fibers. Thus, because PGC-1α is obligatory for the exercise-induced increase in VEGF expression, it is evident that PGC-1α has a particularly prominent role in regulating training-induced VEGF expression.
Importantly, this upregulation in EPCs and CPCs has been implicated in cardiomyocyte healing processes. Kolwicz et al. [80] found that exercise increased CPC proliferation by ∼200% and augmented the presence of KIT-positive cells (a stem cell factor crucial for the mobilization of progenitor cells to sites of injury) in the heart. Together with signal transducer and activator of transcription 3 (STAT3), this exercise-induced increase in CPC proliferation may play a key role in cardiomyocyte proliferation, differentiation, and survival [81]. STAT3 activation has been shown to mediate cardiac hypertrophy and protect cells in response to cardiomyopathy induced by ischemia or drug treatment [82–84]. Significantly, exercise increases STAT3 activation [85, 86], causing release of erythropoietin into the cardiac microenvironment that, in turn, binds to CPCs, causing differentiation into endothelial cells [87]. Presumably, these endothelial cells can then be activated by VEGF to express matrix metalloproteinases, which degrade the vascular basement membrane to form new capillary networks [88]. Collectively, we speculate that aerobic exercise may increase PGC-1α and VEGF secretion and STAT3 activation, with the resultant release, mobilization, and homing of CPCs during angiogenesis inhibitor therapy.
Conclusion
Molecularly targeted therapeutics are the future of cancer systemic therapy; they have already moved from palliative therapy for advanced solid malignancies into the setting of curative-intent treatment for early-stage disease. Cardiotoxicity is a frequent and potentially serious adverse complication of some targeted therapies, leading to a broad range of potentially life-threatening complications, therapy discontinuation, and poor quality of life. Low-cost, multitargeted interventions are therefore urgently required to mitigate these adverse consequences. Evidence reviewed here indicates that aerobic exercise is a nontoxic, pleiotropic therapy that affects diverse cardiac signaling pathways implicated in the cardiotoxicity induced by anti-HER2 and antiangiogenic therapy. It is important to stress that the current evidence base is emergent with a small number of studies; many areas of MTT-induced cardiotoxicity remain to be defined and addressed. A summary of future investigations needed to define the nature and magnitude of the cardioprotective effects of exercise in the setting of MTT is provided in Table 2. Future challenges for research investigating the effects of aerobic exercise in MTT-induced cardiac toxicity are provided in Table 3.
Table 2.
Potential future research directions to assess modulation of molecularly targeted therapeutic cardiotoxicity with aerobic exercise
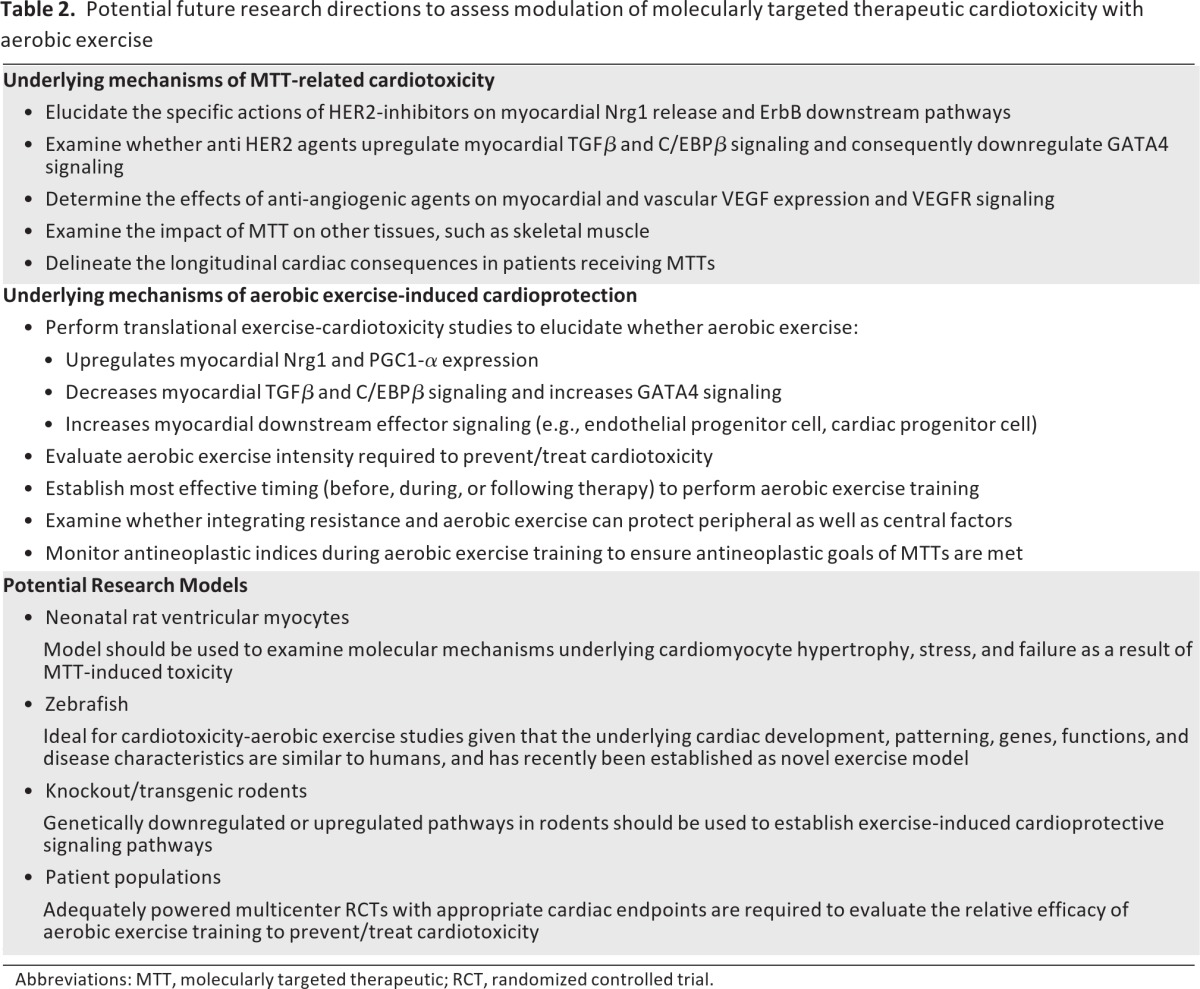
Abbreviations: MTT, molecularly targeted therapeutic; RCT, randomized controlled trial.
Table 3.
Future challenges for research investigating the effects of and mechanisms of aerobic exercise in MTT-induced cardiac toxicity

Abbreviations: MTT, molecularly targeted therapeutic.
Although clinical and research interest in aerobic exercise has increased dramatically over the past decade, a more thorough understanding of the myocardial signaling pathways activated by exercise will be needed to improve cardiovascular outcomes. Collectively, such research will lead to mechanistically driven clinical trials. Importantly, adequately powered multicenter randomized controlled trials are required to evaluate the relative efficacy of aerobic exercise training to prevent/treat cardiotoxicity. These investigations, in turn, will inform exercise prescription rehabilitation guidelines for patients with cancer, whereby the most effective exercise intensity and timing (before, during, or following therapy) are established. We anticipate that exercise will complement molecularly targeted therapeutics to improve the health and longevity of patients with solid malignancies.
Acknowledgments
This work is supported in part by grants from the National Cancer Institute (CA143254, CA142566, CA138634, CA133895, CA164751 to L.W.J.), funds from George and Susan Beischer (L.W.J.), and a Natural Sciences and Engineering Research Council postdoctoral fellowship (J.M.S.).
Author Contributions
Conception/Design: Jessica M. Scott, Lee W. Jones
Manuscript writing: Jessica M. Scott, Susan Lakoski, John R. Mackey, Pamela S. Douglas, Mark J. Haykowsky, Lee W. Jones
Final approval of manuscript: Jessica M. Scott, Susan Lakoski, John R. Mackey, Pamela S. Douglas, Mark J. Haykowsky, Lee W. Jones
Disclosures
John R. Mackey: Roche Oncology; Pamela S. Douglas: Atritech/Boston Scientific, Edwards Lifesciences (RF); CardioDX, Universal Oncology (OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Chen MH, Kerkela R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 3.Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: Incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53:2231–2247. doi: 10.1016/j.jacc.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 4.Force T, Kerkela R. Cardiotoxicity of the new cancer therapeutics: Mechanisms of, and approaches to, the problem. Drug Discov Today. 2008;13:778–784. doi: 10.1016/j.drudis.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones LW, Haykowsky MJ, Swartz JJ, et al. Early breast cancer therapy and cardiovascular injury. J Am Coll Cardiol. 2007;50:1435–1441. doi: 10.1016/j.jacc.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 6.Cardinale D, Salvatici M, Sandri MT. Role of biomarkers in cardioncology. Clin Chem Lab Med. 2011;49:1937–1948. doi: 10.1515/CCLM.2011.692. [DOI] [PubMed] [Google Scholar]
- 7.Cardinale D, Sandri MT. Role of biomarkers in chemotherapy-induced cardiotoxicity. Prog Cardiovasc Dis. 2010;53:121–129. doi: 10.1016/j.pcad.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Khouri M, Douglas P, Mackey J, et al. Cancer therapy-induced cardiac toxicity in early breast cancer: Addressing the unresolved issues. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.112.100560. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neet K, Hunter T. Vertebrate non-receptor protein-tyrosine kinase families. Genes Cells. 1996;1:147–169. doi: 10.1046/j.1365-2443.1996.d01-234.x. [DOI] [PubMed] [Google Scholar]
- 10.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 11.Kolibaba KS, Druker BJ. Protein tyrosine kinases and cancer. Biochim Biophys Acta. 1997;1333:F217–248. doi: 10.1016/s0304-419x(97)00022-x. [DOI] [PubMed] [Google Scholar]
- 12.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 13.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 14.Stephens P, Hunter C, Bignell G, et al. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 15.Lee JW, Soung YH, Seo SH, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006;12:57–61. doi: 10.1158/1078-0432.CCR-05-0976. [DOI] [PubMed] [Google Scholar]
- 16.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 17.Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365:1273–1283. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 19.Hynes NE, Lane HA. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 20.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 21.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 22.Arnal-Estape A, Tarragona M, Morales M, et al. HER2 silences tumor suppression in breast cancer cells by switching expression of C/EBPss isoforms. Cancer Res. 2010;70:9927–9936. doi: 10.1158/0008-5472.CAN-10-0869. [DOI] [PubMed] [Google Scholar]
- 23.Mohsin SK, Weiss HL, Gutierrez MC, et al. Neoadjuvant trastuzumab induces apoptosis in primary breast cancers. J Clin Oncol. 2005;23:2460–2468. doi: 10.1200/JCO.2005.00.661. [DOI] [PubMed] [Google Scholar]
- 24.Dave B, Migliaccio I, Gutierrez MC, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. 2011;29:166–173. doi: 10.1200/JCO.2009.27.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crone SA, Zhao YY, Fan L, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 26.Ky B, Kimmel SE, Safa RN, et al. Neuregulin-1 beta is associated with disease severity and adverse outcomes in chronic heart failure. Circulation. 2009;120:310–317. doi: 10.1161/CIRCULATIONAHA.109.856310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon LI, Burke MA, Singh AT, et al. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J Biol Chem. 2009;284:2080–2087. doi: 10.1074/jbc.M804570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brero A, Ramella R, Fitou A, et al. Neuregulin-1beta1 rapidly modulates nitric oxide synthesis and calcium handling in rat cardiomyocytes. Cardiovasc Res. 88:443–452. doi: 10.1093/cvr/cvq238. [DOI] [PubMed] [Google Scholar]
- 29.Pentassuglia L, Sawyer DB. The role of Neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res. 2009;315:627–637. doi: 10.1016/j.yexcr.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuramochi Y, Guo X, Sawyer DB. Neuregulin activates erbB2-dependent src/FAK signaling and cytoskeletal remodeling in isolated adult rat cardiac myocytes. J Mol Cell Cardiol. 2006;41:228–235. doi: 10.1016/j.yjmcc.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Gu X, Li Z, et al. Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J Am Coll Cardiol. 2006;48:1438–1447. doi: 10.1016/j.jacc.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 32.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 33.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bostrom P, Mann N, Wu J, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 36.Lemmens K, Doggen K, De Keulenaer GW. Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease: Implications for therapy of heart failure. Circulation. 2007;116:954–960. doi: 10.1161/CIRCULATIONAHA.107.690487. [DOI] [PubMed] [Google Scholar]
- 37.Lemmens K, Segers VF, Demolder M, De Keulenaer GW. Role of neuregulin-1/ErbB2 signaling in endothelium-cardiomyocyte cross-talk. J Biol Chem. 2006;281:19469–19477. doi: 10.1074/jbc.M600399200. [DOI] [PubMed] [Google Scholar]
- 38.Esch BT, Scott JM, Warburton DE, et al. Left ventricular torsion and untwisting during exercise in heart transplant recipients. J Physiol. 2009;587:2375–2386. doi: 10.1113/jphysiol.2009.170100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandes T, Hashimoto NY, Magalhaes FC, et al. Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1–7) Hypertension. 2011;58:182–189. doi: 10.1161/HYPERTENSIONAHA.110.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebrasseur NK, Cote GM, Miller TA, et al. Regulation of neuregulin/ErbB signaling by contractile activity in skeletal muscle. Am J Physiol Cell Physiol. 2003;284:C1149–1155. doi: 10.1152/ajpcell.00487.2002. [DOI] [PubMed] [Google Scholar]
- 41.McMullen JR, Shioi T, Zhang L, et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 43.Yoon YS, Uchida S, Masuo O, et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: Restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073–2085. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]
- 44.Zhang KR, Liu HT, Zhang HF, et al. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis. 2007;12:1579–1588. doi: 10.1007/s10495-007-0090-8. [DOI] [PubMed] [Google Scholar]
- 45.McMullen JR, Amirahmadi F, Woodcock EA, et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kikuchi K, Holdway JE, Werdich AA, et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–605. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haykowsky MJ, Mackey JR, Thompson RB, et al. Adjuvant trastuzumab induces ventricular remodeling despite aerobic exercise training. Clin Cancer Res. 2009;15:4963–4967. doi: 10.1158/1078-0432.CCR-09-0628. [DOI] [PubMed] [Google Scholar]
- 48.Haykowsky MJ, Liang Y, Pechter D, et al. A meta-analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: The benefit depends on the type of training performed. J Am Coll Cardiol. 2007;49:2329–2336. doi: 10.1016/j.jacc.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 49.Wisloff U, Stoylen A, Loennechen JP, et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: A randomized study. Circulation. 2007;115:3086–3094. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- 50.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 51.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munoz-Chapuli R, Quesada AR, Angel Medina M. Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci. 2004;61:2224–2243. doi: 10.1007/s00018-004-4070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D'Adamo DR, Anderson SE, Albritton K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J Clin Oncol. 2005;23:7135–7142. doi: 10.1200/JCO.2005.16.139. [DOI] [PubMed] [Google Scholar]
- 54.Choueiri TK, Mayer EL, Je Y, et al. Congestive heart failure risk in patients with breast cancer treated with bevacizumab. J Clin Oncol. 29:632–638. doi: 10.1200/JCO.2010.31.9129. [DOI] [PubMed] [Google Scholar]
- 55.Gerber HP, Hillan KJ, Ryan AM, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 56.Olfert IM, Howlett RA, Tang K, et al. Muscle-specific VEGF deficiency greatly reduces exercise endurance in mice. J Physiol. 2009;587:1755–1767. doi: 10.1113/jphysiol.2008.164384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giordano FJ, Gerber HP, Williams SP, et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A. 2001;98:5780–5785. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lambrechts D, Carmeliet P. Genetics in zebrafish, mice, and humans to dissect congenital heart disease: Insights in the role of VEGF. Curr Top Dev Biol. 2004;62:189–224. doi: 10.1016/S0070-2153(04)62007-2. [DOI] [PubMed] [Google Scholar]
- 59.Chen K, Bai H, Arzigian M, et al. Endothelial cells regulate cardiomyocyte development from embryonic stem cells. J Cell Biochem. 2010;111:29–39. doi: 10.1002/jcb.22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Amende I, Hampton TG, et al. Vascular endothelial growth factor promotes cardiomyocyte differentiation of embryonic stem cells. Am J Physiol Heart Circ Physiol. 2006;291:H1653–1658. doi: 10.1152/ajpheart.00363.2005. [DOI] [PubMed] [Google Scholar]
- 62.Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Facemire CS, Nixon AB, Griffiths R, et al. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Izumiya Y, Shiojima I, Sato K, et al. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain M, Townsend RR. Chemotherapy agents and hypertension: A focus on angiogenesis blockade. Curr Hypertens Rep. 2007;9:320–328. doi: 10.1007/s11906-007-0058-7. [DOI] [PubMed] [Google Scholar]
- 66.Levy D. Left ventricular hypertrophy. Epidemiological insights from the Framingham Heart Study. Drugs. 1988;35(suppl 5):1–5. doi: 10.2165/00003495-198800355-00002. [DOI] [PubMed] [Google Scholar]
- 67.Brucks S, Little WC, Chao T, et al. Contribution of left ventricular diastolic dysfunction to heart failure regardless of ejection fraction. Am J Cardiol. 2005;95:603–606. doi: 10.1016/j.amjcard.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 68.Iemitsu M, Maeda S, Jesmin S, et al. Exercise training improves aging-induced downregulation of VEGF angiogenic signaling cascade in hearts. Am J Physiol Heart Circ Physiol. 2006;291:H1290–1298. doi: 10.1152/ajpheart.00820.2005. [DOI] [PubMed] [Google Scholar]
- 69.Wu G, Rana JS, Wykrzykowska J, et al. Exercise-induced expression of VEGF and salvation of myocardium in the early stage of myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H389–395. doi: 10.1152/ajpheart.01393.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 71.Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 72.Laufs U, Werner N, Link A, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation. 2004;109:220–226. doi: 10.1161/01.CIR.0000109141.48980.37. [DOI] [PubMed] [Google Scholar]
- 73.Mobius-Winkler S, Hilberg T, Menzel K, et al. Time-dependent mobilization of circulating progenitor cells during strenuous exercise in healthy individuals. J Appl Physiol. 2009;107:1943–1950. doi: 10.1152/japplphysiol.00532.2009. [DOI] [PubMed] [Google Scholar]
- 74.Schobersberger W, Hobisch-Hagen P, Fries D, et al. Increase in immune activation, vascular endothelial growth factor and erythropoietin after an ultramarathon run at moderate altitude. Immunobiology. 2000;201:611–620. doi: 10.1016/S0171-2985(00)80078-9. [DOI] [PubMed] [Google Scholar]
- 75.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 76.Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Urbich C, Heeschen C, Aicher A, et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat Med. 2005;11:206–213. doi: 10.1038/nm1182. [DOI] [PubMed] [Google Scholar]
- 78.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 79.Fan Y, Ye J, Shen F, et al. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab. 2008;28:90–98. doi: 10.1038/sj.jcbfm.9600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolwicz SC, MacDonnell SM, Kendrick ZV, et al. Voluntary wheel running and pacing-induced dysfunction in hypertension. Clin Exp Hypertens. 2008;30:565–573. doi: 10.1080/10641960802251891. [DOI] [PubMed] [Google Scholar]
- 81.Cohen T, Nahari D, Cerem LW, et al. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996;271:736–741. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- 82.Kunisada K, Hirota H, Fujio Y, et al. Activation of JAK-STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation. 1996;94:2626–2632. doi: 10.1161/01.cir.94.10.2626. [DOI] [PubMed] [Google Scholar]
- 83.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 84.Kunisada K, Negoro S, Tone E, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:315–319. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trenerry MK, Carey KA, Ward AC, Cameron-Smith D. STAT3 signaling is activated in human skeletal muscle following acute resistance exercise. J Appl Physiol. 2007;102:1483–1489. doi: 10.1152/japplphysiol.01147.2006. [DOI] [PubMed] [Google Scholar]
- 86.Toth KG, McKay BR, De Lisio M, et al. IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS One. 2011;6:e17392. doi: 10.1371/journal.pone.0017392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoch M, Fischer P, Stapel B, et al. Erythropoietin preserves the endothelial differentiation capacity of cardiac progenitor cells and reduces heart failure during anticancer therapies. Cell Stem Cell. 2011;9:131–143. doi: 10.1016/j.stem.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 88.Westenbrink BD, Ruifrok WP, Voors AA, et al. Vascular endothelial growth factor is crucial for erythropoietin-induced improvement of cardiac function in heart failure. Cardiovasc Res. 2010;87:30–39. doi: 10.1093/cvr/cvq041. [DOI] [PubMed] [Google Scholar]
- 89.Tan-Chiu E, Yothers G, Romond E, et al. Assessment of cardiac dysfunction in a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel, with or without trastuzumab as adjuvant therapy in node-positive, human epidermal growth factor receptor 2-overexpressing breast cancer: NSABP B-31. J Clin Oncol. 2005;23:7811–7819. doi: 10.1200/JCO.2005.02.4091. [DOI] [PubMed] [Google Scholar]
- 90.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 91.Baselga J, Cortés J, Kim S, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 93.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 94.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 95.Smith KL, Dang C, Seidman AD. Cardiac dysfunction associated with trastuzumab. Expert Opin Drug Saf. 2006;5:619–629. doi: 10.1517/14740338.5.5.619. [DOI] [PubMed] [Google Scholar]
- 96.Baselga J, Carbonell X, Castaneda-Soto NJ, et al. Phase II study of efficacy, safety, and pharmacokinetics of trastuzumab monotherapy administered on a 3-weekly schedule. J Clin Oncol. 2005;23:2162–2171. doi: 10.1200/JCO.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 97.Perez EA, Koehler M, Byrne J, et al. Cardiac safety of lapatinib: Pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin Proc. 2008;83:679–686. doi: 10.4065/83.6.679. [DOI] [PubMed] [Google Scholar]
- 98.Ross HJ, Blumenschein GR, Jr., Aisner J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. 2010;16:1938–1949. doi: 10.1158/1078-0432.CCR-08-3328. [DOI] [PubMed] [Google Scholar]
- 99.Slamon D, Gomez HL, Kabbinavar FF, et al. Randomized study of pazopanib + lapatinib vs. lapatinib alone in patients with HER2-positive advanced or metastatic breast cancer. J Clin Oncol. 2008;26(15 suppl):1016. [Google Scholar]
- 100.Portera CC, Walshe JM, Rosing DR, et al. Cardiac toxicity and efficacy of trastuzumab combined with pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. Clin Cancer Res. 2008;14:2710–2716. doi: 10.1158/1078-0432.CCR-07-4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baselga J, Gelmon KA, Verma S, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138–1144. doi: 10.1200/JCO.2009.24.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lenihan D, Suter T, Brammer M, et al. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Ann Oncol. 2012;23:791–800. doi: 10.1093/annonc/mdr294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 104.Miller KD, Chap LI, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–799. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 105.Miller K, O'Neill A, Perez E, et al. Phase II feasibility trial incorporating bevacizumab into dose-dense doxorubicin and cyclophosphamide followed by paclitaxel in patients with lymph node-positive breast cancer: a trial of the Eastern Cooperative Oncology Group (E2104) Ann Oncol. 2012;23:331–337. doi: 10.1093/annonc/mdr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bear HD, Tang G, Rastogi P, et al. The effect on pCR of bevacizumab and/or antimetabolites added to standard neoadjuvant chemotherapy: NSABP protocol B-40. J Clin Oncol. 2011;29(15 suppl):1005. [Google Scholar]
- 107.Yardley DA, Hart L, Waterhouse DM, et al. Preliminary safety results: Addition of bevacizumab to 3 docetaxel regimens as adjuvant therapy for early stage breast cancer. Cancer Res. 2009;69(suppl 2) Abstract 4107. [Google Scholar]
- 108.Robert NJ, Dieras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252–1260. doi: 10.1200/JCO.2010.28.0982. [DOI] [PubMed] [Google Scholar]
- 109.Allegra CJ, Yothers G, O'Connell MJ, et al. Initial safety report of NSABP C-08: A randomized phase III study of modified FOLFOX6 with or without bevacizumab for the adjuvant treatment of patients with stage II or III colon cancer. J Clin Oncol. 2009;27:3385–3390. doi: 10.1200/JCO.2009.21.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pande A, Lombardo J, Spangenthal E, Javle M. Hypertension secondary to anti-angiogenic therapy: Experience with bevacizumab. Anticancer Res. 2007;27:3465–3470. [PubMed] [Google Scholar]
- 111.Spratlin JL, Cohen RB, Eadens M, et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010;28:780–787. doi: 10.1200/JCO.2009.23.7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Khakoo AY, Kassiotis CM, Tannir N, et al. Heart failure associated with sunitinib malate: A multitargeted receptor tyrosine kinase inhibitor. Cancer. 2008;112:2500–2508. doi: 10.1002/cncr.23460. [DOI] [PubMed] [Google Scholar]
- 113.Barrios CH, Liu MC, Lee SC, et al. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat. 2010;121:121–131. doi: 10.1007/s10549-010-0788-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bergh J, Greil R, Voytko N, et al. Sunitinib (SU) in combination with docetaxel (D) versus D alone for the first-line treatment of advanced breast cancer (ABC) J Clin Oncol. 2010;28(18 suppl):1010. [Google Scholar]
- 115.Schmidinger M, Zielinski CC, Vogl UM, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26:5204–5212. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 116.Vaklavas C, Lenihan D, Kurzrock R, Tsimberidou AM. Anti-vascular endothelial growth factor therapies and cardiovascular toxicity: What are the important clinical markers to target? The Oncologist. 2010;15:130–141. doi: 10.1634/theoncologist.2009-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 118.Ratain MJ, Eisen T, Stadler WM, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:2505–2512. doi: 10.1200/JCO.2005.03.6723. [DOI] [PubMed] [Google Scholar]
- 119.Wu S, Chen JJ, Kudelka A, et al. Incidence and risk of hypertension with sorafenib in patients with cancer: A systematic review and meta-analysis. Lancet Oncol. 2008;9:117–123. doi: 10.1016/S1470-2045(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 120.Taylor SK, Chia S, Dent S, et al. A phase II study of pazopanib in patients with recurrent or metastatic invasive breast carcinoma: A trial of the Princess Margaret Hospital phase II consortium. The Oncologist. 2010;15:810–818. doi: 10.1634/theoncologist.2010-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 122.Alberts SR, Fitch TR, Kim GP, et al. Cediranib (AZD2171) in patients with advanced hepatocellular carcinoma: A phase II North Central Cancer Treatment Group Clinical Trial. Am J Clin Oncol. 2012;35:329–333. doi: 10.1097/COC.0b013e3182118cdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hyams DM, de Oliveira C, Snyder R, et al. Cediranib in combination with fulvestrant in hormone-sensitive metastatic breast cancer: A phase II randomized study. Cancer Res. 2009;69(suppl 24) doi: 10.1007/s10637-013-9991-2. Abstract 204. [DOI] [PubMed] [Google Scholar]
- 125.Natale RB, Thongprasert S, Greco FA, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:1059–1066. doi: 10.1200/JCO.2010.28.5981. [DOI] [PubMed] [Google Scholar]
- 126.Horti J, Widmark A, Stenzl A, et al. A randomized, double-blind, placebo-controlled phase II study of vandetanib plus docetaxel/prednisolone in patients with hormone-refractory prostate cancer. Cancer Biother Radiopharm. 2009;24:175–180. doi: 10.1089/cbr.2008.0588. [DOI] [PubMed] [Google Scholar]
- 127.de Boer RH, Arrieta O, Yang CH, et al. Vandetanib plus pemetrexed for the second-line treatment of advanced non-small-cell lung cancer: A randomized, double-blind phase III trial. J Clin Oncol. 2011;29:1067–1074. doi: 10.1200/JCO.2010.29.5717. [DOI] [PubMed] [Google Scholar]
- 128.Martin M, Roche H, Pinter T, et al. Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: A phase 2, randomised, double-blind, placebo-controlled study. Lancet Oncol. 2011;12:369–376. doi: 10.1016/S1470-2045(11)70037-7. [DOI] [PubMed] [Google Scholar]
- 129.Rugo HS, Stopeck AT, Joy AA, et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29:2459–2465. doi: 10.1200/JCO.2010.31.2975. [DOI] [PubMed] [Google Scholar]
- 130.Kindler HL, Ioka T, Richel DJ, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomised phase 3 study. Lancet Oncol. 2011;12:256–262. doi: 10.1016/S1470-2045(11)70004-3. [DOI] [PubMed] [Google Scholar]
- 131.Koch LG, Britton SL, Barbato JC, et al. Phenotypic differences in cardiovascular regulation in inbred rat models of aerobic capacity. Physiol Genomics. 1999;1:63–69. doi: 10.1152/physiolgenomics.1999.1.2.63. [DOI] [PubMed] [Google Scholar]
- 132.Barbato JC, Koch LG, Darvish A, et al. Spectrum of aerobic endurance running performance in eleven inbred strains of rats. J Appl Physiol. 1998;85:530–536. doi: 10.1152/jappl.1998.85.2.530. [DOI] [PubMed] [Google Scholar]
- 133.Konhilas JP, Maass AH, Luckey SW, et al. Sex modifies exercise and cardiac adaptation in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2768–2776. doi: 10.1152/ajpheart.00292.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kemi OJ, Loennechen JP, Wisloff U, Ellingsen O. Intensity-controlled treadmill running in mice: Cardiac and skeletal muscle hypertrophy. J Appl Physiol. 2002;93:1301–1309. doi: 10.1152/japplphysiol.00231.2002. [DOI] [PubMed] [Google Scholar]
- 135.Wisloff U, Helgerud J, Kemi OJ, Ellingsen O. Intensity-controlled treadmill running in rats: VO(2 max) and cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2001;280:H1301–1310. doi: 10.1152/ajpheart.2001.280.3.H1301. [DOI] [PubMed] [Google Scholar]
- 136.Hooning MJ, Botma A, Aleman BM, et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst. 2007;99:365–375. doi: 10.1093/jnci/djk064. [DOI] [PubMed] [Google Scholar]
- 137.Wang Y, Wisloff U, Kemi OJ. Animal models in the study of exercise-induced cardiac hypertrophy. Physiol Res. 2010;59:633–644. doi: 10.33549/physiolres.931928. [DOI] [PubMed] [Google Scholar]
- 138.Scott JM, Khakoo A, Mackey JR, et al. Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer: Current evidence and underlying mechanisms. Circulation. 2011;124:642–650. doi: 10.1161/CIRCULATIONAHA.111.021774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brunello A, Roma A, Falci C, Basso U. Chemotherapy and targeted agents for elderly women with advanced breast cancer. Recent Pat Anticancer Drug Discov. 2008;3:187–201. doi: 10.2174/157489208786242313. [DOI] [PubMed] [Google Scholar]
- 140.Jones LW, Liang Y, Pituskin EN, et al. Effect of exercise training on peak oxygen consumption in patients with cancer: A meta-analysis. The Oncologist. 2011;16:112–120. doi: 10.1634/theoncologist.2010-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Betof AS, Dewhirst MW, Jones LW. Effects and potential mechanisms of exercise training on cancer progression: A translational perspective. Brain Behav Immun. 2012 doi: 10.1016/j.bbi.2012.05.001. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]